

Abstract
Background
Domain experts manually construct the Structural Classification of Protein (SCOP) database to categorize and compare protein structures. Even though using the SCOP database is believed to be more reliable than classification results from other methods, it is labor intensive. To mimic human classification processes, we develop an automatic SCOP fold classification system to assign possible known SCOP folds and recognize novel folds for newly-discovered proteins.
Results
With a sufficient amount of ground truth data, our system is able to assign the known folds for newly-discovered proteins in the latest SCOP v1.69 release with 92.17% accuracy. Our system also recognizes the novel folds with 89.27% accuracy using 10 fold cross validation. The average response time for proteins with 500 and 1409 amino acids to complete the classification process is 4.1 and 17.4 seconds, respectively. By comparison with several structural alignment algorithms, our approach outperforms previous methods on both the classification accuracy and efficiency.
Conclusion
In this paper, we build an advanced, non-parametric classifier to accelerate the manual classification processes of SCOP. With satisfactory ground truth data from the SCOP database, our approach identifies relevant domain knowledge and yields reasonably accurate classifications. Our system is publicly accessible at http://ProteinDBS.rnet.missouri.edu/E-Predict.php.
Background
Protein structure classification is well-known to be an important research topic in computational and molecular biology. Through the use of structural classification, life science researchers and biologists are able to study evolutionary evidence from similar proteins that have been conserved in multiple species. In addition, similar 3-D conformations of enzyme active sites and binding sites may correlate with biochemical functions [1]. In recent years, structural genomics projects [2-5] have aimed to link protein sequences to possible functions via high-throughput techniques such as X-ray crystallography and nuclear magnetic resonance (NMR) that determine 3-D protein structures. With a large-scale set of newly-discovered structures, a system that classifies similar protein structures with high efficiency and accuracy becomes an indispensable requirement to the study of structure-to-function relationships.
Several classification systems categorize proteins based on structural similarities. The Class, Architecture, Topology, Homologous Superfamily (CATH) database [6] is constructed by applying the Secondary Structure Alignment Program (SSAP) [7], which consists of a double dynamic programming technique to find the optimal structural alignment of two proteins. The Fold Classification based on Structure-Structure Alignment of Proteins (FSSP) database [8] is built based on the Distance Alignment (DALI) [9] algorithm that applies Monte Carlo heuristics to compare structural similarities from 2-D distance matrices mapped from 3-D protein structures. Generally, these systems rely on the structural alignment algorithms to measure the similarity of two proteins, which is known to be of complexity NP-Hard [10]. To reduce the computational effort of scanning large-scale protein databases, those structural alignment algorithms need to apply heuristics with trade-offs which may return divergent results from the same query protein. At present, the Structural Classification of Protein (SCOP) database [11], which is manually constructed by human experts, is believed to contain the most accurate structural classifications. In the SCOP database, proteins with similar domain structures are usually clustered into the same fold hierarchy. Even though manual classification provides reliable results, it is labor intensive. As of May 30th, 2006, 10864 newly-discovered proteins deposited in the Protein Data Bank (PDB) [12] have not been classified in the latest SCOP v1.69 release. The number of newly-discovered proteins is increasing continuously.
Recent studies [13,14] apply a consensus scheme to classify the SCOP folds for newly-discovered proteins by intersecting multiple classification results from classical structural alignment algorithms such as DALI [9], Combinatorial Extension (CE) [15] and VAST [16]. These consensus approaches yield higher classification accuracies than each individual method. However, a combination of structural alignment algorithms is computationally expensive. To accelerate the manual classification process of SCOP, there is an urgent need to develop a fast, automated SCOP fold classification system with a reasonably high accuracy. By extending our recent works with the real-time tertiary structure retrieval system, ProteinDBS [17-19], we have already studied an efficient model of association rule (AR) mining to identify relevant structural patterns in proteins for SCOP domain and fold classifications [20]. In this paper, we further develop a non-parametric classifier to conduct the SCOP fold classifications with better accuracy and efficiency. Our contribution is to introduce a real-time classification model, E-Predict, that applies the E_Measure metric [21] from the Information Retrieval (IR) field to assign the known SCOP folds and recognize the novel folds for newly-discovered proteins. In the past, a number of systems have been developed to assign a protein structure to an existing fold or recognize it as a novel fold. For example, DALI [22] uses Z-score of the best structural match to either assign a structure to a known fold (Z>2) or novel fold (Z≤2). Other programs, such as CE [15] and VAST [23] can perform similar tasks. However, the computational effort associated with those methods prevents a user from exploring the protein structure database in real time.
Results
There are two important tasks for the SCOP fold classifications. 1) Known SCOP Fold Assignments: the algorithm assigns newly-discovered protein structures into the known SCOP folds. 2) Novel SCOP Fold Recognitions: the algorithm detects whether or not newly-discovered protein structures should be categorized into the novel folds. Given two SCOP database releases v1 and v2 (v1 ⊂ v2), denotes a set of newly-discovered proteins in v2 that have not been identified in v1. The proteins from will be partitioned into either the known SCOP folds of v1 (), or the novel folds that have not been determined prior to v2 (), where . In our experiments, we measure the classification accuracy for proteins from , and then we gauge the accuracy for classifying proteins from . Finally, we report the efficiency of SCOP fold classifications.
Assigning newly-discovered proteins to the known folds
We conduct three experiments for classifying newly-discovered proteins into the known folds. The first experiment compares our classification model, E-Predict, with several methods reported in a recent work [13] such as CE, DALI, VAST and CBOOST. Our test data shown in Table 1 is the same test set used in their work, which has proteins with average sequence identities equal to 16.88% and average sequence similarities equal to 20.76% by conducting all against all pairwise alignments using EMBOSS-Align [24] algorithm. The same ground truth data with their work includes proteins from the entire SCOP vl.59 release. To evaluate the accuracy, we use a general metric, Correct Classification Rate (CCR), which is defined as follows:
Table 1.
A test set that contains 37 protein chains from [13].
| pdb_id | fold_id | pdb_id | fold_id | pdb_id | fold_id | pdb_id | fold_id | pdb_id | fold_id |
| 1gyz_A | 63569 | 1key_A | 48370 | 1key_B | 48370 | 1key_C | 48370 | 1key_D | 48370 |
| 1lkv_X | 48370 | 1ldk_A | 48370 | 1ifr_A | 48725 | 1ivt_A | 48725 | 1gyv_A | 48725 |
| 1gyu_A | 48725 | 1iu1_A | 48725 | 1iu1_B | 48725 | 1gyw_A | 48725 | 1gyw_B | 48725 |
| 1l6p_A | 48725 | 1lpl_A | 50036 | 1k3b_A | 50875 | 1gyh_A | 50933 | 1gyh_B | 50933 |
| 1gyh_C | 50933 | 1gyh_D | 50933 | 1gyh_E | 50933 | 1gyh_F | 50933 | 1gyd_B | 50933 |
| 1gye_B | 50933 | 1jof_A | 50964 | 1jof_B | 50964 | 1jof_C | 50964 | 1jof_D | 50964 |
| 1jof_E | 50964 | 1jof_F | 50964 | 1jof_G | 50964 | 1Jof_H | 50964 | 1l2q_A | 51350 |
| 1ln4_A | 55199 | 1kuu_A | 56234 |
Figure 1 shows that E-Predict outperforms DALI, CE, and VAST, exhibiting an accuracy of 64.86%. Can et al. [13] have proposed a method, named CBOOST, which utilizes a decision tree to integrate DALI, CE, and VAST, achieving the same accuracy of 64.86%. It is worth mentioning that the computationally expensive structural alignment algorithms of CBOOST may not be able to efficiently classify a large number of newly-discovered proteins generated from on-going, high-throughput structure determination projects.
