

Abstract
Bacteriophage display of antibodies provides a method for the generation of immunological reagents against rare and uncharacterized antigens. To ascertain the usefulness of this approach for the characterization of inner-ear proteins, we produced a bacteriophage-displayed antibody-fragment library directed against proteins from the bullfrog's sacculus. This library was probed for bacteriophage that bound to proteins present in a lysate of hair cells, the sensory receptors of the inner ear. The predominant bacteriophage clone after selection expressed an antibody fragment that recognized a single protein in the inner ear. This antigen occurred in both the nonsensory and sensory epithelia of the sacculus. The specificity of the antibody fragment indicates that our bacteriophage-displayed library provides a useful source of immunological tools that should facilitate the identification and biochemical characterization of novel proteins in the inner ear.
Precisely arranged within the inner ear, hair cells detect sounds and both linear and angular accelerations. These epithelial sensory receptors transduce mechanical stimuli into electrical signals; deflection of each hair cell's mechanosensitive organelle, the hair bundle, culminates in the depolarization of the plasma membrane and neurotransmitter release. Hair cells are few in number, with a total of only ≈160,000 in each human ear. In the adult bullfrog's sacculus, a receptor organ for ground-borne vibration and low-frequency sound (1, 2), 2,000–3,000 hair cells are present, each of which is encircled by approximately six supporting cells (3). This mosaic of hair and supporting cells constitutes the saccular sensory epithelium, or macula, which is surrounded by a nonsensory region, or extramacular epithelium.
Although the biophysical properties of saccular hair cells have been studied in detail (reviewed in ref. 4), a corresponding molecular analysis of hair-cell proteins remains in its infancy. The principal impediment to biochemical and molecular-biological studies is the paucity of starting material that can be obtained from the inner ear (5). Cell lines with some supporting-cell characteristics have been reported, but no immortalized cells faithfully represent hair cells (6–8). Because access to hair-cell proteins therefore depends on the isolation of the few thousands of hair cells found in each ear, conventional biochemical approaches for the identification of novel hair-cell proteins are virtually impossible without the availability of highly specific reagents.
An immunological approach provides tools for characterizing proteins even when nothing is known a priori about their properties. Because a relatively large amount of purified antigen is necessary to elicit an immune response and to screen the resulting hybridomas, however, the limited quantity of inner-ear protein makes the production of conventional mAbs unattractive (9). Recombinant-antibody technology provides alternative methods for the generation of immunological reagents, including libraries of antibody fragments expressed by filamentous bacteriophage. The principal advantage of this methodology is that each bacteriophage both displays multiple copies of a single antibody fragment on its surface and contains the gene encoding that antibody fragment. This linkage permits the selective enrichment, from a primary library of high complexity, of bacteriophage based on their ability to bind antigen (reviewed in refs. 10–12). In addition, the generation, selection, and screening of antibody fragments in principle require far less antigen than that necessary for conventional mAb production.
Although prior immunization enhances the probability of acquiring immunological reagents that recognize a particular antigen, the absolute requirement for an immune response is obviated by the generation during library construction of antibody fragments that are not expressed in vivo and that recognize new epitopes (13). This feature should permit the isolation of immunological reagents when minimal antigen is available for immunization, when it is necessary to immunize with a heterogenous complex of proteins, when proteins are highly conserved between species, or when antibodies must be isolated against uncharacterized proteins. All of these features commend the recombinant method for the study of inner-ear proteins. To isolate tools useful in the study of the inner ear, we therefore chose to produce a bacteriophage-displayed expression library of antibody fragments directed against inner-ear proteins from the bullfrog's sacculus.
Materials and Methods
Immunization.
Thirty bullfrog (Rana catesbeiana) saccular maculae were isolated, homogenized in adjuvant (Ribi Immunochem), and injected s.c. near the ankle of a BALB/c mouse. After repetition of the procedure 3 days later, the immune response was augmented every 2–3 days by the injection without adjuvant of enzymatically isolated hair cells (14) from eight bullfrog sacculi. Two days after the fifth hair-cell injection, the mouse was sacrificed, and the spleen and popliteal lymph nodes were harvested and frozen.
Ig Variable-Domain PCR Amplification.
