

Abstract
The homeodomain is a highly conserved DNA-binding motif that is found in numerous transcription factors throughout a large variety of species from yeast to humans. These gene-specific transcription factors play critical roles in development and adult homeostasis, and therefore, any germline mutations associated with these proteins can lead to a number of congenital abnormalities. Although much has been revealed concerning the molecular architecture and the mechanism of homeodomain-DNA interactions, the study of disease-causing mutations can further provide us with instructive information as to the role of particular residues in a conserved mode of action. In this paper, I have compiled the homeodomain missense mutations found in various human diseases and re-examined the functional role of the mutational “hot spot” residues in light of the structures obtained from crystallography. These findings should be useful in understanding the essential components of the homeodomain and in attempts to design agonist or antagonists to modulate their activity and to reverse the effects caused by the mutations.
Homeodomains and inherited human diseases
The regulation of gene transcription is based on specific interactions between transcription factors and their target genes. These transcription factors play central roles in all developmental processes and also in adult homeostasis (Duboule 1994). Thus, numerous congenital syndromes have been shown to be caused by mutations in genes encoding transcription factors, and the numbers of mutations and congenital defects are expected to grow (Semenza 1989; Engelkamp and van Heyningen 1996). Indeed, a recent analysis of the complete human genome has revealed that transcription factors represent one of the four major functional groups of proteins whose germline mutations are the causes of known human diseases (Jimenez-Sanchez et al.2001).
Among these transcription factors, the homeodomain has become one of the most studied eukaryotic DNA-binding motifs since its discovery when homeotic mutations, i.e., mutations leading to segmental transformations, were observed in Drosophila (Gehring 1966; Lewis 1978) and later localized in genes encoding a stable domain of about 60 residues (McGinnis et al. 1984; Scott and Weiner 1984). Since then, hundreds of homeodomains in a large variety of species have been found at all levels of the developmental hierarchy, establishing that genetic control based on homeoboxes is common both to various levels of the development of an organism and to a wide range of species (Duboule 1994).
Many human diseases ranging from developmental abnormalities to metabolic disorders have been linked to mutations in the genes encoding these homeodomaincontaining proteins (D’Elia et al. 2001; Goodman and Scambler 2001; Zhao and Westphal 2002). Mutations affecting transcription factors lead to the breakdown or abnormal control of the transcriptional machinery because of loss of function, either as a hafloinsufficiency or in a dominant negative fashion (Seidman and Seidman 2002). These mutations include nonsense or frameshift mutations that result in truncated and non-functional proteins, or missense mutations giving rise to single amino acid substitutions that can cause subtle, yet detrimental effects in individuals. Whereas nonsense or frameshift mutations are readily understandable, disease-causing missense mutations and the encoded single amino acid substitutions can be more instructive as to the requirement and specific role of that particular residue for protein function. These missense mutations could affect protein expression levels, protein stability, protein localization, post-translational modification, and/or the specific activity of a protein including its physical interactions (Wang and Moult 2001). In this paper, I have compiled and re-examined the diseasecausing mutations in homeodomains from a structural viewpoint and have addressed the role of key residues that are more frequently mutated in patients and the effects of these mutations.
Method of data mining
There are 155 homeodomain-containing proteins in the UCSC Human Genome Browser (http://genome.ucsc. edu/cgi-bin/hgGateway), many of which contain more than one isoform. Many disease-causing mutations are found in these proteins, and the current information has been obtained from the available resources on the web, including the Human Gene Mutations Database and other bioinformatics databases. Among these, I have used the Online Mendelian Inheritance in Man in Baltimore (http://www.ncbi.nlm.nih.gov/Omim), the Human Gene Mutation Database in Cardiff (http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html), the Bioinformatic Harvester in Heidelberg (http://harvester.embl.de), and the NIH Homeodomain Resources (http://research.nhgri.nih.gov/homeodomain/), in addition to the literature. The list of missense mutations, gene products, associated human diseases, homeobox classes, and mutational effects on protein stability and functions are tabulated in Table 1. A total of 119 independent homeodomain missense mutations has been documented for 26 different genes giving rise to various inherited human diseases. These are all monogenic causes of their respective diseases in which the direct relationship between sequence and protein function (missense mutations) and between protein function and disease state (monogenic causes) can be addressed. Throughout this text, the conventional numbering system for homeodomain residues has been used.
Table 1.
List of human mutations found in homeodomains and their associated diseases.Abbreviations used in the ‘Mutational effects’ DB disruption of DNA binding, NL disruption of nuclear localization, PS disruption of protein stability, DI disruption of domain-domain interactions, ? not explainable from the structure). The references for each mutation can be found in the databases available online, especially in Online Mendelian Inheri-column: tance in Man in Baltimore (http://www.ncbi.nlm.nih.gov/omim) ( and the Bioinformatic Harvester in Heidelberg (http://harvester.embl.de)
| Disease/syndrome | Gene product | OMIM number (gene) | Homeobox class | Missense mutation | Mutational effects |
|---|---|---|---|---|---|
| Parietal foramina | ALX4 | 605420 | Homeodomain (214-273) | R218Q | DB/NL |
| R272P | PS | ||||
| Infantile spasm syndrome | ARX | 300382 | Homeodomain (328-387) | R332H | DB |
| L343Q | PS | ||||
| P353L | PS | ||||
| Microphthalmia | CHX10 | 142993 | Homeodomain (148-207) | R200P | DB |
