

Abstract
Advances in proteomics technology offer great promise in the understanding and treatment of the molecular basis of disease. The past decade of proteomics research, the study of dynamic protein expression, post-translational modifications, cellular and sub-cellular protein distribution, and protein-protein interactions, has culminated in the identification of many disease-related biomarkers and potential new drug targets. While proteomics remains the tool of choice for discovery research, new innovations in proteomic technology now offer the potential for proteomic profiling to become standard practice in the clinical laboratory. Indeed, protein profiles can serve as powerful diagnostic markers, and can predict treatment outcome in many diseases, in particular cancer. A number of technical obstacles remain before routine proteomic analysis can be achieved in the clinic; however the standardisation of methodologies and dissemination of proteomic data into publicly available databases is starting to overcome these hurdles. At present the most promising application for proteomics is in the screening of specific subsets of protein biomarkers for certain diseases, rather than large scale full protein profiling. Armed with these technologies the impending era of individualised patient-tailored therapy is imminent. This review summarises the advances in proteomics that has propelled us to this exciting age of clinical proteomics, and highlights the future work that is required for this to become a reality.
Introduction
The successful completion of the human genome project has led to a tremendous increase in our understanding of the molecular basis of diseases. However, a comprehensive understanding of the dynamic protein pathways involved in normal and disease states, and in response to medical treatment, is required if we are to effectively treat disease. The next major challenge toward this aim is to identify the constituents of the human proteome in order to understand the human genome. Of particular importance will be to decipher protein alterations between health and disease to enable the identification and prioritisation of pharmaceutically relevant targets. Indeed, from a therapeutics perspective, the majority of drug targets are proteins and not nucleic acids. Technologies available to date such as microarray that can identify large numbers of differentially expressed genes, fail to take into account the multiple protein products of these genes and their functional significance. Proteome analyses aim to not only identify changes in protein expression, but also post-translational modifications, protein-protein interactions, cellular and sub-cellular distribution, and temporal patterns of expression. The purpose of differential and functional proteomics is to obtain this information that will then lead to improved understanding of the cellular pathways and their inter-relationships in cells and living organisms. The power of proteomics as a tool for discovery of biological pathways and disease processes is now well established. Indeed, proteomics has already uncovered many potential new drug targets for varying diseases. The current era of proteomics is now beginning to investigate how this technology can serve the clinician for high-throughput diagnostic and prognostic applications. This report reviews the current status of clinical proteomics with a particular emphasis on cancer biology and treatment.
Power of Multiple Biomarkers of Disease
Proteomics was initially defined by Dr Marc Wilkins, at the time a PhD student of Macquarie University, as the “protein complement of a given genome” and thus refers to all proteins expressed by a cell or tissue. Since then, the term proteomics has come to encompass the systematic analysis of protein populations with a goal of concurrently identifying, quantifying, and analysing large numbers of proteins in a functional context. As such, the ultimate goal of most proteomic studies is to determine which proteins or groups of proteins are responsible for a specific function or phenotype. Proteomics thus has enormous potential in identifying proteins associated with different disease states. Traditional biomarker analysis has concentrated on identifying one marker of a particular disease. However there is now general agreement of the statistical argument that a panel of independent disease-related proteins considered in an aggregate should be less prone to the influence of genetic and environmental ‘noise’ than is the level of a single marker protein,1 and proteomics has the power to identify such panels of proteins in a high-throughput manner. For example, Rai et al. identified three potential biomarkers that could differentiate ovarian cancer from healthy individuals and compared their performance against the tumour marker, cancer antigen 125 (CA125).2 Each biomarker individually did not out-perform CA125, however the combination of two of the new biomarkers together with CA125 significantly improved their performance.2,3 Thus identification of new protein biomarkers should substantially improve our ability to diagnose and treat human disease.
DNA Microarrays for Disease Profiling
Advancements in gene expression profiling are beginning to allow for correlations of clinical data with genome-wide expression.4 DNA microarrays are being used to uncover associations between gene expression and specific subtypes of disease. For example, a study of breast cancer found that gene expression data could be used to classify tumours into a basal epithelial-like group, an ErbB2 overexpressing group, and a normal breast group,5 and later studies showed significantly different outcomes for patients belonging to the various groups.6 Such studies have major importance when it comes to molecularly targeted treatments. The monoclonal antibody inhibitor of ErbB2, trastuzumab (HerceptinR) has been used successfully as monotherapy and in combination with chemotherapy in women with ErbB2 (HER-2) overexpressing metastatic breast cancer.7–10 However, response rates are generally less than 50%, indicating that patients either do not respond or have disease progress after an initial response. Identification by microarray of the other genes altered in the ErbB2 subtype are now highlighting other drug targets that can be used for combination therapy to improve these response rates.5,6 DNA microarrays can then provide sophisticated multiplex panels for various diseases. A large microarray study on breast cancer was able to distinguish between patients with the same stage of disease but different responses to treatment and overall outcome,11,12 and this has led to a nationwide clinical trial in the Netherlands in which gene expression profiles for 70 classifier genes are being collected on all breast cancer patients and used as an adjunct to classical clinical staging.