Figure 1.
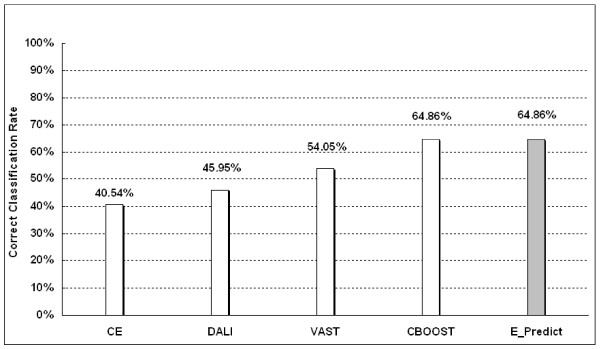
The Correct Classification Rate of assigning the known folds for test proteins in Table 1.
The second experiment exhaustively evaluates the accuracy of E-Predict on several general test sets from to . In Table 2 and Table 3, our test proteins in are selected from the known SCOP folds of v2, which also maintain at least one protein chain and 10 proteins in v1, respectively. Figure 2(a) shows that E-Predict achieves 72% to 82% classification accuracies for the general test sets of seven SCOP releases. According to Figure 3, there exists a large number of SCOP folds with small sizes. When a newly-discovered protein belongs to a small-size fold, there is a limited amount of ground truth data available. In machine learning, classifiers usually require sufficient ground truth data to guarantee the accuracy. Figure 2(b) demonstrates that E-Predict is able to achieve much higher accuracies, 90% to 96%, for the general test sets of seven SCOP releases with more than 10 ground truth proteins. In the future, when newly-discovered protein structures are categorized into those small-size SCOP folds, the accuracy of E-Predict could be further improved.
Table 2.
The number of proteins in a test set of novel folds, general and non-redundant test sets in which are selected from the known SCOP folds of v2 with at least one protein chain in v1.
| test set | size (#proteins) | test set | size (#proteins) | test set | size (#proteins) |
| 4192 | 442 | - | - | ||
| 4047 | 431 | 94 | |||
| 4547 | 468 | 10 | |||
| 5226 | 491 | 190 | |||
| 5445 | 494 | 48 | |||
| 10521 | 736 | 215 | |||
| 5604 | 585 | 86 |
Table 3.
The number of proteins in general and non-redundant test sets in which are selected from the known SCOP folds of v2 with at least 10 protein chains in v1.
| test set | size (#proteins) | test set | size (#proteins) |
| 1832 | 158 | ||
| 1901 | 168 | ||
| 2136 | 166 | ||
| 1947 | 189 | ||
| 2062 | 198 | ||
| 4735 | 302 | ||
| 2298 | 263 |
Figure 2.
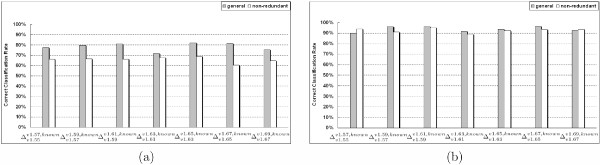
The Correct Classification Rate of assigning the known folds for various SCOP releases using E-Predict on (a) general and non-redundant test set in which are selected from the known SCOP folds of v2 with at least one protein chain in v1 (Table 2) (b) general and non-redundant test set in which are selected from the known SCOP folds of v2 with at least 10 protein chains in v1 (Table 3).
Figure 3.
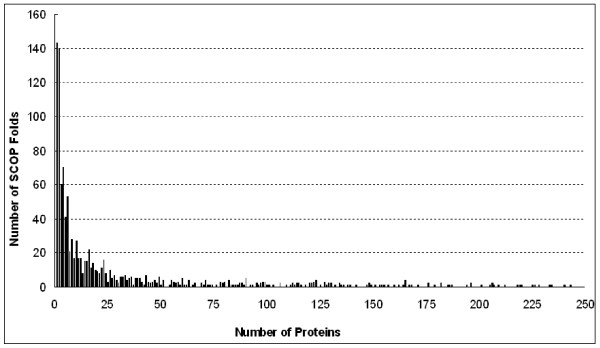
The amount of proteins in the folds against the number of SCOP folds in the SCOP v1.69 release.
The third experiment evaluates the accuracy of E-Predict on non-redundant test sets, which are obtained from randomly sampling one protein chain among each SCOP superfamily. In Table 2 and Table 3, a non-redundant test set is defined by randomly selecting one protein from each SCOP superfamily of the general test set . According to SCOP [11], proteins between two different SCOP superfamilies have low sequence similarities, which suggest that test proteins in our non-redundant sets should maintain low sequence similarities. Table 4 measures the degree of sequence redundancy for 10 pairs of proteins, which are randomly sampled from the non-redundant set with the average sequence identity and sequence similarity equal to 12.55% and 21.17%, respectively. In addition, the experiment using the non-redundant test sets avoids the case that some folds in the general test sets predominate the classification accuracy with relatively more test proteins. For example, there are 900 out of 1000 test proteins in a general test from the same SCOP fold f1. The quantity of this fold may affect the accuracy significantly when a majority of these 900 proteins are correctly classified. In Figure 2(a), E-Predict presents a reduction of accuracies on several sets of non-redundant proteins in comparison with the general test sets in Table 2, which includes small-size folds. This gap demonstrates that the impact of some SCOP folds with outnumbered proteins in the general test sets improves the overall accuracy. Figure 2(b) shows that E-Predict exhibits similar accuracies on seven sets of the non-redundant proteins in comparison with the general test sets in Table 3, which have at least 10 ground truth proteins. This suggests that with a sufficient amount of ground truth data non-redundant proteins can still be classified with a reasonably high accuracy.
Table 4.
The sequence redundancy in a set that contains 10 pairs of proteins, which are randomly sampled from
| pairs | pdb_id1 | super family_id1 | pdb_id2 | super family_id2 | sequence identity | sequence similarity |
| 01 | 1osd_A | 55008 | 1uta_A | 110997 | 2.10% | 3.50% |
| 02 | 1ug8_A | 82708 | 1vm0_A | 82704 | 12.80% | 26.80% |
| 03 | 1v5n_A | 57889 | 1rq8_A | 75471 | 13.60% | 23.50% |
| 04 | 1veu_B | 103196 | 1j3m_A | 103247 | 22.40% | 34.20% |
| 05 | 1tu1_B | 55724 | 1smb_A | 55797 | 6.80% | 10.80% |
| 06 | 1thq_A | 56925 | 1xfs_B | 55961 | 18.10% | 28.40% |
| 07 | 1vki_B | 55826 | 1sk3_A | 55846 | 17.70% | 30.50% |
| 08 | 1tf1_D | 55781 | 1pp6_E | 55676 | 10.30% | 17.50% |
| 09 | 1ucd_A | 55895 | 1vkw_A | 55469 | 9.00% | 14.70% |
| 10 | 1tt4_A | 55931 | 1vkp_A | 55909 | 12.70% | 21.80% |
| Avg. 12.55% | Avg. 21.17% |
Recognizing the novel folds for newly-discovered proteins
We measure the accuracies of classifying six sets of proteins with the novel folds from to , which are listed in Table 2. We accumulate labeled proteins from the prior SCOP releases to obtain more ground truth data. For example, when an experiment is conducted with test proteins from , our ground truth data is composed of new proteins from . We compare our E-Predict algorithm with two prevalent classification methods, Nearest Neighbor search (NN) [25] and C4.5 Decision Tree (DT) [26]. Figure 4 presents a plot of CCR against six test sets to , which are listed in Table 2. From computational results, E-Predict outperforms NN and C4.5 DT. There is a noticeable reduction in accuracy when classifying proteins in . This is probably because the test set, , is harder to be correctly predicted than the other sets. To address the issue that accuracies may be biased by particular new structures, we conduct 10 fold cross validation that sequentially selects 10% of ground truth data from as a test set and the rest of 90% of ground truth data as a training set for 10 times. In the 10 fold experiment, our approach achieves 89.27% accuracy of the novel fold recognitions.