| R200Q | DB | ||||
| Cone-rod dystrophy | CRX | 602225 | Homeodomain (39-98) | R41Q | NL |
| R41W | NL | ||||
| E80A | ? | ||||
| R90W | PS/DB | ||||
| Septooptic dysplasia | HESX1 | 601802 | Homeodomain (108-167) | N125S | PS(?) |
| R160C | DB | ||||
| Currarino syndrome | HLXB9 | 142994 | Homeodomain (242-301) | R246G | DB/NL |
| R246H | DB/NL | ||||
| T247S | PS | ||||
| W289G | PS | ||||
| W289L | PS | ||||
| Q291P | PS/DB | ||||
| R293W | PS/DB | ||||
| R294Q | DB | ||||
| R294W | DB | ||||
| Hand-foot genital syndrome | HOXA13 | 142959 | Homeodomain (322-381) | Q371L | PS |
| N372H | DB | ||||
| Synpolydactyly | HOXD13 | 142989 | Homeodomain (268-327) | S308C | PS |
| I314L | PS/DB | ||||
| MODY4 (diabetes) | IPF-1/PDX-1 | 600733 | Homeodomain (146-205) | R197H | PS/DB |
| Hypodontia | MSX1 | 142983 | Homeodomain (166-225) | R196P | PS/DB |
| Parietal foramina craniosynostosis | MSX2 | 123101 | Homeodomain (142-201) | P148H | PS |
| L154P | PS | ||||
| R172H | PS/DB | ||||
| R194S | DB | ||||
| Atrila septal defect | NKX2.5 (CSX) | 600584 | Homeodomain (138-197) | T178M | PS |
| Q187H | PS/DB | ||||
| N188K | DB | ||||
| R189G | PS/DB | ||||
| Y191C | DB | ||||
| Rieger syndrome | PITX2 | 601542 | Homeodomain (85-144) | R43W | DB/NL |
| L54Q | PS | ||||
| T68P | PS | ||||
| R69H | PS/DB | ||||
| V83L | PS | ||||
| R84W | DB | ||||
| K88E | PS/DB | ||||
| R90C | PS/DB | ||||
| R91P | DB | ||||
| Leri-Weill dyschondrosteosis | SHOX | 312865 | Homeodomain (117-176) | Q128L | PS |
| L132V | PS | ||||
| Langer mesomelic dysplasia | R153L | PS(?) | |||
| R153S | PS(?) | ||||
| L154P | PS | ||||
| V163F | PS/DB | ||||
| R168W | PS/DB | ||||
| A170P | PS/DB | ||||
| R173H | PS | ||||
| R173C | PS | ||||
| Holoprosencephaly | SIX3 | 603714 | Homeodomain (206-266) | L226V | PS(?) |
| V250A | PS | ||||
| R257P | PS/DB | ||||
| Holoprosencephaly | TGIF | 602630 | Homeodomain (35-97) | P63R | PS/DB |
| R90C | DB | ||||
| Keratoconus | VSX1 | 605020 | Homeodomain (164-223) | R166W | NL |
| Waardenburg syndrome | PAX3 | 606597 | Paired box-homeodomain (219-278) | F238S | PS/DB |
| 193500 | V265F | PS/DB | |||
| W266C | PS | ||||
| R270C | PS/DB | ||||
| R271G | DB | ||||
| R271H | DB | ||||
| Coloboma of optic nerve | PAX6 | 607108 | Pairedbox-homeodomain (210-269) | F258S | PS |
| Neuroblastoma | PHOX2B | 603815 | Paired-like homeodomain (98-157) | R100L | NL |
| R141G | DB | ||||
| Pituitary hormone deficiency | PROP-1 | 601538 | Paired-like homeodomain (69-128) | R73H | DB/NL |
| R73C | DB/NL | ||||
| F88S | PS/DB | ||||
| F117I | PS | ||||
| R120C | PS/DB | ||||
| Nail-patella syndrome | LMX1B | 602575 | LIM-homeodomain (196-255) | R200Q | DB/NL |
| 161200 | A213P | PS(?) | |||
| S218P | PS(?) | ||||
| R226P | PS/DB | ||||
| L229P | PS | ||||
| A230V | PS | ||||
| W243C | PS | ||||
| N246K | DB | ||||
| A249P | PS/DB | ||||
| Pituitary hormone deficiency | PIT-1/POU1F1 | 173110 | POU-homeodomain (214-273) | K216E | NL |
| D227Y | ? | ||||
| E230K | PS | ||||
| P239S | PS/DB | ||||
| R265W | PS/DB | ||||
| R271W | DI | ||||
| Deafness | POU3F4 | 300039 | POU-homeodomain (278-337) | A312V | PS |
| L317W | PS | ||||
| R323G | DB | ||||
| R330S | DB | ||||
| K334E | PS/DI | ||||
| Diabetes ( MODY5) | HNF1b (TCF-2) | 604284 | POU-homeodomain (231-311) | A241T | PS |
| Diabetes ( MODY3) | HNF1a | 142410 | POU-homeodomain (199-279) | R200W | NL |
| R200Q | NL | ||||
| R203C | DB/NL | ||||
| R203H | DB/NL | ||||
| K205 | DB/DI | ||||
| Y218C | PS/DB | ||||
| R229Q | PS/DB | ||||
| R229P | PS/DB | ||||
| E240Q | PS/DI | ||||
| E240H | PS/DI | ||||
| C241G | PS/DI | ||||
| C241 | PS/DI | ||||
| L254M | DI | ||||
| S256T | DI | ||||
| V259D | PS | ||||
| V259G | PS | ||||
| T260M | PS/DB | ||||
| R263C | DB | ||||
| R271W | PS/DB | ||||
| R271G | PS/DB | ||||
| R272H | DB | ||||
| R272C | DB |
Molecular architecture and DNA-binding mode of homeodomain
Remarkable features of structural and functional conservation have been observed within homeodomain family members. Compared with primary sequences, their three-dimensional structures are more conserved, which indicates the importance of proper architecture for correct functioning with respect to, for example, DNA recognition and protein-protein interactions. Some amino acids, such as Trp48, Phe49, Asn51, and Arg53, which are invariant among almost all homeodomains, are essential in maintaining structural integrity and/or making contacts with DNA, whereas other residues vary in order to provide DNA-binding specificity and other protein functions.
This high degree of conservation between the sequence and structure makes the homeodomain an ideal model for studying protein-DNA interactions and gene regulation. The homeodomain is composed of three helices, which are folded around a hydrophobic core in which the second and third helix adopt a helix-turnhelix motif for DNA recognition, and a flexible N-terminal arm with additional important functional roles (Gehring et al. 1994; Billeter 1996; Wolberger 1996). The third (recognition) helix and the N-terminal arm recognize the major groove and the adjacent minor groove of target DNAs, respectively. The N-terminal arm also contains a stretch of basic residues known as the nuclear localization signal (NLS). Unlike conventional helixturn-helix motifs, which use the residues on the turn and the first loop of the third helix to contact DNA, homeodomains make these contacts with residues that are located toward the C-terminal end of the third helix. This structure is highly conserved among otherwise highly different species and different ways of recognizing target genes. These homeodomains are either found alone as a DNA-binding motif or in tandem with another module, such as paired-homeodomains (Wilson et al. 1995), LIM-homeodomains (Hobert and Westphal 2000), POU-homeodomains (Ryan and Rosenfeld 1997), or cut-homeodomains (Harada et al. 1994).
A vast body of knowledge about homeodomains has accumulated over the last 15 years, including the results from extensive in vitro binding studies of various DNA fragments and from crystallographic structures of both free and DNA-bound forms of homeodomains revealing the molecular mode of their interactions with DNA (Gehring et al. 1994; Billeter 1996; Wolberger 1996). In addition, some of these conjugate-homeodomain structures show diverse and subtle variations in homeodomain architecture and homeodomain-DNA interactions but display the highly conserved and universal mode of DNA recognition (Jacobson et al. 1997; Xu et al. 1999; Chi et al. 2002).