From a mixture of the popliteal lymph nodes and one-third of the spleen, total RNA was isolated by acid/phenol extraction followed by isopycnic centrifugation through a cesium chloride gradient (15). The reverse-transcription reaction used 100 μg of total RNA and 200 pmol each of degenerate primers to Ig heavy-chain variable domains and κ light-chain variable domains in a volume of 400 μl (Superscript Preamplification System, Life Technologies, Gaithersburg, MD). To reverse-transcribe the Ig heavy-chain variable domains (16), we used the primer VHIFor2 (TGAGGAGACGGTGACCGTGGTCCCTTGGCCCC; all primers are given in the 5′ → 3′ orientation). Two primers were used to reverse-transcribe the light-chain variable domains, Vk4ForI (CCGTTTTATTTCCARCTTKGTCCC) and Vk4ForII (CCGTTTSAKYTCCAGCTTGGTSCC).
Ig heavy- and light-chain variable-domain repertoires were amplified by the PCR (Fig. 1A). Amplification of the heavy-chain variable domains used the primers VHIFor2 and VHIBack (AGGTSMARCTGCAGSAGTCWGG). Amplification of light-chain variable domains used either the Vk4ForI or the Vk4ForII primer and the primer VkNewBack (GACATTGWGMTSACMCAGTCT). Each PCR was performed with 1.5 μl of product from the reverse-transcription reaction, 50 pmol of each primer, and 5 units of Taq polymerase (Promega) in a reaction volume of 50 μl; the reaction was cycled 23 times at 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min. A total of 17 reactions were performed to amplify the heavy-chain variable-domain repertoire. After we had conducted 11 PCRs each with the Vk4ForI primer and the VK4ForII primer, the PCR products were combined to create a single light-chain variable-domain repertoire.
Figure 1.

Construction of the scFv insert and assessment of library diversity. (A) Lymphoid RNA, isolated from a mouse immunized with bullfrog sacculi and isolated hair cells, was reverse-transcribed and used as a template for PCR amplification of Ig heavy-chain (VH) and κ light-chain (VL) variable-domain repertoires. Heavy-chain primer VHIBack annealed to the 5′ end of the variable region (V) and VHIFor2 to the 3′ end of the joining region (J) of Ig heavy-chain cDNAs, resulting in products encoding the variable region, diversity region (D), and joining region of Ig heavy-chain proteins. Degenerate light-chain primers, VkNewBack and Vk4ForI or Vk4ForII, likewise annealed to kappa cDNAs, generating products encoding light-chain variable and joining regions. PCR products were gel-purified and randomly combined through an oligonucleotide encoding the (Gly4Ser)3 linker by splicing-by-overlap extension. Finally, restriction-endonuclease sites were appended to the assembled construct by the PCR and the purified product was ligated into an expression vector for bacteriophage display. This panel is based on an illustration in ref. 21. (B) Ig light-chain and heavy-chain variable domains were PCR-amplified from RNA present in the popliteal lymph nodes and spleen of a mouse immunized with bullfrog sacculi and isolated hair cells. Two pairs of primers were used to amplify the light-chain variable-domain repertoire; the amplified products generated by the VkNewBack and Vk4ForI primers are denoted LCI and the products generated by the VkNewBack and Vk4ForII primers are denoted LCII. The heavy-chain repertoire was amplified using the primers VHIBack and VHIFor2; the products are designated HC. (C) scFv fragments were constructed using PCR-sewing techniques to randomly join heavy-chain and light-chain variable domains. The sewn products of two reactions are indicated by an arrow. (D) Library diversity was ascertained by selecting bacterial colonies at random and examining their scFv insert sequences by PCR amplification and digestion with BstNI restriction endonuclease. Each lane contains the digestion products from the insert of a single bacterial colony; 17 representative clones are depicted here. This analysis indicates that the starting library was diverse, with no observed replication of digestion patterns. The lanes denoted M in B–D contain size markers of 1,114, 900, 692, 500, 404, 320, 242, 190, 147, 124, and 110 bp.
Assembly of Single-Chain Antibody-Fragment Constructs.