Figure 4.
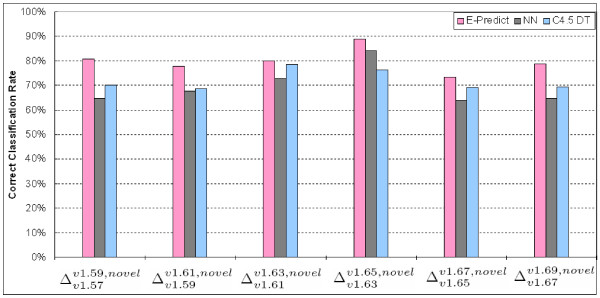
The Correct Classification Rates of recognizing the novel SCOP folds for proteins in various SCOP releases.
Efficiency
For efficiency, we measure the average response time of the entire classification process, including feature extraction, nearest neighbor search on an M-tree [27], and the computation of the SCOP folds by the E-Predict algorithm. The classification process performs one-against-all structural comparisons by scanning the entire SCOP database. Our system runs on a Fedora-Core Linux system with Dual Xeon IV 2.4 GHz processors and 2 GB RAM. A large-scale test set is chosen from the SCOP vl.69 release with 51911 protein chains which have more than 20 amino acids. Figure 5 shows the average response time of fold classifications for various protein chain sizes. When the protein size increases, the E-Predict algorithm demands more computational resources to extract features from larger distance matrices. When the protein chain size reaches a certain threshold, the Linux system may swap huge distance matrices into the virtual memory resulting in a significant I/O time. This effect is reflected in Figure 5 with long computation times for the protein chain size larger than 1099 amino acids, where more memory is required to prevent page swapping. On average, classifying a newly-discovered protein to a SCOP fold takes 3.5 seconds. In our test set, the longest protein chain, comprised of 1409 amino acids, completes the classification process in 17.4 seconds.
Figure 5.
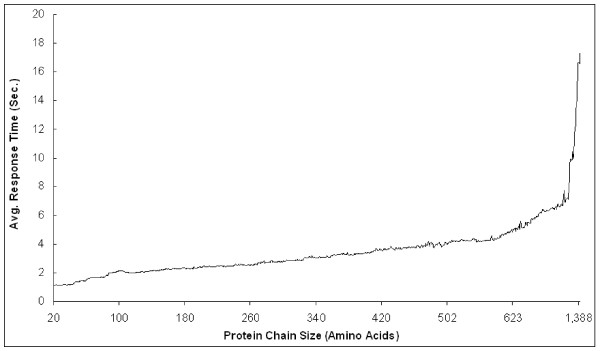
The protein chain sizes against the average response time of classifying test proteins.
Discussion
Our approach yields better accuracy and efficiency compared to the structure alignment algorithms. The accuracy is achieved by analyzing the ranked SCOP folds of a nearest neighbors search using the E-Predict algorithm. In addition, efficiency results from using an M-tree [27] for fast nearest neighbor searches. In the following subsections, we compare our performance with the structural alignment algorithms in terms of efficiency and accuracy.
Performance in efficiency
Since structural alignment algorithms usually apply dynamic programming techniques to align each pair of amino acids in two proteins, they demand a huge amount of computational resources. Instead of aligning amino acids, our E-Predict model transforms relevant protein structure information into high-level features, and similar protein structures are then retrieved from a high-dimensional feature space by a nearest neighbors search in the M-tree. Our approach is able to return the classification result in seconds. Since performing the structural alignment algorithms with multiple pairwise alignments of a newly-discovered structure against the known protein structures from the SCOP database is known to be computationally expensive [10], the response times for the structural alignment algorithms are not plotted in Figure 5.
The accuracy of assigning newly-discovered proteins to the known folds
For the assignment of proteins to the known SCOP folds, the E-Predict algorithm mainly contributes to the accuracy. Traditional structural alignment methods usually apply heuristics to reduce computational efforts of aligning a large combination of amino acids in two proteins. Different heuristics could return diverse results from the same set of proteins since these algorithms might be trapped in local optimal solutions. Even though a consensus method that combines classification results of multiple structural alignment algorithms outperforms each individual structural alignment approach [13], it is computationally expensive. Instead of performing structural alignments, our model maps both known proteins from the SCOP database and newly-discovered protein structures into 33-D feature vectors. With a nearest neighbor search for a newly-discovered structure t in the high-dimensional feature space, there may exist multiple candidate folds, which are associated with nearest neighbor proteins in the vicinity of t. One way to assign a SCOP fold to t is to choose the fold of the nearest neighbor protein in the high-dimensional feature space. Since it is possible that hundreds of folds are partially overlapped in the high-dimensional feature space, the nearest neighbor of t may be an outlier that deviates from the majority of proteins in its fold. To avoid selecting an outlier, we apply the E_Measure metric that considers the ranks of at least two nearest neighbor proteins for each fold. The algorithm rewards a SCOP fold in which proteins are highly ranked and penalizes a fold with proteins in the lower ranks. Hence, when the SCOP fold includes only a single highly ranked protein with the other proteins from this fold ranked much lower, the algorithm is able to avoid assigning this fold to t based on the penalty of low ranking. From computational results, E_Measure has a vital impact on the classification accuracy.
Misclassifications of assigning newly-discovered proteins to the known folds
Within the framework of ProteinDBS [17-19], our model, E-Predict, transforms a 3-D protein structure into a 33-D feature vector that represents the geometric properties of folded proteins. Applying these features to measure the structural similarity of proteins, E-Predict outperforms several classification methods that apply the structural alignment algorithm using the test set in Table 1. E-Predict also yields reasonably high accuracy for several test sets in Table 3 with sufficient ground truth data. However, misclassifications still exist. The limited amount of 33-D ground truth data available for training contributes to the classification errors. As more ground truth data becomes available in small-size SCOP folds, a higher classification accuracy is expected. The second reason for misclassifications is due to the overlapping of folds in the high-dimensional feature space. To further separate overlapping folds, our system needs more relevant features to detect the protein 3-D folding with sufficient discriminating power. Another possible reason for misclassifications is that SCOP may categorize a partial segment of a PDB protein chain (substructure) into a domain. Since our approach measures the global similarity of distance matrices for classification, users need to submit the portion of the protein chain identified in the SCOP domain to ensure a correct classification. In Figure 6, we measure the correlation between the classification accuracy and a structure variation value, S, for a query protein t and the best matched protein of t in our classified SCOP fold. For a pair of proteins (p1, p2), the structural variation S is defined as follows:
Figure 6.