Sequence conservation in human homeodomains and their mutational “hot spots”
The trinity of the sequence-structure-function relationship is the core element of the natural world of proteins, and some degree of variation might be expected to be allowed for these proteins, particularly for transcription factors during adaptive evolution, in order to ensure DNA and protein-binding specificities for recognizing a larger spectrum of gene promoters and co-regulators. However, selective pressure maintains vital functions, and thus, the degree and pattern of sequence conservation among various members of a protein family is highly informative regarding the functional requirement of each residue.
The signature motif of the homeodomain is found within the DNA-recognition helix in which hydrophobic core aromatic residues and DNA-binding core residues are strictly conserved (Fig. 1). Two aromatic residues in helix 3, Trp48 and Phe49, are almost absolutely conserved and have become markers for identifying divergent homeoboxes. Likewise, Asn51 and Arg53, which are also in helix 3, are strictly conserved and form bidentate contacts with adenine and nonspecific interactions with backbone atoms, respectively. In addition, throughout the sequence, other residues are highly conserved for structural and functional roles, in accordance with findings related to the frequency of disease-causing mutations (Fig. 1). These findings indicate that the mutational “hot spot” residues serve as core residues for maintaining the overall architecture of the homeodomain and for optimally recognizing the site-specific target genes.
Fig. 1.

Disease-causing missense mutations found in homeodomains and their frequencies. a Individual unique mutations are indicated by stars. Numbers given before each sequence indicate the beginning number of the amino acids, but the residue numbers used in the text follow the conventional numbering system of 1-60 shown top. In the sequence alignment, strictly conserved and highly conserved residues are red and green, respectively. b Mutation frequencies are determined by the number of independent missense mutations found on a particular residue and indicated by a yellow-to-red color scheme. The HNF1α sequence, as a prototype, is shown bottom with the atypical 21 residue insertion between helix 2 and 3
When the database information was complied, three mutational “hot spots” were identified along the sequence of the homeodomain (Fig. 1a, b): Arg5, which recognizes the minor groove of DNA (Fig. 2a, b), and the successive residues of Arg52 and the strictly conserved Arg53, which recognize the major groove (Fig. 2a, c). These are all surface residues that are either directly or indirectly involved in DNA binding. These findings are contrary to a general observation that the relative probability of disease-causing mutations is highest in the protein interior and lowest on the protein surface, and that the dominant mechanism by which disease mutations damage protein function is a decrease in protein stability (Wang and Moult 2001; Ferrer-Costa et al. 2002), validating the significance of these residues for protein function. Structural descriptions and the functional roles for each “hot spot” residue are provided in the following sections.
Fig. 2.
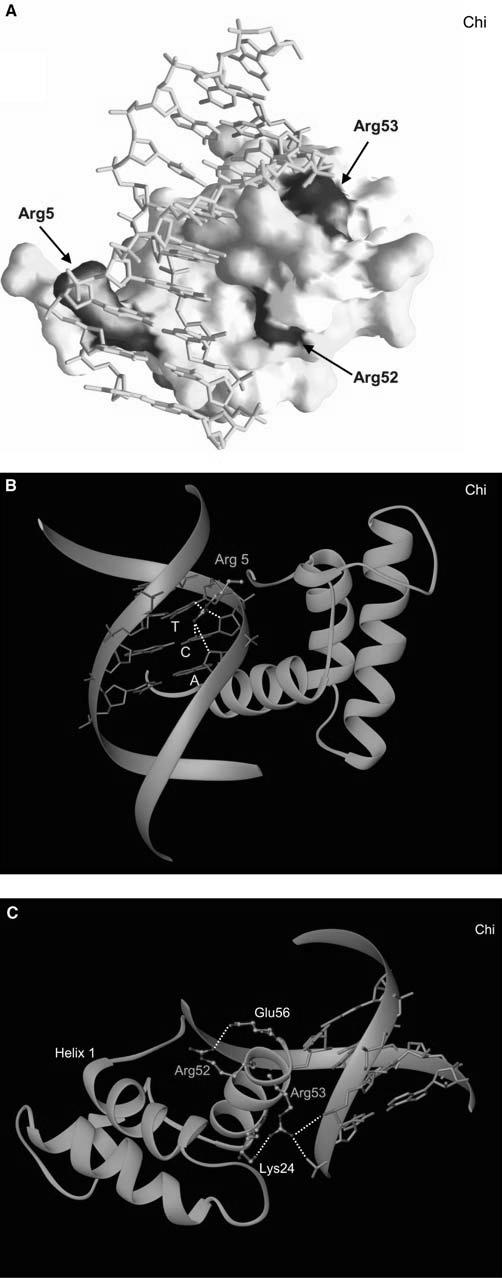
Mapping of mutations to the homeodomain structure and a detailed structural view of each mutational “hot spot” in HNF1α. a Surface representation of the homeodomain bound to DNA, with mutation sites colored according to the frequencies shown in Fig. 1. b A close-up view of Arg5 and its interactions with DNA. c A close-up view displaying Arg 52 and Arg 53 and their interactions with DNA and neighboring residues. In the majority of homeodomains, there is an additional salt bridge between Arg52 and Glu17 on the first helix to assist the anchoring of the recognition helix at the major groove
Hot spot 1: arginine 5
A significant contribution to the optimal DNA binding of the homeodomains comes from the N-terminal arm (Gehring et al. 1994; Shang et al. 1994). The Arg5 residue is located in the middle of this N-terminal extension. Arg5 has the dual function of binding DNA through the minor groove and serving as part of the NLS. Even though the exact details of the DNA-binding mode vary among different homeodomains (Table 2), Arg5 always protrudes deep into the minor groove and makes an extensive and nondiscriminatory hydrogen bonding network with the base and sugar atoms (Fig. 2b). These interactions are often further stabilized by the basic residues at positions 2 and 3, which make additional hydrogen bonds with DNA backbone atoms. Thus, Arg5 appears to serve as a core element of the N-terminal arm in recognizing the minor groove without imposing DNA specificity. This structural finding is consistent with the biochemical data in which proteins mutated at this position are expressed at levels similar to wild-type proteins but have markedly reduced DNAbinding activity (McIntosh et al. 1998; Qu et al. 1998; Yamada et al. 1999). Partially impaired nuclear localization has also been observed for the R203C (R5C by the conventional numbering) mutant of HNF1α (Yamada et al. 1999).
Table 2.