To obtain efficient PCR sewing of the heavy- and light-chain variable domains, we found it essential to use Pfu polymerase (Stratagene) to render the amplified products blunt-ended. To assemble the single-chain variable-region antibody-fragment (scFv) construct (Fig. 2A), we subjected equimolar amounts of the gel-purified light- and heavy-chain variable-domain repertoires and of linker DNA encoding (Gly4Ser)3 (Amersham Pharmacia) to 16 cycles at 94°C for 1 min, 63°C for 1 min, and 72°C for 2 min in the presence of 1 mM dNTPs and Taq polymerase (Promega). Because the sequences at the ends of the linker DNA were complementary to those of the heavy-chain and light-chain products, the three fragments were assembled into a single product in this sewing step. The product was further amplified, and restriction endonuclease sites were introduced by the addition to the reaction mixture of supplemental deoxynucleotides, Taq polymerase, 0.5 μM each primers NotIVK4ForI (TTCTCGACTTGCGGCCGCCCGTTTTATTTCCARCTTKGTCCC) and NotIVK4ForII (TTCTCGACTTGCGGCCGCCCGTTTSAKYTCCAGCTTGGTSCC) and 1 μM primer SfiIVH1Back (CGCAACTGCGGCCCAGCCGGCCATGGCCCAGGTSMARCTGCAGSAGTCWGG). The reaction mixture was cycled 26 times to 94°C for 1 min, 63°C for 1 min, and 72°C for 2 min. Excess primers were removed from the assembled products (QiaQuick, Qiagen, Chatsworth, CA) before digestion with NotI and SfiI restriction endonucleases. The digested products were gel-purified and ≈150 ng of insert was ligated into the pCANTAB 5E expression vector (Amersham Pharmacia) and electroporated into the Escherichia coli supE strain TG1. The resultant library was amplified in soft agarose (17) and stored as glycerol stocks.
Figure 2.

Selection from the scFv-bearing bacteriophage library. (A) The library of bacteriophage-displayed scFvs was propagated and scFv expression was induced. For each round of selection, 1011-1012 bacteriophage were placed in a selection tube whose surface had been coated with a hair-cell lysate. After incubation, extensive washing removed unbound bacteriophage. Bound bacteriophage were eluted and used to infect bacteria for propagation and expression of the enriched clones. The eluted bacteriophage subsequently were used in the next round of selection. For simplicity, we do not depict the preincubation step for removal of bacteriophage that bound to plastic or blocking reagent. (B) BstNI digestion patterns of 24 randomly selected clones indicate that a single clone, indicated by *, was enriched as a result of the antibody fragment that it expressed. Only those clones that had inserts amplifiable by the PCR are shown. Size markers are as indicated in Fig. 1.
Expression of Bacteriophage-Displayed Antibody Fragments.
To express single-chain antibody fragments as gene III fusion proteins, we used 4 × 108 cells, initially from the original library and subsequently from prior rounds of selection, to inoculate a 100-ml culture of 2× YT medium (Bio 101) containing 150 mg⋅l−1 carbenicillin and 2% (wt/vol) glucose. On reaching the logarithmic growth phase, the cells were harvested by centrifugation, washed in 2× YT medium without glucose, and resedimented. Cell pellets were resuspended in 200 ml of 2× YT medium with 150 μg/ml carbenicillin and infected with 1.5 × 1011 VCSM helper bacteriophage (New England Biolabs) for 1 hr at 37°C without shaking followed by 1 hr at 37°C with shaking. After infection, the culture was supplemented with kanamycin to 50 mg⋅l−1 and isopropylthio-β-d-galactoside to 0.4 mM. The culture was grown for an additional 16–20 hr at 30°C with shaking at 250 rpm. Bacteriophage were harvested from the medium by two successive precipitations with a solution of 20% (wt/vol) polyethylene glycol-8000 and 2.5 M NaCl. The final bacteriophage precipitate was resuspended in sterile PBS (ECL-PBS; 100 mM NaCl, 80 mM Na2HPO4, and 20 mM NaH2PO4 at pH 7.5) and titered by infection of TG1 cells.
Bacteriophage Selection.