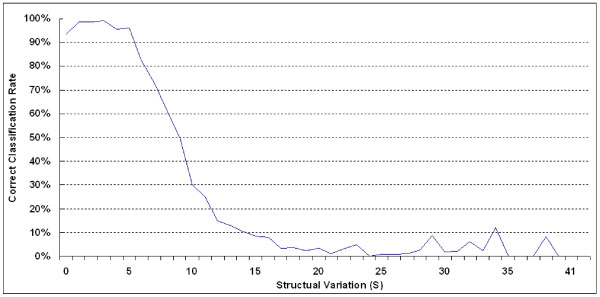
Correct Classification Rates of classifying test proteins against structural variation values.
where RMSD means the root mean square deviation of aligned segments, and NA denotes the number of amino acids in the aligned segments of two proteins. Np1 and Np2 represent the number of amino acid residues in p1 and p2, respectively. These measurements are computed using SARF [28]. The smaller S value can be interpreted as a better structural match for two proteins p1 and p2. Two proteins that have a high structural similarity can usually be superimposed with longer aligned residue segments and a small RMSD value, resulting in a small S value. For example, the SARF algorithm aligns a query protein t with 100 amino acids and its best matched protein p1 with 100 amino acids and returns structurally similar segments with 90 amino acid residues and 0.3 Å of RMSD. Their structure variation value S is computed as 0.3/() = 0.67. When S is smaller than 6, we expect the E-Predict algorithm to maintain above 90% classification accuracy. This statistic is obtained from the classification of 41262 testing proteins.
The accuracy of recognizing the novel folds for newly-discovered proteins
Since no protein has been labeled with the novel folds in our 33-D ground truth data, the novel fold recognition becomes a challenging problem. To address this issue, we introduce three features: E_Measure evaluation score, structural variation value, and Euclidean distance measurement. These features measure structural similarity between a newly-discovered protein and the nearest neighbor protein in a candidate known fold suggested by the E-Predict algorithm. Then, our method applies the E-Predict algorithm as a classifier to identify meaningful patterns from ground truth data, which has been obtained by the aggregation of proteins in several prior SCOP releases. Computational results show that using these three features benefits the classification accuracy.
Misclassifications of recognizing the novel folds for newly-discovered proteins
To recognize the novel folds for newly-discovered protein structures, our classification model exploits three relevant features. With the assumption that protein structures in the novel folds usually present low structural similarities to proteins in the known folds, a high E_Measure evaluation score, a high Euclidean distance, and a high structural variation value are expected for newly-discovered protein structures from the novel folds. Due to noise in ground truth data and imperfect features, a few proteins in the novel folds may have a low structural variation value, a low E_Measure score, or a low Euclidean distance measurement. Even though our approach presents an improved accuracy over NN and C4.5 DT, there is still a need to discover more relevant features for better recognition performance.
Conclusion
We have developed an automatic SCOP fold classification system that is able to assign the known SCOP folds and recognize the novel folds for newly-discovered proteins. For the known fold assignments, the algorithm transforms protein structures into 33-D feature vectors and constructs an M-tree to index these feature vectors for fast retrievals. The E-Predict algorithm is then applied to classify newly-discovered proteins in the known SCOP folds. For the novel fold recognitions, the algorithm utilizes three relevant features that are related to structural similarity of proteins. From the computational results, our method outperforms several structural alignment algorithms such as DALI, CE and VAST, achieving reasonably high classification accuracy and efficiency. This research can help accelerate the classification process of the SCOP database and benefit the biomedical research community to further study biochemical functions of proteins with similar 3-D structures.
Methods
Our classification model, E-Predict, contains three primary functions. First, E-Predict assigns newly-discovered protein structures to the known SCOP folds. Second, E-Predict recognizes the novel folds for newly-discovered protein structures. Third, E-Predict indexes the high-dimensional protein data for a fast nearest neighbors search.
Assigning newly-discovered proteins to the known folds
According to the SCOP hierarchical setting, proteins that share similar secondary structure arrangements are usually classified in the fold level [11]. The entire process of assigning newly-discovered proteins to the known folds is shown in Figure 7. The labeling procedure transforms protein structures from the SCOP database into 33-D feature vectors, which are labeled with their corresponding SCOP folds. These labeled proteins are then used as our ground truth data. The testing procedure converts newly-discovered proteins into feature vectors and submits these unlabeled vectors into a classifier to obtain possible SCOP fold assignments. In the following, we discuss several components of the entire process such as distance matrix generation, feature extraction, and classifier design.
Figure 7.
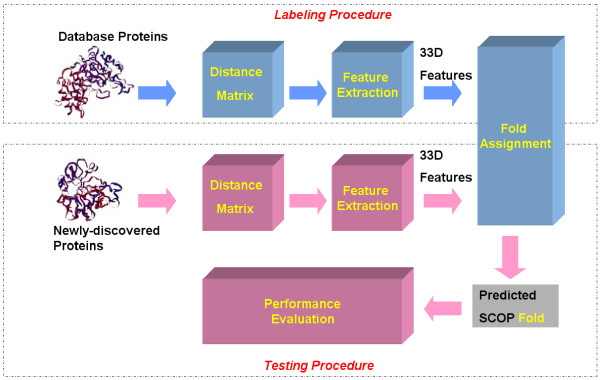
E-Predict model for assigning newly-discovered proteins to the known folds.
Mapping 3-D backbone structures into 2-D distance matrices
Proteins are polypeptide chains, which are chained by 20 types of amino acids. Instead of considering the side chains of amino acids, many computational biology papers [6,9,15] use the Cα atom to describe each amino acid. In our model, the Cα backbone of the kth protein chain with n amino acids can be represented by a set of vectors, Ωk = {}, where denotes the 3-D coordinate of the ith Cα atom. Protein backbones can be transformed into 2-D distance matrices. For Ωk, the corresponding distance matrix, Dk, is defined as D[i, j] = dist(, ), 1 ≤ i, j ≤ n, in a Euclidean space. A distance geometry method [29] shows that the 2-D distance matrix is generally sufficient to recover the original 3-D structure in polynomial time. Several examples in the literature convert protein backbone structures into distance matrices and then detect the structural similarity from them [9,30,31]. Since 2-D distance matrices maintain sufficient 3-D structural information, similar protein backbones are expected to have similar distance matrices. Figure 8 shows 3-D protein backbone structures and their corresponding 2-D distance matrices sampled from the SCOP Heme-dependent peroxidases and Acid proteases folds. Within each SCOP fold, the proteins maintain high similarities in both 3-D backbone structures and 2-D distance matrices. Variations in distance matrices are detectable by comparing structures that belong to different folds.
Figure 8.

The 3-D backbone structures and distance matrices of four protein chains, which are selected from the SCOP folds: (1)Heme-dependent peroxidases: 1kta_A(a-b), 1ekv_A(c-d), (2)Acid proteases : 1lee_A(e-f), 1lf2_A(g-h).