List of homeodomain proteins and details of interactions made by mutational “hot spot” residyes (PDB accession code Protein database accession codes can be found at http://www.rcsb.org/pdb/). Crystal structures of native proteins complexed with DNA were used to complete this table. Each residue has distinctive and highly conserved roles. In some proteins, Arg52 is replaced by Lys52 but similar hydrogen bonding patterns are maintained.
| Protein | Species | Homeodomain family | PDB accession code | Arg5 (hydrogen bond with) | Arg52 (salt bridge with) | Arg53 (salt bridge with) |
|---|---|---|---|---|---|---|
| MATα2 | Yeast | Homeodomain | 1APL | N3 on A | E17 | PO4 on C |
| 1AKH | O2 on T | E56 | PO4 on G | |||
| 1K61 | O4′1 | on T PO4 | on A | |||
| 1MNM | O4′ | on | A | |||
| 1YRN | O4′ | on | G | |||
| MATa1 | Yeast | Homeodomain | 1AKH | O4 on T | Replaced by Lys | PO4 on A |
| 1YRN | PO4 on T | |||||
| UBX | Fruit fly | Homeodomain | 1B8I | N3 on A | E17 | PO4 on C |
| O2 on T | PO4 on G | |||||
| O4′ | on | T | ||||
| O4′ | on | A | ||||
| PBX | Fruit fly | Hemeodomain | 1B8I | N3 on A | Replaced by Lys | PO4 on A |
| N3 on G | PO4 on T | |||||
| N7 on G | ||||||
| O6 on G | ||||||
| O2 on T | ||||||
| O4′ | on | T | ||||
| O4′ | on | G | ||||
| Engrailed | Fruit fly | Homeodomain | 3HDD | N2 on G | Replaced by Lys | PO4 on G |
| O2 on T | PO4 on T | |||||
| O4′ | on T PO4 | on C | ||||
| O4′ | on | A | ||||
| Antennapedia | Fruit fly | Homeodomain | 9ANT | N2 on G | E17 | PO4 on C |
| N3 on A | PO4 on G | |||||
| O2 on T | ||||||
| O4′ | on | T | ||||
| O4′ | on | A | ||||
| Even-skipped | Fruit fly | Homeodomain | 1JGG | N3 on A | E17 | PO4 on A |
| O2 on T | E56 | PO4 on A | ||||
| O4′ | on | T | ||||
| Paired-box | Fruit fly | Paired-homeo | 1FJL | N2 on G | E17 | PO4 on C |
| N3 on A | PO4 on T | |||||
| O2 on T | PO4 on A | |||||
| O4′ | on | T | ||||
| O4′ | on | A | ||||
| MSX-1 | Mouse | Homeodomain | 1IG7 | O2 on T | E17 | PO4 on C |
| O4′ | on A PO4 | on T | ||||
| O4′ | on | T | ||||
| HOXa9 | Mouse | Homeodomain | 1PUF | N3 on A | E17 | PO4 on G |
| O2 on T | PO4 on T | |||||
| O2 on C | ||||||
| O4′ | on | C | ||||
| PBX1 | Mouse | Homeodomain | 1PUF | N3 on A | Replaced by Lys | PO4 on A |
| O2 on T | PO4 on T | |||||
| O4′ | on | T | ||||
| O4′ | on | A | ||||
| HOXB1 | Human | Homeodomain | 1B72 | O2 on T | E17 | PO4 on C |
| O4′ | on T PO4 | on T | ||||
| O4′ | on | G | ||||
| PBX1 | Human | Homeodomain | 1B72 | O2 on T | Replaced by Lys | PO4 on C |
| O4′ | on T PO4 | on C | ||||
| O4′ | on | A | ||||
| O4′ | on | G | ||||
| OCT-1 | Human | POU-homeo | 1OCT | N2 on G | E17 | PO4 on C |
| 1CQT | N4 on C | E56 | PO4 on T | |||
| 1E3O | N7 on G | PO4 on A | ||||
| 1HF0 | O4 on T | |||||
| 1GT0 | O6 on G | |||||
| O4′ | on | A | ||||
| PIT-1 | Human | POU-homeo | 1AU7 | N3 on A | E17 | PO4 on C |
| O2 on T | E56 | PO4 on T | ||||
| O2 on C | ||||||
| O4′ | on | A | ||||
| HNF1α | Human | POU-homeo | 1IC8 | O2 on C | E56 | PO4 on C |
| O2 on T | PO4 on T | |||||
| O4′ | on | C | ||||
| O4′ | on | A |
Hot spot 2: arginines 52 and 53
This major mutational “hot spot” found on the recognition helix includes Arg52 and Arg53. Arg53 is strictly conserved in all homeodomains and makes direct hydrogen bonds with DNA backbones from two nonspecific nucleotides at the 5′ flanking region of the promoter recognition sequence in all cases (Table 2 and Fig. 2c). This acts as a claw hooking onto a rope and holding it tightly and serves as a clamp to anchor the recognition helix from one side for optimal interactions in the major groove. In addition, Arg52 is highly conserved and tethers the recognition helix for optimal DNA binding by forming a salt bridge with the Glu17 on the first helix, except in hepatocyte nuclear factor 1 a (HNF1α) in which the closest residue Glu21 is 4.16 Å away and beyond the acceptable hydrogen bonding distance. In many cases, Arg52 further stabilizes the recognition helix by forming an additional salt bridge with Glu56 (Table 2, Fig. 2c). Thus, Agr52 appears to be required both for the conformational stability of the recognition helix and the entire homeodomain (Weiler et al. 1998) and for optimal DNA interactions. In some homeodomains, Arg52 is replaced by Lys52 (Table 2), but similar hydrogen bonding patterns are still maintained. This intricate network of interactions by Arg52 and Arg53 has been evolutionally conserved to ensure the correct positioning of the recognition helix but does not dictate the sequence-specific promoter recognition. Biochemical data have confirmed greatly reduced DNA binding and transcriptional activity in “hot spot 2” mutants, despite normal protein expression levels and protein stability (Dattani et al. 1998; Wu et al. 1998; Swaroop et al. 1999; Vaxillaire et al. 1999; Yoshiuchi et al. 1999; Wilkie et al. 2000; Quentien et al. 2002).
Other residues contributing to stability and DNA-binding affinity and specificity
Protein stability and correct folding are the foundations of protein function. The compact homeodomain is stabilized by the hydrophobic core, which holds all of its helices together. Highly conserved Val/Ile45 and strictly conserved Trp48 and Phe49 on the recognition helix take part in the formation of the core. Other notable highly conserved amino acids forming the hydrophobic core include Leu/Trp/Phe16 and Tyr/Phe20 from the first helix and Leu/Ile/Phe/Met34 from the second helix. A recent phase-display shotgun scanning method used on the engrailed homeodomain has revealed the importance of additional hydrophobic residues such as Phe20 and Tyr25 (Sato et al. 2004; Wolfe 2004). However, the frequencies of diseasecausing mutations on these hydrophobic core residues are low compared with those occurring in DNA-binding domains (Fig. 1a, b). A similar pattern has been observed in p53, another well-known transcription factor in which the largest number of human disease mutations have been found within a single gene product (Bullock and Fersht 2001). These findings are in contrast to the generally believed observation that the majority of human disease-causing mutations disrupt protein stability (Wang and Moult 2001; Ferrer-Costa et al. 2002).