For each round of selection, hair cells were enzymatically isolated from ≈40 bullfrog sacculi by a slight modification of a published method (14): protease treatment used 25 mg⋅liter−1 subtilisin Carlsberg (protease XXIV, Sigma) and DNAse I digestion was omitted. Isolated hair cells were collected and sonicated in ECL-PBS containing a mixture of protease inhibitors (Complete, Boehringer Mannheim) at 25-fold the recommended concentration, as well as 40 μM 4-(2 aminoethyl)-benzenesulfonyl fluoride (Pefabloc SC, Boehringer Mannheim), 0.7 μg/ml pepstatin (Boehringer Mannheim), and 0.5 mg⋅liter−1 leupeptin (Boehringer Mannheim). Particulate matter was removed by centrifugation at 16,000 × g for 15 min at 4°C. The supernatant was used to coat a selection tube (Maxisorb Startube, Nalge Nunc) for at least 18 hr at room temperature. An identical tube was incubated with ECL-PBS and protease inhibitors alone to serve as a preincubation tube. After coating, the tubes were rinsed with ECL-PBS and blocked for 2 hr with ECL-PBS containing 2% (wt/vol) nonfat dry milk powder (Carnation, Nestlé Food Company, Glendale, CA).
Meanwhile, 1011-1012 bacteriophage were suspended in 250 μl of an ECL-PBS solution containing 2% (wt/vol) nonfat dry milk powder and 0.1% (vol/vol) polyoxyethylene sorbitan monolaurate (Tween-20). To remove bacteriophage that bound to nonfat dry milk or plastic, the suspension was first incubated in the preincubation tube for 2 hr. The suspension then was added to the selection tube and incubated with gentle rotation for at least 4 hr at room temperature. The selection tube was washed 20 times with ECL-PBS containing 0.1% (vol/vol) Tween-20 and 20 times with ECL-PBS alone. After bound bacteriophage had been eluted for 10 min by the addition of 72 mM triethylamine at pH 11.75, the solution was neutralized with 1 M Tris⋅HCl at pH 7.4. Eluted bacteriophage were used to infect logarithmically growing TG1 cells. After elution, TG1 cells also were added directly to the selection tube to recover any high-affinity binders still attached to the target antigen. After each round, infected TG1 cells were amplified in soft agarose, titered, and frozen as glycerol stocks for use as the inoculum for subsequent selection rounds.
Analysis of scFv Inserts.
To ascertain the diversity of the original library and of clones after rounds of selection, we randomly picked infected TG1 colonies and amplified their scFv inserts with the PCR primers and conditions specified for library construction. To obtain a DNA fingerprint of the insert sequences (18), we digested the products with BstNI (New England Biolabs) and resolved them on 3% agarose gels (Nusieve GTG, FMC).
Isolation of Soluble Antibody Fragments.
Soluble scFvs were harvested from the bacterial periplasm by osmotic shock without phosphorylcholine (19) but with 40 μM Pefabloc SC, 0.7 mg⋅liter−1 pepstatin, and 0.5 mg⋅liter−1 leupeptin in all solutions. The scFvs often were used without further purification. For some applications, scFvs were purified from the periplasmic-shock fraction or from the bacterial growth medium by affinity to an epitope-tag antibody column (anti-E tag antibody, Amersham Pharmacia).
Light-Microscopic Immunocytochemistry on Sections.
Bullfrog sacculi were fixed overnight at 4°C with 4% (wt/vol) formaldehyde in PBS (68 mM NaCl, 58 mM Na2HPO4, and 17 mM NaH2PO4 at pH 7.4). After a rinse in PBS, the tissues were cryoprotected with 30% (wt/vol) sucrose in PBS and embedded (Tissue-Tek, Sakura Finetek) for sectioning. Sections 17 μm in thickness were blocked for 2 hr with PBS containing 5% (wt/vol) nonfat dry milk and 3% (vol/vol) heat-inactivated goat serum and incubated overnight at 4°C with 50% (vol/vol) periplasmic extract in blocking solution containing 3 mg⋅liter−1 anti-epitope tag antibody (Amersham Pharmacia) or with anti-epitope tag antibody alone. The antibody solutions were prepared at least 15 min in advance to allow the scFv and anti-epitope tag antibody to interact before incubation with the sample. Unbound antibodies were removed by three 10-min washes with PBS containing 0.1% (vol/vol) Tween-20. To detect bound antibodies, we incubated the samples for 2 hr with 5 mg⋅liter−1 BODIPY-FL goat anti-mouse IgG conjugate (Molecular Probes) in PBS containing 5% (wt/vol) nonfat dry milk powder, washed as indicated above, and observed with a Zeiss LSM-410 confocal microscope.
Immunoblotting of Bullfrog Inner-Ear Homogenate.