Feature extraction
In the area of content-based image retrieval (CBIR), several computational techniques have been developed to retrieve visually similar images from databases for a query image [32-34]. When each element of the distance matrix is interpreted as a grayscale pixel, the distance matrix can be converted into a 2-D image. After preprocessing protein structures into grayscale images, we apply the CBIR technique to retrieve similar distance matrices in ranked order. In our previous work [17,19], we extract 24 local and 9 global features that are relevant to the visual content of distance matrices using a suite of computer vision algorithms such as histograms and textures [35-37]. To obtain local features, each distance matrix is partitioned into six band regions, which are parallel to the diagonal of the matrix. In each band, histograms are computed by four bins of distance ranges: [0–5], [6–10], [11–15], and [16-∞]. Since the distance matrix is symmetric, global features such as Entropy, Homogenity and Contrast are computed for the entire upper triangle of each distance image. Interested readers are referred to our previous publications [17,19] for details of the feature extraction algorithms applied in this work. The transformation of a distance matrix into a 33-D feature vector ensures each feature vector uniquely identifies a protein chain. Table 5 and Table 6 list the 24 local features and 9 global features extracted for proteins from the SCOP Heme-dependent peroxidases and Acid proteases folds, respectively. For a large-scale protein database such as the SCOP database, it is important to develop a classifier that groups proteins within the same fold and separates proteins from different folds.
Table 5.
Local features of proteins from the SCOP folds: (1)Heme-dependent peroxidases : 1stq_A, 1sog_A, (2) Acid proteases: 1lee_A, 1lf2_A. Histogram [a,b] denotes the distance histogram for the ath band region and the bth grayscale bin.
| Image Features | 1stq_A | 1sog_A | 1lee_A | 1lf2_A |
| Histogram [1,1] | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| Histogram [l,2] | 0.0002 | 0.0002 | 0.0000 | 0.0000 |
| Histogram [l,3] | 0.0018 | 0.0020 | 0.0001 | 0.0002 |
| Histogram [1,4] | 0.0050 | 0.0053 | 0.0009 | 0.0011 |
| Histogram [2,1] | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| Histogram [2,2] | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| Histogram [2,3] | 0.0023 | 0.0022 | 0.0000 | 0.0001 |
| Histogram [2,4] | 0.0044 | 0.0043 | 0.0012 | 0.0010 |
| Histogram [3,1] | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| Histogram [3,2] | 0.0004 | 0.0004 | 0.0003 | 0.0004 |
| Histogram [3,3] | 0.0020 | 0.0019 | 0.0017 | 0.0019 |
| Histogram [3,4] | 0.0092 | 0.0080 | 0.0048 | 0.0055 |
| Histogram [4,1] | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| Histogram [4,2] | 0.0006 | 0.0006 | 0.0015 | 0.0012 |
| Histogram [4,3] | 0.0040 | 0.0042 | 0.0056 | 0.0053 |
| Histogram [4,4] | 0.0132 | 0.0130 | 0.0172 | 0.0166 |
| Histogram [5,1] | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| Histogram [5,2] | 0.0014 | 0.0015 | 0.0036 | 0.0035 |
| Histogram [5,3] | 0.0124 | 0.0128 | 0.0133 | 0.0134 |
| Histogram [5,4] | 0.0423 | 0.0425 | 0.0298 | 0.0304 |
| Histogram [6,1] | 0.0203 | 0.0201 | 0.0179 | 0.0180 |
| Histogram [6,2] | 0.0392 | 0.0386 | 0.0291 | 0.0289 |
| Histogram [6,3] | 0.0503 | 0.0496 | 0.0474 | 0.0485 |
| Histogram [6,4] | 0.0845 | 0.0833 | 0.0796 | 0.0795 |
Table 6.
Global features of proteins from the SCOP folds: (1)Heme-dependent peroxidases: 1stq_A, 1sog_A, (2)Acid proteases: 1lee_A, 1lf2_A.
| Image Features | 1stq_A | 1sog_A | 1lee_A | 1lf2_A |
| Dimension | 291.00 | 294.00 | 331.00 | 329.00 |
| Binary_Threshold | 23.0000 | 23.0000 | 26.0000 | 26.0000 |
| Texture_Energy | 0.0155 | 0.0153 | 0.0107 | 0.0107 |
| Texture_Entropy | 51.7143 | 51.8067 | 54.4426 | 54.4139 |
| Texture_Homogenity1 | 2.5344 | 2.5261 | 2.2184 | 2.2192 |
| Texture_Homogenity2 | 1.7608 | 1.7529 | 1.4467 | 1.4485 |
| Texture_Contrast | 0.0027 | 0.0027 | 0.0041 | 0.0041 |
| Texture_Correlation | 6.8883 | 6.8914 | 6.7682 | 6.7659 |
| Texture_Cluster_Tendency | 0.0387 | 0.0392 | 0.0517 | 0.0515 |
Fold assignment
To ensure high accuracy when classifying a newly-discovered protein, we have designed a novel method that extends algorithms from Information Retrieval (IR) [38]. For the assignment of newly-discovered proteins to the known folds, we first discuss two well-recognized methods, C4.5 Decision Tree (DT) [26] and Nearest Neighbor (NN) [25], and then our new approach, E-Predict, which achieves a better classification accuracy than C4.5 DT or NN.
Decision tree approaches have been developed for classification in supervised machine learning [26]. Using a set of ground truth data that contains feature vectors of proteins and their associated fold labels, a classifier usually divides the high-dimensional feature space, discussed previously, into multiple subspaces, which are normally in the form of hyper-cubes or hyper-spheres. In the labeling process using C4.5 DT, the majority of proteins from the same fold are expected to be clustered into a small number of subspaces. Proteins from different folds are separated into different subspaces based on minimization of entropy. A newly-discovered protein can then be classified into one of the known subspaces for fold assignment by following decision features of internal tree nodes and their corresponding thresholds. However, a small number of proteins from the same SCOP fold with similar feature values may be partitioned into different leaf nodes by C4.5 DT due to their feature values, which are distributed around thresholds of internal nodes. With hundreds of folds in the SCOP database, the more proteins from different folds that have been grouped into a leaf node, the higher the probability of misclassification.
Instead of partitioning the high-dimensional space, Nearest Neighbor (NN) [25] assigns a SCOP fold for a newly-discovered protein by searching for its nearest neighbor with the Euclidean distance measurement. Figure 9(a) shows that NN outperforms C4.5 DT by 13%, on average, for fold assignments using the test sets in , which are selected from the SCOP folds in the SCOP v2 release that have at least one protein from the SCOP v1 release. Figure 9(b) shows that NN also outperforms C4.5 DT by 8.45%, on average, for fold assignments using the test sets in , which are selected from the SCOP folds in the SCOP v2 release that have at least 10 proteins from the SCOP v1 release. Even though NN yields a better classification performance than C4.5 DT, there still exists an important issue to consider: misclassifications from an outlier in NN search. An outlier is defined as a protein chain whose feature vector deviates greatly from the majority of proteins in the same SCOP fold. In the high-dimensional feature space with multiple overlapping SCOP folds, NN search may assign an incorrect SCOP fold to a newly-discovered protein by selecting an outlier as the nearest neighbor. For instance, we assume that the true fold of a newly-discovered protein t is F2. From Result F1, shown in the second row of Figure 10, the nearest neighbor of t is p1, which is an outlier to the majority of proteins in fold F1. When NN search is used for classification, the algorithm falsely classifies that t is in fold F1. One possible way to address this issue is to assign the newly-discovered protein to the SCOP fold that has the majority in the top k Nearest Neighbor (k-NN). In Figure 9(b), 3-NN yields a better accuracy than 5-NN in six test sets. Also, we find that 3-NN achieves a better accuracy than NN in . Unfortunately, 3-NN does not perform as well as NN on the other test sets due to the existence of two or more outliers in the 3-NN selection. In general, the k-NN classification method simply takes the majority of the top k nearest neighbors without considering the ranking information of nearest neighbor proteins.