An exhaustive survey of transcription-factor-DNA interactions reveals a number of forces contributing to their strength and specificity. Some of these forces act locally in distinct regions of the interacting surfaces, whereas others exert a more global influence on complex formation. Local forces include hydrogen bonds, ionic salt bridges, hydrophobic interactions, and van der Waals contacts, whereas global forces include plasticity and sequence-dependent folding, conformational changes, and cooperativity gained through simultaneous DNA recognition by multiple protein modules (Ogata et al. 2003). As a rule, DNA-binding domains mediate: (1) nonspecific or “positioning contacts” that provide a general moderate affinity and (2) base-specific contacts that ensure high-affinity binding to specific target sequences. Nonspecific contacts are principally interactions with the DNA backbone of phosphate and sugar moieties and frequently involve electrostatic attractions (ionic salt bridges) between basic protein residues and the polyanionic DNA phosphoskeleton. Base specificity is governed by a network of local contacts of the types outlined above between flexible amino acid side chains that emanate from the binding domain and the exposed edges of the base pairs, primarily in the major groove of the DNA target sequence. The difference between the binding energies for the sequence-dependent and sequence-independent components of the interaction is the measure of the sequence selectivity of a DNA-binding domain (Ogata et al. 2003).
Homeodomainsarenoexceptiontothesegeneralrulesof protein-DNA interactions. Key residues for specific and nonspecific interactions have been well characterized. Whereas target DNA sequences of respective homeodomains differ from each other, they share some common features such as the “TAAT” core sequences. In the major groove, base-specific recognitions are made primarily by residues Val/Ile47, Gln/Lys50, and Asn51 (Gehring et al. 1994;Billeter 1996;Wolberger 1996).Amongthese,the side chain of theinvariant Asn51 from the recognition helix specifically contacts A3 of the TAAT core by accepting a hydrogen bond from adenine N6 and donating a hydrogen bond to adenine N7. This is conserved in all homeodomain proteins,andtheN51Amutationinengrailedhomeodomain abrogatesbindingtoDNA(Ades and Sauer 1994).
DNA-binding specificity appears to be conferred primarily by Val/Ile47 and Gln/Lys50 (Ades and Sauer 1994; Pomerantz and Sharp 1994; Connolly et al. 1999), and earlier studies have indicated that mutations in the Val/Ile47 and Gln/Lys50 residues alter DNA target specificity (Treisman et al. 1989; Ades and Sauer 1994; Tucker-Kellogg et al. 1997; Grant et al. 2000; Simon and Shokat 2004). For example, in HNF1α, Val/Ile47 is replaced by Asn, which recognizes cytosine (lacking a methyl group) instead of thymine, and Gln/Lys50 is replaced by Ala, which does not take part in DNA binding. Val/Ile47 mostly recognizes T4 of the TAAT core via a van der Waals contact with the methyl group at the C5 position, whereas Gln/Lys50 mostly recognizes the nucleotides 3′ to the TAAT core. However, these residues do not appear to be essential for DNA binding because the replacement of Val/Ile47 by Arg, Asn, His, or Gly residues still renders compatible or better DNA bindings (Pomerantz and Sharp 1994), and Q50K replacement enhances DNA-binding affinity (Ades and Sauer 1994). Furthermore, the crystal structures of the Q50K and Q50A mutants reveal only subtle changes at the protein-DNA interface (Tucker-Kellogg et al. 1997; Grant et al. 2000). Consistent with these findings, only a few numbers of mutations are found at these residues (Fig. 1a, b). DNA-binding specificity appears to be more tolerant of mutation than the binding affinity governed mostly by nonspecific interactions.
Nonspecific DNA interactions in homeodomains are made by the basic residues on the N-terminal arm, on the loop between the first and the second helices, and on the recognition helix (Gehring et al. 1994; Billeter 1996). The mutational “hot spot” residues are found among these nonspecific DNA-contacting residues. Arg5 is found at the N-terminal, and Arg52 and Arg53 are located on the recognition helix, and these residues are highly intolerant of any substitutions (Fig. 1a, b). Additionally, a moderate frequency of mutation is also observed at Arg31, which is located at the beginning of helix 2, serves as an anchor for the DNA recognition helix, and directly or indirectly participates in DNA backbone interactions. Whereas it makes a direct hydrogen bond with the DNA backbone atom in the MSX1 structure (PDB accession code 1IG7), it is not close enough (greater than 6 Å) to accept a hydrogen in many other homeodomains, including that of HNF1α. Instead, it provides an overall positively charged environment favorable for DNA interactions. It also makes a salt bridge with the carbonyl backbone atom Glu42 at the beginning of helix 3, which serves to anchor the recognition helix properly for optimal DNA binding and local stabilization. Thus, Arg31 at the beginning of the second helix also appears to have a significant structural and functional role as part of the general homeodomain-DNA backbone interactions.
Large collection of mutations in HNF1α
Among the proteins listed in Table 1, HNF1α represents the most number of mutations found in a single protein, and the mutational “hot spot” residues are well represented (Table 1, Fig. 1a, b). Mutations in Hnf-1a are the most common monogenic causes of the form of diabetes known as maturity onset diabetes of the young (MODY). The recent crystal structure of HNF1α bound to DNA has revealed that HNF1α belongs to the POU transcription factor family, despite the lack of sequence homology in the POUSpecific domain region, and has unveiled the way in which HNF1α confers site-specific promoter recognition, thus telling us why function is lost by MODY3 mutations (Chi et al. 2002). Unlike nonsense and frameshift mutations that are found sporadically throughout the HNF1α sequence, missense mutations are clustered into DNA-binding domains and are almost evenly distributed between the POUHomeo and POUSpecific domain. However, because information about diseasecausing mutations of other POUSpecific domains is limited, I intend to confine the discussion in this review to homeodomains in which mutation information is more abundant.
Even though HNF1α displays moderate variation from other homeodomains in that a 21 amino acid insertion, important for extensive domain-domain interactions, has occurred between the second and the third helix (Chi et al. 2002), it still retains the conserved DNA-binding mode and can serve as a prototype for discussion and graphical representations of homeodomain-DNA interactions (Fig. 2).
The hallmarks of DNA-homeodomain interactions are present in HNF1α (Chi et al. 2002). The recognition helix is situated in the major groove, oriented perpendicular to the long axis of the DNA. As in all homeodomains, Asn51 (Arg270 in human HNF1α) forms bidentate contacts with adenine at the TAAT core, whereas Arg53 (Arg272) within the conserved WFXNXR motif of the recognition helix makes nonspecific interactions with the backbone atoms. In addition, Arg5 (Arg203) in the N-terminal arm of the POUHomeo domain forms hydrogen bonds with thymine, cytosine, and adenine in the minor groove.