To determine the specificity of scFv-278, an entire bullfrog inner ear, including the eight major receptor organs containing hair cells, was sonicated in 8 M urea, 5 mM EDTA, 0.2% (vol/vol) 2-mercaptoethanol, 50 mM Tris⋅Cl, 40 μM Pefabloc SC, 0.7 mg⋅liter−1 pepstatin, and 0.5 mg⋅liter−1 leupeptin at pH 9.2. Particulate matter was sedimented by centrifugation at 16,000 × g for 15 min at 4°C and discarded. The supernatant was retained and the equivalent of 1/10 of the protein from an ear was separated on a 12% SDS/PAGE gel (NOVEX, San Diego), transferred to a nitrocellulose membrane (Hybond ECL; Amersham Pharmacia), and processed for immunodetection with a mixture of 1.8 mg⋅liter−1 affinity-purified scFv-278, 1.5 mg⋅liter−1 anti-epitope tag antibody, and 2.5% (vol/vol) Liquid Block (Amersham Pharmacia) in ECL-PBS that was prepared at least 15 min in advance. The blot was immersed in the antibody solution for at least 12 hr at 4°C. After three washes, bound scFv was detected with a 1:7,000 dilution of anti-mouse antibody conjugated to horseradish peroxidase (Amersham Pharmacia) followed by enhanced chemiluminescence (ECL, Amersham Pharmacia). In a control experiment, omission of the scFv eliminated detection of the band.
Results
Recombinant scFvs, chimeric polypeptides each consisting of an Ig heavy-chain and light-chain variable region joined by a 15-aa linker (20, 21), bind antigens with affinities comparable to those of conventional monoclonal antibodies (10). We used recombinant methods to generate an expression library of scFvs directed against bullfrog saccular proteins and displayed on the surface of filamentous bacteriophage (21).
Generation of a Bacteriophage-Displayed scFv Library.
To augment the number of scFvs directed against inner-ear proteins, we immunized a mouse with bullfrog saccular maculae and with isolated hair cells. After immunization, the RNA from this animal's lymphoid organs served as the template for reverse transcription and amplification by the PCR of the murine Ig heavy-chain and light-chain variable-domain repertoires (Fig. 1 A and B; ref. 16). scFv constructs (Fig. 1A) were generated by randomly joining individual heavy-chain variable domains with light-chain variable domains through gene splicing by overlap junction, or PCR sewing (Fig. 1C; refs. 20 and 22). Such random pairings should not only have recapitulated original combinations that had been optimized by the murine immune system, but also should have created new conjunctions of heavy- and light-chain variable domains that yielded antibodies with novel specificities. Joined products were expressed as fusion proteins with the gene III coat protein of the filamentous bacteriophage fd.
The primary scFv library contained 4 × 107 independent clones. To characterize the library's diversity, we examined the insert sequences of ≈100 randomly chosen clones by PCR amplification of the Ig sequences followed by digestion with a frequently cutting restriction endonuclease (18). This analysis, which revealed that all examined clones contained inserts and that each was unique (Fig. 1D), suggested that little bias occurred during the PCR amplification steps. An analysis of scFv expression from ≈300 randomly selected clones indicated that >60% produced antibody fragments at various levels (data not shown).
Selection of scFv-Bearing Bacteriophage.
The heterogeneity of sequences encoding scFvs suggested that our library was highly diverse and therefore should have contained scFvs directed against numerous antigens, including proteins of the internal ear. To isolate antibody fragments directed against saccular proteins, we selected for bacteriophage that bound to a saccular hair-cell lysate, a heterogeneous mixture of proteins. For each round of selection, a lysate from 50,000 to 100,000 saccular hair cells was used to coat the surface of a selection tube and thereby to serve as the target material for selection (Fig. 2A).
The bacteriophage recovered after each round of selection were used to infect bacteria and subjected to another round. To determine the diversity of clones after the fourth round, we amplified the scFv sequences of 123 randomly selected clones by the PCR. Twenty-nine of these clones contained inserts with an identical restriction-endonuclease digestion pattern, indicating that this clone, number 278, had been enriched based on the scFv that it expressed (Fig. 2B). Twenty-two clones displayed other digestion patterns, some of which occurred two or three times. The remaining clones, which lacked amplifiable inserts, presumably arose through the growth advantage conferred on bacteriophage that did not express heterologous proteins.