Figure 9.
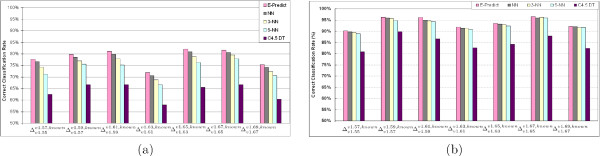
A comparison of classification performance between E-Predict, NN, 3-NN, 5-NN, and C4.5 DT classifiers using (a) testing proteins in which are selected from the SCOP folds in v2 that have at least one protein in v1 (b) testing proteins in which are selected from the SCOP folds in v2 that have at least 10 proteins in v1.
Figure 10.
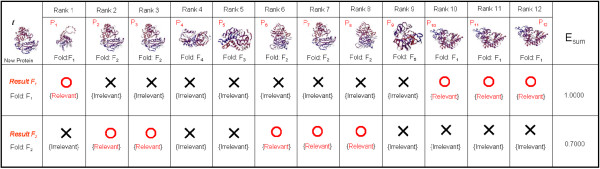
An example of E_Measure calculations for two SCOP folds in a list of nearest neighbor proteins.
In this work, we have developed the E-Predict algorithm which applies the E_Measure metric [21] to calculate the ranking information of nearest neighbor proteins. E_Measure was originally developed to evaluate the effectiveness of retrieval systems in IR. The more relevant documents retrieved with high ranks, the higher the retrieval accuracy. In the context of IR, Precision and Recall are two commonly used metrics for evaluating the retrieval performance. Let nt be the total number of relevant documents in the database for a certain query t and s(R,i) be the rank of the top ith relevant document in the retrieved document set R with 1 ≤ i ≤ nt. Precision can be obtained by computing the ratio of the number of relevant documents retrieved to the total number of documents retrieved.
For example, if the second relevant document is ranked seventh in R, (i.e. s(R, 2) = 7), then Precision(2) = . Recall is the ratio of the number of relevant documents i retrieved to the total number of relevant documents nt in the database.
For example, if there exist 11 relevant documents for query t, the second relevant document in the results will have Recall(2) = . E_Measure takes into consideration both Precision and Recall to evaluate the retrieval accuracy with a weighting factor b as shown in the following equation:
When a relevant document is highly ranked, a low E_Measure is expected. The effectiveness of a retrieval system ς can be evaluated by the summation of E_Measures for all nt relevant documents.
In practice, the best IR system is the one with the smallest E_sum(ς).
Instead of directly applying the above-mentioned evaluation method for our SCOP fold classification task, our E-Predict algorithm extends the method by visiting candidate folds in the top k nearest neighbor results R, and then ranking the folds using E_Measure. The E-Predict algorithm is shown in Appendix 1. From lines 2 to 16, the algorithm collects the SCOP folds of retrieved proteins in R into a set of candidate SCOP folds, II, with each candidate fold having at least nt proteins appearing in R. The algorithm then computes an evaluation score (F) for each candidate SCOP fold, F ∈ ∏, by accumulating E_Measures of the top nt proteins labeled with F, as shown from lines 17 to 26. Our approach assumes that the most relevant SCOP fold assigned to a newly-discovered protein t should have proteins that are highly ranked in R. For example, if F1 ∈ ∏ is the candidate SCOP fold to be evaluated, we revisit R by assigning the label 'relevant' to proteins that are from F1 and the label 'irrelevant' to those from folds other than F1. Among these relevant proteins, we select the top nt proteins and form RF1 for our classification process. The Result F1 in Figure 10 shows that the top two proteins (nt = 2) labeled with F1 are ranked at 1 and 10.
For fold F1, the pairs of (precision, recall) for these two proteins are (Precision(1) = , Recall(1) = ) and (Precision(2) = , Recall(2) = ). Applying Eq.(5) with b = 1.0, we obtain E_Measure(1, 1.0) = and E_Measure(2, 1.0) = . Substituting these two values into Eq.(6), we compute = 1.00. Similarly, for candidate fold F2, using Result F2 of Figure 10, the effectiveness of F2 is = 0.70.
According to Figure 3, there exists a significant number of small-size folds in the SCOP v1.69 release with 143 folds containing only one protein chain and 140 folds with two protein chains. When a newly-discovered protein belongs to a small-size fold, the algorithm might give a false positive due to insufficient ground truth data. To classify proteins in these small-size folds, we expect the NN search to retrieve a correct fold in the high-dimensional space by turning on a parameter λ in the E-Predict algorithm. Let P0 be the nearest neighbor protein of a query t in R and be the nearest neighbor protein in the candidate fold with the minimum score (see line 28 of Appendix 1). The algorithm computes the structural variation values, S, for one pair (t,P0) and the other pair (t, ) using the function in Eq.(2). The algorithm finally assigns the candidate fold with the minimum S value to the newly-discovered protein.
In the E-Predict algorithm, there exist two parameters, b and nt, that affect classification results. From our empirical observations, the best setting for the latest SCOP v1.69 release has b = 1.5 and nt = 6 with λ = on and k set to 500 nearest neighbors. Figure 9 shows comparisons of classification accuracies among E-Predict, NN, 3-NN, 5-NN, and C4.5 DT across seven test sets from to . For all test sets, E-Predict always outperforms k-NN and C4.5 DT with an improved classification accuracy.
Recognizing the novel folds for newly-discovered proteins
Classifying newly-discovered proteins into either the novel folds or the known folds has been identified as a two-class recognition problem [13]. Let v1, v2 and v3 denote three different SCOP releases in chronological order. To classify proteins from , our algorithm relies on ground truth data from with three features, which are derived from the result of E-Predict algorithm and will be discussed in great detail in the following section. In the labeling procedure of Figure 11, the algorithm first extracts the three features from proteins in . These proteins are then categorized into either the known folds of v1 or the novel folds as our ground truth data. In the testing procedure, proteins in the novel folds of are selected as our test data and are disjoint with our ground truth data. Once the three features are extracted from the testing proteins, we apply the E-Predict algorithm to classify test proteins into either the novel folds or the known folds.
Figure 11.
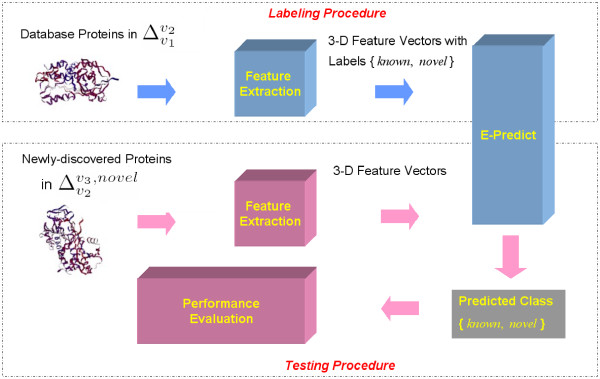
E-Predict model for recognizing the novel folds for newly-discovered proteins.