Many of the mutated residues in the POUHomeo domain are involved directly in DNA recognition, including those that normally create hydrogen-bonding networks with DNA, viz., basic residues Arg5 (Arg203) and Arg53 (Arg272). Other mutations appear to disrupt DNA recognition indirectly through perturbations in the local environment. The cluster of basic residues at the amino-terminus of the POUHomeo domain serves as an NLS. Mutation of Arg2 (Arg200) and Arg5 (Arg203) within the putative NLS probably hinders nuclear translocation: Thus, the substitution of residues such as Arg5 (Arg203) have dual potential consequences on HNF1α function. Additional mutations interfere with intramolecular interactions between its POUSpecific and POUHomeo domains; these would distort their relative orientations and ability to interact cooperatively with DNA. Others are predicted to disrupt protein folding or stability, which may lead to the accumulation of misfolded protein or premature degradation.
Discussion
Mutations are of fundamental importance for gene diversity and evolution but are also associated with diseases and death when they occur at critical sites. The study of naturally occurring missense mutations on protein-coding genes can be instructive. Even though mutations in a single protein might not be definitively informative because human mutations are not random and are influenced by the local DNA sequence environment (Antonarakis et al. 2000; Krawczak et al. 2000; Zhang and Gerstein 2003), accumulated occurrences on many functionally related proteins or a group of family members can yield information on the importance of each residue and the underlying functional mechanism.
Many residues of the homeodomain participate in DNA recognition, and the analysis of disease-causing mutations has revealed Arg5, Arg52, and Arg53 as key functional elements in this vital function. These mutation-intolerant arginine residues make nonspecific interactions with DNA backbone atoms, indicating that nonspecific DNA binding is a prerequisite for any further sequence-specific recognition and binding. Homeodomains have been shown to be capable of binding to DNA nonspecifically or atypically with reasonable binding affinity (Aishima and Wolberger 2003). Thus, these mutational “hot spot” residues appear to recognize DNA nonspecifically anywhere along the chain and maintain stable homeodomain-DNA complexes while translocating to their target sites at which point specific interactions can be made by other residues (Kalodimos et al. 2004).
All of these “hot spot” mutations appear to be arginine residues. A similar finding has been made on p53 in which five out of six mutational “hot spot” residues are arginine residues that either directly or indirectly affect DNA binding (Bullock and Fersht 2001). Assuming that each base-pair has the same chance of naturally becoming modified, arginine would not be expected to be the amino acid with the highest mutation rate in a protein, because arginine has the highest number of possible codons. Nevertheless, arginine residues account for almost 15% of all human disease mutations (Vitkup et al. 2003). This high mutational recurrence of arginine residues is not unexpected and could be partially attributable to the high mutability of cytosine present in the CpG dinucleotide. CpG dinucleotides are believed to be hypermutable because of deamination when they are methylated (Cooper et al. 1997; Pfeifer 2000). However, several mutations of arginine codons of human homeodomain genes are not C to T transitions (D’Elia et al. 2001). This is true for many other proteins. Thus, the high frequency of arginine substitutions are believed to reflect their functional requirements as surface residues that play vital roles in catalysis, protein-protein interactions, and protein-DNA interactions, as in the case of the homeodomains.
Homeodomain-containing transcription factors often interact with other transcription factors binding to adjacent recognition sites, in addition to coactivators, to enhance transcriptional activity (Di Palma et al. 2003; Okada et al. 2003). These synergistic interactions with other transcription factors and coactivators serve as additional elements that control the specificity of the homeodomains (Gehring et al. 1994). Even though the molecular details of the combinatorial synergism and recruitment by each homeodomain-containing transcription factor are ill-defined, those residues found on the putative protein-protein interaction surfaces seem to have higher mutational tolerances than the core residues affecting nonspecific DNA-binding affinity.
A protein is made up of a large number of amino acid residues with unequal contributions to protein stability and various other functions. Even though alanine scanning mutagenesis (Shang et al. 1994; Acton et al. 2000; Morrison and Weiss 2001) or phase display (Connolly et al. 1999; Pabo et al. 2001; Sato et al. 2004; Simon et al. 2004) can be used systematically to assess the contributions of individual amino acid side chains to protein properties, better and more definite indications can be obtained from naturally occurring monogenic mutations resulting in altered phenotypes. Therefore, these findings should be valuable in attempts to design homeodomains with high affinity for the targeting of specific genes; similar approaches have been made for zinc-finger DNA-binding proteins with clinically important applications (Choo and Isalan 2000; Wolfe et al. 2000; Jamieson et al. 2003). These findings should also be useful in the design of small agents that can modulate the function of homeodomain-containing transcription factors and that can reverse the effects caused by disease-causing mutations.
Acknowledgments
Acknowledgments I wish to thank S. Shoelson for initiating the HNF1α project and for insightful discussions. I also thank K. Sarge and members of the Chi laboratory for comments on the manuscript. This work was funded in part by fellowships from the Juvenile Diabetes Research Foundation and the Mary Iacocca Foundation to Y.-I. Chi.