DNA profiling has also proved a powerful tool for classifying subtypes of leukaemia patients.13–18 Microarray profiling has been used to determine treatment-specific changes in acute lymphoblastic leukaemia (ALL) patients treated with methotrexate and mercaptopurine.19 Recently, Holleman et al. reported gene expression patterns in drug-resistant ALL.20 The in vitro sensitivity to vincristine, prednisolone, asparaginase and daunorubucin was determined for each patient, and the gene expression profiles correlated to drug sensitivity. Forty differentially expressed genes were identified in vincristine-resistant ALL, only one of which has been previously associated with drug resistance. Importantly, the gene expression signatures associated with resistance to the individual anticancer agents were also related to the patients’ responses to treatment, suggesting that the expression of genes associated with drug resistance is an independent predictor for outcome of treatment in ALL.20 This study has highlighted a number of genes that are potential targets for new therapies.
While gene expression profiling highlights the potential of individualised patient-tailored therapy, gene expression analysis does not correlate well with protein expression.21 In addition, mRNA analysis does not uncover any information on the post-translational modifications, activity, sub-cellular and tissue distribution, and interactions of proteins. Indeed, as most drug targets are proteins, a proteome profile is much more informative than simply gene expression. Traditionally, DNA microarrays have had limited use for the analysis of biological fluids, however, recent studies highlight the presence of circulating nucleic acids and their potential use as diagnostic tools.22,23 Indeed, for a biomarker to be useful in routine clinical tests its detection in samples that are easy to obtain, such as plasma or urine, is a major advantage.
Proteome Technologies
A range of techniques are now available for the analytical separation and identification of proteins from complex mixtures (Figure 1). One and two dimensional gel electrophoresis, or high-performance liquid chromatography (HPLC) are the most common separation methods, while mass spectrometry (MS) is now the gold standard for protein identification.
Figure 1.
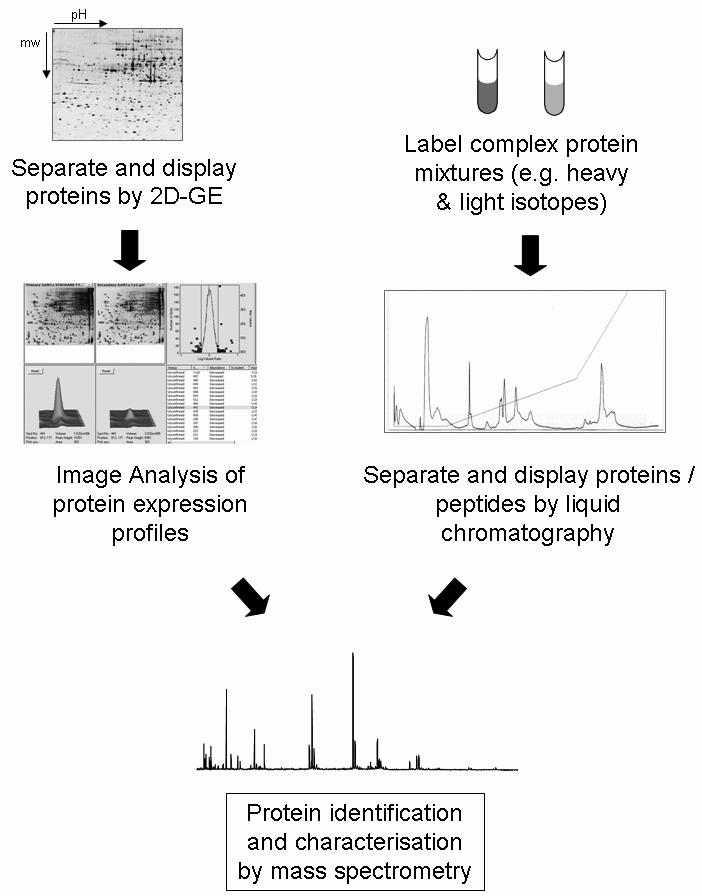
A standard proteome approach. The most common approach to separate complex protein mixtures is by 2-dimensional gel electrophoresis (2D-GE), which separates proteins in a pH gradient according to isoelectric point in the first dimension, and in an acrylamide matrix according to molecular weight in the second dimension. Relative levels of expression are compared between gels of different samples using computer algorithms to determine differential protein changes. Proteins of interest are excised from the gel, trypsin digested, and subjected to mass spectrometry for identification and characterisation. An alternative approach is to pre-label protein mixtures and separate proteins, or more often peptides, by multidimensional liquid chromatography. Differences in peptide levels and protein identification are then performed by mass spectrometry.