Characterization of a Selected scFv.
As an initial step toward determining the specificity of the dominant clone enriched by our selection procedure, we used the corresponding antibody fragment, scFv-278, to characterize the cognate antigen. The phagemid expression vector used in library construction allows both the bacteriophage display of a scFv and the expression of the scFv in a soluble form that is directed to the bacterial periplasm. The soluble 30-kDa scFv can be detected by virtue of an epitope tag located at its carboxyl terminus.
The tissue distribution of the antigen was confirmed by immunocytochemical labeling of cryosections of bullfrog organs including the sacculus, skin, heart, lung, spinal cord, eye, esophagus, and skeletal muscle (data not shown). Intense labeling was detectable only in the sacculus (Fig. 3). Immunoreactivity also was exhibited by scattered cells of the peripheral corneal epithelium, iris, and epicardium, and to a greater extent of the esophagus, but this labeling was far less pronounced than that in the sacculus.
Figure 3.

Immunolabeling of saccular cryosections with the enriched scFv. To characterize the enriched clone, we immunolabeled cryosections of the bullfrog's sacculus with the soluble scFv and examined them by confocal microscopy. (A) A differential-interference–contrast image reveals the components of the saccular epithelium and the underlying saccular nerve. (B) Immunolabeling of the cryosection depicted in A indicates that the antibody fragment recognizes an antigen in the nonsensory epithelium and to a lesser extent in the sensory epithelium of the sacculus. No labeling is seen in the saccular nerve and associated connective tissue. (C) The components of the sacculus are apparent in a second, adjacent cryosection. (D) Omission of the scFv eliminates the intense labeling of the sensory and nonsensory epithelia.
In sections through the sacculus, immunoreactivity was intense in the nonsensory epithelium and was weaker in the sensory epithelium, the location of hair cells and supporting cells (Fig. 3 A and B). No labeling was observed in the connective tissue or saccular nerve. This labeling pattern was not encountered with six other scFv fragments tested (data not shown). No immunolabeling was seen when the scFv was omitted (Fig. 3 C and D).
The labeling of cells in the sacculus, including cells in the sensory epithelium, led us to further examine the specificity of scFv-278 by immunoblotting. A single band, corresponding to a molecular mass of slightly less than 50 kDa, was detected by the scFv on an immunoblot of inner-ear proteins (Fig. 4). These data indicate that scFv-278 recognizes an epitope present on a single protein in the inner ear and is thus a highly specific immunological reagent.
Figure 4.
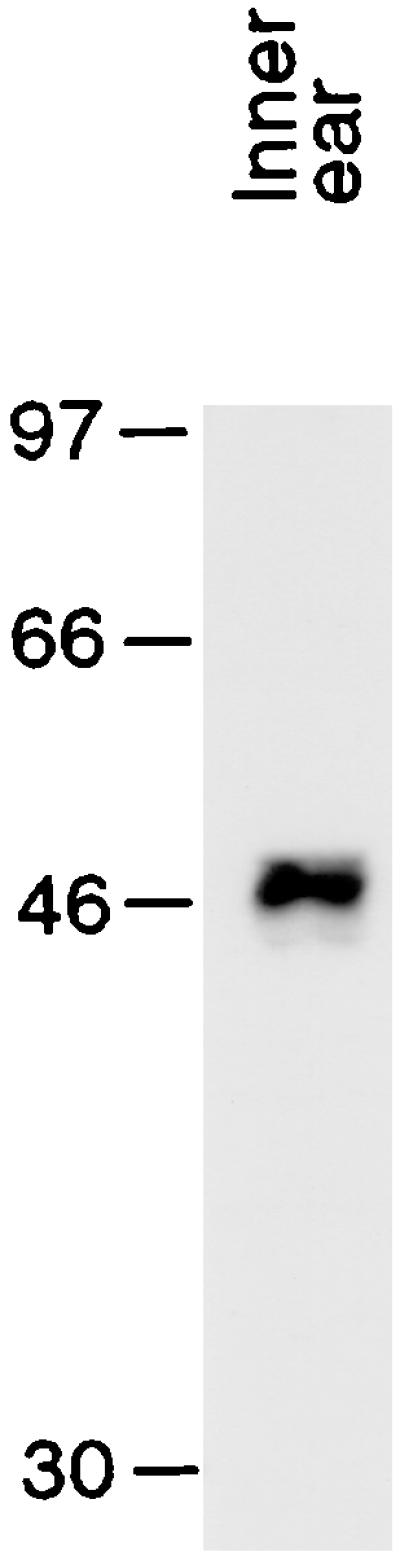
To determine the specificity of the enriched scFv, we subjected a protein homogenate of the bullfrog's inner ear to SDS/PAGE and immunoblotting. A single protein of mass slightly less than 50 kDa was detected, indicating that the scFv is highly specific for a particular antigen. Occasionally, other bands were detected corresponding to molecular masses of ≈100 kDa and ≈150 kDa (data not shown). In conjunction with their sizes, the sporadic detection of these bands suggests that they represented multimers of the ≈50-kDa protein. Molecular-mass markers, denominated in kDa, are indicated at the left.