Feature extraction
For a newly-discovered protein PN that does not belong to the known folds, we assume this protein has a low structural similarity to those proteins in the known folds. Under this assumption, we identify the three features that are used to achieve the novel fold recognition task. Figure 12 illustrates an example showing the three features for PN. The first feature, , is the minimum evaluation score of PN using the E-Predict algorithm with a suggested known fold F*. The second feature, Dist, represents the Euclidean distance between PN and , which denotes the nearest neighbor protein of PN labeled with fold F*. The third feature, S, is the structural variation value between PN and using the function defined in Eq.(2). After feature extraction, these feature values are normalized between 0 and 1; each protein is then represented by a 3-D feature vector. The rationale for using these three features is in the following. Let PK be a newly-discovered protein that has been classified in the known folds. If PN is structurally dissimilar to all known protein structures from the SCOP database, then the Euclidean distance between PN and its nearest neighbor protein in a known fold suggested by E-Predict is expected to be greater than the distance between PK and its nearest neighbor protein . Similarly, the structural variation value of PN and its nearest neighbor protein is expected to be higher than the structural variation value of PK and its nearest neighbor protein. Also, the minimum evaluation score of PN, , is expected to be higher than the score of PK. Table 7 lists a brief summary of expected properties of the three features for proteins in the novel folds and the known folds.
Figure 12.
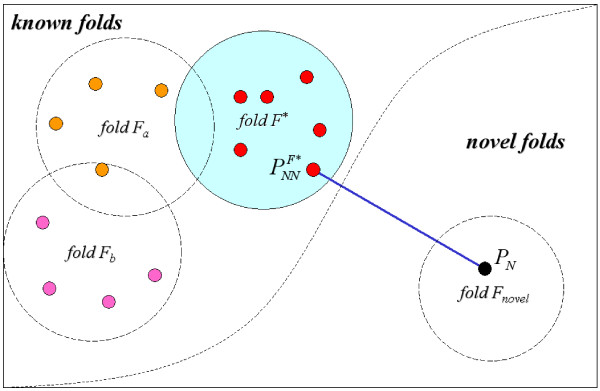
An example of identifying for a newly-discovered protein PN in the novel folds by selecting the nearest neighbor protein in a fold F* derived from the E-Predict algorithm.
Table 7.
A comparison of the three features for proteins in the novel folds and the known folds.
| (f1) (ςF*) | (f2)Dist | (f3)S | |
| novel folds | High | High | High |
| known folds | Low | Low | Low |
Novel fold recognition
With the three features, labeling and testing procedures can be conducted to recognize the novel folds for newly-discovered proteins. According to the statistics in Table 2 for a certain release of the SCOP database, the majority of proteins are from the known folds. From our empirical observations, the classifier is biased to favor the known folds in a 3-D feature space with two overlapping classes. To reduce noise from the known folds, our model randomly selects an equal number of proteins from the known folds and the novel folds in the labeling procedure. We then apply the E-Predict algorithm to classify test proteins into either the novel folds or the known folds.
Online index using the M-tree
Exhaustively searching for the nearest neighbors within a large-scale database such as the SCOP database is known to be computationally expensive. To improve the efficiency of k-NN search, we use an M-tree to index the high-dimensional feature vectors of proteins. The M-tree [27] scales well to support dynamic operations such as insert, delete and update. Each root node in a subtree maintains two values, a radius Rs and a prototype protein, that create a hyper-sphere in the high-dimensional feature space, As, to include all proteins within the subtree. During the Depth-First-Search(DFS) traversal, the M-tree algorithm maintains a priority queue with k slots to record the current k nearest neighbors. In addition, a radius centered at the query protein, Rq, defines a search space Aq. Initially, both Rq and Aq are set to ∞. Once a protein has been inserted into the queue, Rq is then updated by the maximum Euclidean distance between the current proteins in the queue and the query protein, resulting in a much smaller search space Aq. A fast nearest neighbor search is achieved by: 1) Applying the triangle inequality, the M-tree algorithm avoids traversing subtrees which do not overlap with the search space Aq. 2) Concurrently, Aq shrinks at a rapid rate due to the insertion of proteins in the queue. In our implementation, the M-tree indices have been properly organized into memory on several servers for a robust, fast nearest neighbor search.
Web interface
We have implemented a web interface to suggest a set of known SCOP folds and to recognize the novel folds for newly-discovered proteins. Users are allowed to submit 3-D protein structures in the PDB format. Our system first converts protein structures into 33-D feature vectors. Then, an evaluation score for each fold is computed from the ranked results of a nearest neighbors search. In seconds, a ranked list of SCOP folds is displayed to the user. To aid in visually inspecting the classification results, a tool is provided to superimpose a known protein from a suggested fold on the query structure using the KiNG (Kinemage, Next Generation) graphic package http://kinemage.biochem.duke.edu/software/king.php. In Figure 13, a 3-D superimposition view shows that the query protein is structurally similar to a known protein 9xim_A in the suggested fold. Our system, ProteinDBS-predict, is publicly accessible at http://ProteinDBS.rnet.missouri.edu/E-Predict.php.
Figure 13.
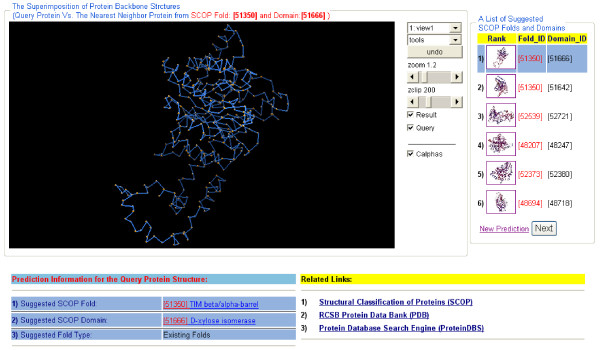
The superimposition of a newly-discovered protein and a known protein chain from the top ranked SCOP fold.
Authors' contributions
Both CS and PC designed the algorithm. PC implemented related programs and a web-based interface. CS supervised the whole project. DX contributed technical advice and helped test the system. PC drafted the manuscript and all authors finalized it. All authors read and approved the final manuscript.
Appendix
Appendix 1 E-Predict Algorithm
| Require: t, R, b, nt, λ |
| 1: ∏ = ∅ |
| 2: for each protein p ∈ R do |
| 3: if p·fold ∉ ∏ then |
| 4: ∏ = ∏ ∪ {p·fold} |
| 5: Count[p·fold] ← 1 |
| 6: else |
| 7: Count[p·fold] ← Count[p·fold] + 1 |
| 8: end if |
| 9: end for |
| 10: for i ← 0 to |∏| - 1 do |
| 11: if Count[i] <nt then |
| 12: ∏ ← ∏ - {i} |
| 13: end if |
| 14: [i] ← 0 |
| 15: Count[i] ← 0 |
| 16: end for |
| 17: for each candidate SCOP fold F∈ ∏ do |
| 18: for each p ∈ R starting from the top ranked protein do |
| 19: if p·fold = F then |
| 20: Count[F] ← Count[F] + 1 |
| 21: if Count[F] <nt then |
| 22: [F] ← [F] + E_Measure(p, b) |
| 23: end if |
| 24: end if |
| 25: end for |
| 26: end for |
| 27: F* ← arg minf [f] |
| 28: if (λ = on) AND (S(t,P0) < S(t,)) then |
| 29: F* ← P0.fold |
| 30: end if |
| 31: return F* |
Acknowledgments
Acknowledgements
The authors would like to thank Dr.Tolga Can of University of Middle East Technical University, Ankara, Turkey for the classification results of DALI, CE, and VAST algorithms. This research was supported by the University of Missouri-Columbia Research Council.