References
- Acton TB, Mead J, Steiner AM, Vershon AK. Scanning mutagenesis of Mcm1: residues required for DNA binding, DNA bending, and transcriptional activation by a MADS-box protein. Mol Cell Biol. 2000;20:1–11. doi: 10.1128/mcb.20.1.1-11.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ades SE, Sauer RT. Differential DNA-binding specificity of the engrailed homeodomain: the role of residue 50. Biochemistry. 1994;33:9187–9194. doi: 10.1021/bi00197a022. [DOI] [PubMed] [Google Scholar]
- Aishima J, Wolberger C. Insights into nonspecific binding of homeodomains from a structure of MATalpha2 bound to DNA. Proteins. 2003;51:544–551. doi: 10.1002/prot.10375. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, Krawczak M, Cooper DN. Disease-causing mutations in the human genome. Eur J Pediatr. 2000;159(Suppl 3):S173–S178. doi: 10.1007/pl00014395. [DOI] [PubMed] [Google Scholar]
- Billeter M. Homeodomain-type DNA recognition. Prog Biophys Mol Biol. 1996;66:211–225. doi: 10.1016/s0079-6107(97)00006-0. [DOI] [PubMed] [Google Scholar]
- Bullock AN, Fersht AN. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1:68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- Chi YI, Frantz JD, Oh BC, Hansen L, Dhe-Paganon S, Shoelson SE. Diabetes mutations delineate an atypical POU domain in HNF-1alpha. Mol Cell. 2002;10:1129–1137. doi: 10.1016/s1097-2765(02)00704-9. [DOI] [PubMed] [Google Scholar]
- Choo Y, Isalan M. Advances in zinc finger engineering. Curr Opin Struct Biol. 2000;10:411–416. doi: 10.1016/s0959-440x(00)00107-x. [DOI] [PubMed] [Google Scholar]
- Connolly JP, Augustine JG, Francklyn C. Mutational analysis of the engrailed homeodomain recognition helix by phage display. Nucleic Acids Res. 1999;27:1182–1189. doi: 10.1093/nar/27.4.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DJ, Krawczak M, Antonarakis SE. The nature and mechanisms of human gene mutation. In: Scriver CD, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. 7th edn. McGraw-Hill; New York: 1997. pp. 259–291. [Google Scholar]
- Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19:125–133. doi: 10.1038/477. [DOI] [PubMed] [Google Scholar]
- D’Elia AV, Tell G, Paron I, Pellizzari L, Lonigro R, Damante G. Missense mutations of human homeoboxes: a review. Hum Mutat. 2001;18:361–374. doi: 10.1002/humu.1207. [DOI] [PubMed] [Google Scholar]
- Di Palma T, Nitsch R, Mascia A, Nitsch L, Di Lauro R, Zannini M. The paired domain-containing factor Pax8 and the homeodomain-containing factor TTF-1 directly interact and synergistically activate transcription. J Biol Chem. 2003;278:3395–3402. doi: 10.1074/jbc.M205977200. [DOI] [PubMed] [Google Scholar]
- Duboule D. Guidebook to the homeodomain genes. Oxford University Press; Oxford: 1994. [Google Scholar]
- Engelkamp D, Heyningen van V. Transcription factors in disease. Curr Opin Genet Dev. 1996;6:334–342. doi: 10.1016/s0959-437x(96)80011-6. [DOI] [PubMed] [Google Scholar]
- Ferrer-Costa C, Orozco M, Cruz de la X. Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J Mol Biol. 2002;315:771–786. doi: 10.1006/jmbi.2001.5255. [DOI] [PubMed] [Google Scholar]
- Gehring W. Cell heredity and changes of determination in cultures of imaginal discs in Drosophila melanogaster. J Embryol Exp Morphol. 1966;15:77–111. [PubMed] [Google Scholar]
- Gehring WJ, Qian YQ, Billeter M, Furukubo-Tokunaga K, Schier AF, Resendez-Perez D, Affolter M, Otting G, Wuthrich K. Homeodomain-DNA recognition. Cell. 1994;78:211–223. doi: 10.1016/0092-8674(94)90292-5. [DOI] [PubMed] [Google Scholar]
- Goodman FR, Scambler PJ. Human HOX gene mutations. Clin Genet. 2001;59:1–11. doi: 10.1034/j.1399-0004.2001.590101.x. [DOI] [PubMed] [Google Scholar]
- Grant RA, Rould MA, Klemm JD, Pabo CO. Exploring the role of glutamine 50 in the homeodomain-DNA interface: crystal structure of engrailed (Gln50 → ala) complex at 2.0 Å. Biochemistry. 2000;39:8187–8192. doi: 10.1021/bi000071a. [DOI] [PubMed] [Google Scholar]
- Harada R, Dufort D, Denis-Larose C, Nepveu A. Conserved cut repeats in the human cut homeodomain protein function as DNA binding domains. J Biol Chem. 1994;269:2062–2067. [PubMed] [Google Scholar]
- Hobert O, Westphal H. Functions of LIM-homeobox genes. Trends Genet. 2000;16:75–83. doi: 10.1016/s0168-9525(99)01883-1. [DOI] [PubMed] [Google Scholar]
- Jacobson EM, Li P, Leon-del-Rio AM, Rosenfeld MG, Aggarwal AK. Structure of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. Genes Dev. 1997;11:198–212. doi: 10.1101/gad.11.2.198. [DOI] [PubMed] [Google Scholar]
- Jamieson AC, Miller JC, Pabo CO. Drug discovery with engineered zinc-finger proteins. Nat Rev Drug Discov. 2003;2:361–368. doi: 10.1038/nrd1087. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez G, Childs B, Valle D. Human disease genes. Nature. 2001;409:853–855. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- Kalodimos CG, Biris N, Bonvin AMJJ, Levandoski MM, Guennuegues M, Boelens R, Kaptein R. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 2004;305:386–389. doi: 10.1126/science.1097064. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Chuzhanova NA, Stenson PD, Johansen BN, Ball EV, Cooper DN. Changes in primary DNA sequence complexity influence the phenotypic consequences of mutations in human gene regulatory regions. Hum Genet. 2000;107:362–365. doi: 10.1007/s004390000393. [DOI] [PubMed] [Google Scholar]
- Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- McGinnis W, Levine MS, Hafen E, Kuroiwa A, Gehring WJ. A conserved DNA sequence in homoeotic genes of the Drosophila antennapedia and bithorax complexes. Nature. 1984;308:428–433. doi: 10.1038/308428a0. [DOI] [PubMed] [Google Scholar]
- McIntosh I, Dreyer SD, Clough MV, Dunston JA, Eyaid W, Roig CM, Montgomery T, Ala-Mello S, Kaitila I, Winterpacht A, Zabel B, Frydman M, Cole WG, Francomano CA, Lee B. Mutation analysis of LMX1B gene in nail-patella syndrome patients. Am J Hum Genet. 1998;63:1651–1658. doi: 10.1086/302165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison KL, Weiss GA. Combinatorial alanine-scanning. Curr Opin Chem Biol. 2001;5:302–307. doi: 10.1016/s1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- Ogata K, Sato K, Tahirov TH, Tahirov T. Eukaryotic transcriptional regulatory complexes: cooperativity from near and afar. Curr Opin Struct Biol. 2003;13:40–48. doi: 10.1016/s0959-440x(03)00012-5. [DOI] [PubMed] [Google Scholar]
- Okada Y, Nagai R, Sato T, Matsuura E, Minami T, Morita I, Doi T. Homeodomain proteins MEIS1 and PBXs regulate the lineage-specific transcription of the platelet factor 4 gene. Blood. 2003;101:4748–4756. doi: 10.1182/blood-2002-02-0380. [DOI] [PubMed] [Google Scholar]
- Pabo CO, Peisach E, Grant RA. Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem. 2001;70:313–340. doi: 10.1146/annurev.biochem.70.1.313. [DOI] [PubMed] [Google Scholar]