Discussion
We have demonstrated that the production of a bacteriophage-displayed scFv library is a useful technique for the isolation of a monoclonal immunological reagent when conventional biochemical or immunological approaches are difficult. We chose to generate our library from the lymphoid organs of a mouse immunized with a heterogeneous mixture of proteins, a saccular homogenate and isolated hair cells. Using this approach, we have created a source for immunological reagents that can be repeatedly used for the isolation of reagents against proteins of the inner ear.
The experiments described here constitute an initial investigation of the efficacy of the recombinant-antibody technique for characterization of proteins in small populations of cells from the nervous system. Although this methodology has been quite successful for the isolation of antibody fragments against purified antigens, selection strategies using heterogeneous mixtures of proteins, including cell-surface antigens, have proven more challenging (23–26) and rarely have been used on relatively small numbers of acutely dissected cells (27).
The scFv identified in this study represents the first product of the recombinant-antibody approach in the study of inner-ear proteins; further exploitation of the scFv library should yield additional quarry. The initial experiments also have brought to light several considerations that should be borne in mind in the design of future experiments. Although the recombinant technique yields antibody fragments with a wide variety of specificities, its value depends on the selection procedure by which bacteriophage clones are enriched. In the experiment described here, the selection used the complex mixture of proteins in a lysate of saccular hair cells that was absorbed to the surface of a tube. Because the immunogen and the target material were heterogeneous mixtures, enrichment of a single scFv-bearing bacteriophage was unexpected. This approach doubtlessly biased the selection toward bacteriophage bearing scFvs against a protein that is especially antigenic, abundant, or adherent. This bacteriophage also may have enjoyed a growth advantage over others that bound different saccular antigens. By examining bacteriophage clones from the earlier selective rounds on the hair-cell lysate and by conducting the selection step with different cellular preparations, such as isolated hair bundles (5), we should be able to isolate scFv-bearing bacteriophage directed against other hair-cell proteins.
Our results demonstrate the efficacy of our library for the generation of highly specific reagents with which to study proteins of the inner ear. With these immunological tools in hand, it should now be possible to more thoroughly characterize inner-ear proteins and to ascertain their roles in our senses of hearing and balance.
Acknowledgments
We thank Dr. E. S. Ward for advice and reagents, Dr. E. A. Lumpkin for help with confocal microscopy, and Mss. A. Bell, L. Henry, S. Mellody, R. Orman, and C. S. Denis for technical assistance. Drs. R. A. Baird, P. G. Gillespie, E. A. Lumpkin, and members of our research group provided suggestions on drafts of the manuscript. This work was supported by National Institutes of Health Grant DC00241. A.J.H. is an Investigator of the Howard Hughes Medical Institute.