Contributor Information
Pin-Hao Chi, Email: pinhao@diglib1.cecs.missouri.edu.
Chi-Ren Shyu, Email: shyuc@missouri.edu.
Dong Xu, Email: xudong@missouri.edu.
References
- Zarembinski TI, Hung LW, Mueller-Dieckmann HJ, Kim KK, Yokota H, Kim R, Kim SH. Structure-based assignment of the biochemical function of a hypothetical protein: A test case of structural genomics. Proc Natl Sci USA. 1998;95:15189–15193. doi: 10.1073/pnas.95.26.15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley SK. An overview of structural genomics. Nat Struct Biol. 2000;7:932–934. doi: 10.1038/80697. [DOI] [PubMed] [Google Scholar]
- Stevens RC, Yokoyama S, Wilson IA. Global efforts in structural genomics. Science. 2001;294:89–92. doi: 10.1126/science.1066011. [DOI] [PubMed] [Google Scholar]
- Chen L, Oughtred R, Berman HM, Westbrook J. TargetDB: a target registration database for structural genomics projects. Bioinformatics. 2004;20:2860–2862. doi: 10.1093/bioinformatics/bth300. [DOI] [PubMed] [Google Scholar]
- von Grotthuss M, Plewczynski D, Ginalski K, Rychlewski L, Shakhnovich EI. PDB-UF: database of predicted enzymatic functions for unannotated protein structures from structural genomics. BMC Bioinformatics. 2006;7 doi: 10.1186/1471-2105-7-53. doi:10.1186/1471-2105-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl FM, Bennett CF, Bray JE, Harrison AP, Martin N, Shepherd A, Sillitoe I, Thornton J, Orengo CA. The CATH database: an extended protein family resource for structural and functional genomics. Nucl Acids Res. 2003;31:452–455. doi: 10.1093/nar/gkg062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WR, Orengo CA. Protein structure alignment. J Mol Biol. 1989;208:1–22. doi: 10.1016/0022-2836(89)90084-3. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–602. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Godzik A. The structural alignment between two proteins: Is there a unique answer? Protein Science. 1996;5:1325–1338. doi: 10.1002/pro.5560050711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, Kramer Green R, Flippen-Anderson JL, Westbrook J, Berman HM, Bourne PE. The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucl Acids Res. 2005;33:D233–D237. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can T, Camoglu O, Singh AK, Wang YF. Automated Protein Classification Using Consensus Decision. Proceedings of the Third Int IEEE Computer Society Computational Systems Bioinformatics Conference: 16–19 August 2004; Stanford. 2004. pp. 224–235. [DOI] [PubMed]
- Cheek S, Qi Y, Krishna SS, Kinch LN, Grishin NV. SCOPmap: Automated assignment of protein structures to evolutionary superfamilies. BMC Bioinformatics. 2004;5:197–197. doi: 10.1186/1471-2105-5-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindyalov HN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Engineering. 1998;9:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- Madej T, Gibrat JF, Bryant SH. Threading a database of protein cores. Proteins. 1995;23:356–369. doi: 10.1002/prot.340230309. [DOI] [PubMed] [Google Scholar]
- Chi PH, Scott G, Shyu CR. A fast protein structure retrieval system using image-based distance matrices and multidimensional index. International Journal of Software Engineering and Knowledge Engineering, Special Issue on Software and Knowledge Engineering Support in Bioinformatics. 2005;15:527–545. [Google Scholar]
- Leslie M. Protein Matchmaking. Science. 2004;305:1381. [Google Scholar]
- Shyu CR, Chi PH, Scott G, Xu D. ProteinDBS – A content-based retrieval system for protein structure databases. Nucl Acids Res. 2004;32:W572–W575. doi: 10.1093/nar/gkh436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi PH, Shyu CR. Predicting Ranked SCOP Domains by Mining Associations of Visual Contents in Distance Matrices. Proceedings of The Fourth Asia Pacific Bioinformatics Conference. 2006. pp. 49–58.
- van Rijsbergen CJ. Information Retrieval, Butterworths. 2 1979. [Google Scholar]
- Holm L, Sander C. The FSSP database of structurally aligned protein fold families. Nucl Acids Res. 1994;22:3600–3609. [PMC free article] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/S0959-440X(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R. Discriminant adaptive nearest neighbor classification. IEEE Trans, on Pattern Analysis and Machine Intelligence. 1996;18:607–616. doi: 10.1109/34.506411. [DOI] [Google Scholar]
- Quinlan JR. C4-5: programs for machine learning, Morgan Kaufmann. 1993.
- Ciaccia P, Patella M, Zezula P. M-tree: an efficient access method for similarity search in metric spaces. Proceedings of the International Conference on Very Large Databases. 1997. pp. 426–435.
- Alexandrov NN. SARFing the PDB. Protein Engineering. 1996;9:727–732. doi: 10.1093/protein/9.9.727. [DOI] [PubMed] [Google Scholar]
- Havel TF, Kuntz ID, Crippen GM. The theorey and practice of geometry. Bull Math Biol. 1983;45:665–720. doi: 10.1016/S0092-8240(83)80020-2. [DOI] [Google Scholar]
- Zaki MJ, Jin S, Bystroff C. Mining Residue Contacts in Proteins Using Local Structure Predictions. IEEE Trans, on Systems, Man and Cybernetics – Part B, special issue on Bio-imaging and Bio-informatics. 2003;33:789–801. doi: 10.1109/TSMCB.2003.816916. [DOI] [PubMed] [Google Scholar]
- Kolodny R, Linial N. Approximate protein structural alignment in polynomial time. Proc Natl Acad Sci. 2004:12201–12206. doi: 10.1073/pnas.0404383101. DOI:10.1073/pnas.0404383101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SK, Kunii TL. Pictorial dataBase systems. IEEE Computer. 1981;14:13–21. [Google Scholar]
- Smeulders AWM, Worring M, Santini S, Gupta A, Jain R. Content-based image retrieval at the end of the early years. IEEE Trans, on Pattern and Machine Intell. 2000;2:1349–1380. doi: 10.1109/34.895972. [DOI] [Google Scholar]
- Smeulders AWM, Huang TS, Gevers T. Special Issue on Content-Based Image Retrieval. International Journal of Computer Vision. 2004;56:5–6. doi: 10.1023/B:VISI.0000004865.97704.b9. [DOI] [Google Scholar]
- Rosenfeld A, Kak AC. Digital picture processing. New York: Academic Press; 1982. [Google Scholar]
- Otsu N. A threshold selection method from gray-level histogram. IEEE Trans, on Systems, Man and Cybernetics. 1979;9:62–66. [Google Scholar]
- Haralick RM, Shanmugam K, Dinstein I. Textural features for image classification. IEEE Trans, on Systems, Man and Cybernetics. 1973;3:610–621. [Google Scholar]
- Baeza-Yates R, Ribeiro-Neto B. Modern Information Retrieval, Addison Wesley. 1999.