- Pfeifer GP. p53 mutational spectra and the role of methylated CpG sequences. Mutat Res. 2000;450:155–166. doi: 10.1016/s0027-5107(00)00022-1. [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Sharp PA. Homeodomain determinants of major groove recognition. Biochemistry. 1994;33:10851–10858. doi: 10.1021/bi00202a001. [DOI] [PubMed] [Google Scholar]
- Qu S, Tucker SC, Ehrlich JS, Levorse JM, Flaherty LA, Wisdom R, Vogt TF. Mutations in mouse aristaless-like4 cause Strong’s luxoid polydactyly. Development. 1998;125:2711–2721. doi: 10.1242/dev.125.14.2711. [DOI] [PubMed] [Google Scholar]
- Quentien M-H, Pitoia F, Gunz G, Guillet M-P, Enjalbert A, Pellegrini I. Regulation of prolactin, GH, and Pit-1 gene expression in anterior pituitary by Pitx2: an approach using Pitx2 mutants. Endocrinology. 2002;143:2839–2851. doi: 10.1210/endo.143.8.8962. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Rosenfeld MG. POU domain family values: flexibility, partnerships, and developmental codes. Genes Dev. 1997;11:1207–1225. doi: 10.1101/gad.11.10.1207. [DOI] [PubMed] [Google Scholar]
- Sato K, Simon MD, Levin AM, Shokat KM, Weiss GA. Dissecting the engrailed homeodomain-DNA interaction by phage-displayed shotgun scanning. Chem Biol. 2004;11:1017–1023. doi: 10.1016/j.chembiol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Scott MP, Weiner AJ. Structural relationships among genes that control development: sequence homology between the antennapedia, ultrabithorax, and fushi tarazu loci of Drosophila. Proc Natl Acad Sci USA. 1984;81:4115–4119. doi: 10.1073/pnas.81.13.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman JG, Seidman C. Transcription factor haploinsufficiency: when half a loaf is not enough. J Clin Invest. 2002;109:451–455. doi: 10.1172/JCI15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Transcription factors and human disease. Oxford University Press; Oxford: 1989. [Google Scholar]
- Shang Z, Isaac VE, Li H, Patel L, Catron KM, Curran T, Montelione GT, Abate C. Design of a “minimAl” homeodomain: the N-terminal arm modulates DNA binding affinity and stabilizes homeodomain structure. Proc Natl Acad Sci USA. 1994;91:8373–8377. doi: 10.1073/pnas.91.18.8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Shokat KM. Adaptability at a protein-DNA interface: re-engineering the engrailed homeodomain to recognize an unnatural nucleotide. J Am Chem Soc. 2004;126:8078–8079. doi: 10.1021/ja048113w. [DOI] [PubMed] [Google Scholar]
- Simon MD, Sato K, Weiss GA, Shokat KM. A phage display selection of engrailed homeodomain mutants and the importance of residue Q50. Nucleic Acids Res. 2004;32:3623–3631. doi: 10.1093/nar/gkh690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaroop A, Wang Q, Wu W, Cook J, Coats C, Xu S, Chen S, Zack D, Sieving P. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of photoreceptor function. Hum Mol Genet. 1999;8:299–305. doi: 10.1093/hmg/8.2.299. [DOI] [PubMed] [Google Scholar]
- Treisman J, Gonczy P, Vashishtha M, Harris E, Desplan C. A single amino acid can determine the DNA binding specificity of homeodomain proteins. Cell. 1989;59:553–562. doi: 10.1016/0092-8674(89)90038-x. [DOI] [PubMed] [Google Scholar]
- Tucker-Kellogg L, Rould MA, Chambers KA, Ades SE, Sauer RT, Pabo CO. Engrailed (Gln50 → Lys) homeodomain- DNA complex at 1.9 A resolution: structural basis for enhanced affinity and altered specificity. Structure. 1997;5:1047–1054. doi: 10.1016/s0969-2126(97)00256-6. [DOI] [PubMed] [Google Scholar]
- Vaxillaire M, Abderrahmani A, Boutin P, Bailleul B, Froguel P, Yaniv M, Pontoglio M. Anatomy of a homeoprotein revealed by the analysis of human MODY3 mutations. J Biol Chem. 1999;274:35639–35646. doi: 10.1074/jbc.274.50.35639. [DOI] [PubMed] [Google Scholar]
- Vitkup D, Sander C, Church GM. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003;4:R72. doi: 10.1186/gb-2003-4-11-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Moult J. SNPs, protein structure, and disease. Hum Mutat. 2001;17:263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- Weiler S, Gruschus JM, Tsao DH, Yu L, Wang LH, Nirenberg M, Ferretti JA. Site-directed mutations in the vnd/NK-2 homeodomain. Basis of variations in structure and sequence- specific DNA binding. J Biol Chem. 1998;273:10994–11000. doi: 10.1074/jbc.273.18.10994. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Tang Z, Elanko N, Walsh S, Twigg SR, Hurst JA, Wall SA, Chrzanowska KH, Maxson RE., Jr Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat Genet. 2000;24:387–390. doi: 10.1038/74224. [DOI] [PubMed] [Google Scholar]
- Wilson DS, Guenther B, Desplan C, Kuriyan J. High resolution crystal structure of a paired (Pax) class cooperative homeodomain dimer on DNA. Cell. 1995;82:709–719. doi: 10.1016/0092-8674(95)90468-9. [DOI] [PubMed] [Google Scholar]
- Wolberger C. Homeodomain interactions. Curr Opin Struct Biol. 1996;6:62–68. doi: 10.1016/s0959-440x(96)80096-0. [DOI] [PubMed] [Google Scholar]
- Wolfe SA. Mapping key elements of a protein motif. Chem Biol. 2004;11:889–891. doi: 10.1016/j.chembiol.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183–212. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- Wu W, Cogan JD, Pfaffle RW, Dasen JS, Frisch H, O’Connell SM, Flynn SE, Brown MR, Mullis PE, Parks JS, Phillips JA, 3rd, Rosenfeld MG. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. 1998;18:147–149. doi: 10.1038/ng0298-147. [DOI] [PubMed] [Google Scholar]
- Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy- terminal subdomain in DNA binding. Genes Dev. 1999;13:1263–1275. doi: 10.1101/gad.13.10.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Tomura H, Nishigori H, Sho K, Mabe H, Iwatani N, Takumi T, Kito Y, Moriya N, Muroya K, Ogata T, Onigata K, Morikawa A, Inoue I, Takeda J. Identification of mutations in the hepatocyte nuclear factor-1alpha gene in Japanese subjects with early-onset NIDDM and functional analysis of the mutant proteins. Diabetes. 1999;48:645–648. doi: 10.2337/diabetes.48.3.645. [DOI] [PubMed] [Google Scholar]
- Yoshiuchi I, Yamagata K, Yang Q, Iwahashi H, Okita K, Yamamoto K, Oue T, Imagawa A, Hamaguchi T, Yamasaki T, Horikawa Y, Satoh T, Nakajima H, Miyazaki J, Higashiyama S, Miyagawa J, Namba M, Hanafusa T, Matsuzawa Y. Three new mutations in the hepatocyte nuclear factor-1alpha gene in Japanese subjects with diabetes mellitus: clinical features and functional characterization. Diabetologia. 1999;42:621–626. doi: 10.1007/s001250051204. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gerstein M. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 2003;31:5338–5348. doi: 10.1093/nar/gkg745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Westphal H. Homeobox genes and human genetic disorders. Curr Mol Med. 2002;2:13–23. doi: 10.2174/1566524023363077. [DOI] [PubMed] [Google Scholar]