Abbreviation
- scFv
single-chain variable-region antibody fragment
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.030535797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.030535797
References
- 1.Koyama H, Lewis E R, Leverenz E L, Baird R A. Brain Res. 1982;250:168–172. doi: 10.1016/0006-8993(82)90964-7. [DOI] [PubMed] [Google Scholar]
- 2.Yu X, Lewis E R, Feld D. J Comp Physiol A. 1991;169:241–248. doi: 10.1007/BF00215871. [DOI] [PubMed] [Google Scholar]
- 3.Jacobs R A, Hudspeth A J. Cold Spring Harbor Symp Quant Biol. 1990;55:547–561. doi: 10.1101/sqb.1990.055.01.053. [DOI] [PubMed] [Google Scholar]
- 4.Hudspeth A J. Neuron. 1997;19:947–950. doi: 10.1016/s0896-6273(00)80385-2. [DOI] [PubMed] [Google Scholar]
- 5.Gillespie P G, Hudspeth A J. J Cell Biol. 1991;112:625–640. doi: 10.1083/jcb.112.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holley M C, Nishida Y, Grix N. Int J Dev Neurosci. 1997;15:541–552. doi: 10.1016/s0736-5748(96)00109-8. [DOI] [PubMed] [Google Scholar]
- 7.Rivolta M N, Grix N, Lawlor P, Ashmore J F, Jagger D J, Holley M C. Proc R Soc London Ser B. 1998;265:1595–1603. doi: 10.1098/rspb.1998.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng J L, Lewis A K, Gao W-Q. Hear Res. 1998;117:13–23. doi: 10.1016/s0378-5955(97)00205-0. [DOI] [PubMed] [Google Scholar]
- 9.Holley M C. Tissue Cell. 1992;24:613–624. doi: 10.1016/0040-8166(92)90032-3. [DOI] [PubMed] [Google Scholar]
- 10.Marks J D, Hoogenboom H R, Griffiths A D, Winter G. J Biol Chem. 1992;267:16007–16010. [PubMed] [Google Scholar]
- 11.Winter G, Griffiths A D, Hawkins R E, Hoogenboom H H. Annu Rev Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 12.McCafferty J, Hoogenboom H R, Chiswell D J, editors. Antibody Engineering: A Practical Approach. Oxford: Oxford Univ. Press; 1996. [Google Scholar]
- 13.Sheets M D, Amersdorfer P, Finnern R, Sargent P, Lindqvist E, Schier R, Hemingsen G, Wong C, Gerhart J C, Marks J D. Proc Natl Acad Sci USA. 1998;95:6157–6162. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lumpkin E A, Hudspeth A J. Proc Natl Acad Sci USA. 1995;92:10297–10301. doi: 10.1073/pnas.92.22.10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okayama H, Kawaichi M, Brownstein M, Lee F, Yokota T, Arai K. Methods Enzymol. 1987;154:3–28. doi: 10.1016/0076-6879(87)54067-8. [DOI] [PubMed] [Google Scholar]
- 16.Orlandi R, Güssow D H, Jones P T, Winter G. Proc Natl Acad Sci USA. 1989;86:3833–3837. doi: 10.1073/pnas.86.10.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perez C, Kriegler M. Resolutions. 1990;6:1–2. [Google Scholar]
- 18.Marks J D, Hoogenboom H R, Bonnert T P, McCafferty J, Griffiths A D, Winter G. J Mol Biol. 1991;222:581–597. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- 19.Skerra A, Plückthun A. Science. 1988;240:1038–1041. doi: 10.1126/science.3285470. [DOI] [PubMed] [Google Scholar]
- 20.McCafferty J, Griffiths A D, Winter G, Chiswell D J. Nature (London) 1990;348:552–554. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 21.Clackson T, Hoogenboom H R, Griffiths A D, Winter G. Nature (London) 1991;352:624–628. doi: 10.1038/352624a0. [DOI] [PubMed] [Google Scholar]
- 22.Horton R M, Hunt H D, Ho S N, Pullen J K, Pease L R. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 23.De Kruif J, Terstappen L, Boel E, Logtenberg T. Proc Natl Acad Sci USA. 1995;92:3938–3942. doi: 10.1073/pnas.92.9.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kupsch J-M, Tidman N H, Kang N V, Truman H, Hamilton S, Patel N, Bishop J A N, Leigh I M, Crowe J S. Clin Cancer Res. 1999;5:925–931. [PubMed] [Google Scholar]
- 25.Ridgway J B B, Ng E, Kern J A, Lee J, Brush J, Goddard A, Carter P. Cancer Res. 1999;59:2718–2723. [PubMed] [Google Scholar]
- 26.Tordsson J, Abrahmsén L, Kalland T, Ljung C, Ingvar C, Brodin T. J Immunol Methods. 1997;210:11–23. doi: 10.1016/s0022-1759(97)00165-8. [DOI] [PubMed] [Google Scholar]
- 27.Palmer D B, George A J T, Ritter M A. Immunology. 1997;91:473–478. doi: 10.1046/j.1365-2567.1997.00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]