Two-Dimensional Gel Electrophoresis (2D-GE)
Recent advancements in proteome technology now allow the protein complement of a given genome to be analysed. Since the introduction of 2D-GE in 197524 improvements such as immobilised pH gradients (IPGs), that separate proteins according to charge in the first dimension prior to separation of proteins according to size in the second dimension, have rendered the technique reproducible between laboratories.25 This has greatly facilitated higher throughput analyses, and high-resolution separation of proteins from cells or tissue extracts. Another important advancement is the utilisation of single pH unit IPGs for greater separation, and sub-cellular fractionation that not only increases sensitivity but also determines the localisation of proteins in a cell.26,27 Such information, e.g. translocation of proteins from one compartment to another under experimental conditions, is an important component of protein function, and this information is not attainable with genomic techniques. For differential proteome analysis, protein expression profiles are compared (for example between normal and disease tissue, sensitive and resistant cells, or control and drug-treated cells) using powerful computer algorithms that detect the expression levels of each protein spot. Whilst large scale protein profiling preceded genomic profiling, the power of proteomics was not fully realised for many years due to inherent limitations of traditional protein identification tools, most notably Edman sequencing, which is not amenable to high-throughput automation and requires large amounts of purified protein. Protein identification through MS based techniques now overcomes many of these limitations. Matrix-assisted ionisation-time of flight (MALDI-TOF) MS has fast-tracked the identification of proteins isolated by 2D-GE and other methods due to the exquisite speed and sensitivity of this tool.28 Combined with sequence database searching using the increasing publicly available nucleotide and protein databases, it is apparent why the identification and discovery component of proteomics is becoming the tool of choice in drug discovery.
Although 2D-GE is a powerful technique, one of its limitations is that 2D gels remain relatively low throughput and require large amounts of starting material (~50μg) with low sensitivity for detection of low abundance proteins such as cytokines and signalling molecules. In addition, certain basic proteins, and very high- or very low-molecular weight proteins are not separated well by 2D-GE. Techniques such as free-flow electrophoresis (FFE) have been developed to help resolve complex protein mixtures using a combination of FFE (liquid based IEF method) and 2D-GE.29 The use of narrow range, overlapping pH gradients in the first dimension also improves the number of proteins visualised on 2D gels.30 Until recently, a limitation in 2D-gel technology was the reproducibility, necessitating the use of multiple gels to obtain statistical validity. A major advance in this area has come from the introduction of Cy dye fluorophores for pre-labelling of protein samples. Two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) technology adds a quantitative component to conventional 2D-gel analyses, allowing for comparison of protein expression changes across multiple samples simultaneously without gel-to-gel variation, and hence with statistical confidence.31–33 2D-DIGE utilizes Cy dye fluorophores for protein labelling prior to fractionation. This facilitates multiplexing of protein samples, allowing for direct comparison of different samples within the one gel,32 and more importantly, enables the introduction of a standardised internal control (Figure 2).34 The power of 2D-DIGE has been demonstrated for a number of biological applications, including studies on cancer35–41 and analyses utilising mouse models.42–45
Figure 2.
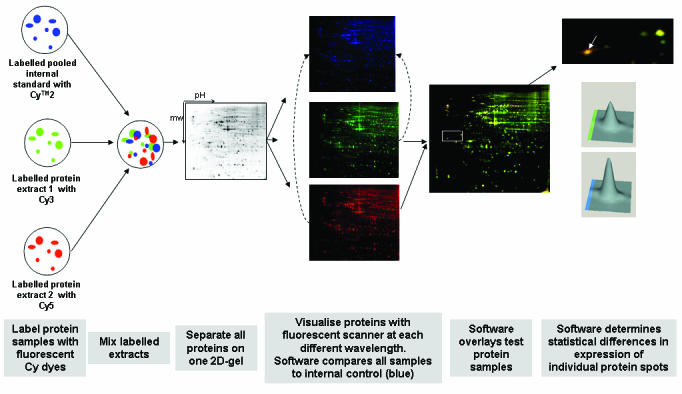
Two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) workflow. Protein samples to be compared are covalently labelled with either Cy3 or Cy5 fluorescent dyes. An internal control, to be run on every single gel in the experiment, is labelled with Cy2. All three samples are combined and separated on the one 2D-gel, thus eliminating gel-to-gel variation. The single gel is scanned at three different wavelengths to generate an image specific for each CyDyeTM fluore. The DeCyderTM software (Amersham Biosciences, GE Healthcare) normalises the test samples to the internal control, and then overlays the two test samples to identify changes in expression levels of individual protein spots. A 3-dimensional view of matched proteins is generated to ensure correct detection of protein spots. As all gels are run with the same internal standard, multiple gels from numerous experiments can all be compared with statistical confidence. Figure adapted from Amersham Biosciences (http://www1.amershambiosciences.com) and reference 42.
Mass-Spectrometry Based Proteomics
Whilst 2D-GE remains the most widely used tool for separating proteins, many new technologies have emerged over the past few years that complement and enhance the proteomic armoury. A mass spectrometer can be considered as a highly accurate weighing scale for extremely low mass particles. Importantly, mass spectrometers can be automated and can achieve sensitivity down to the femtomole level. Proteins are usually cleaved into smaller fragments (peptides) with an enzymatic protease (in most cases trypsin), and the peptide masses are detected by the mass spectrometer. Peptides can be combined with an acidic matrix and applied to a stainless steel plate (MALDI), or may be introduced through a needle in liquid form (electrospray, ES), and are ionised in the mass spectrometer. Different mass spectrometers detect the mass of peptides in various ways. In time of flight (TOF) instruments peptides fly down a flight tube and the time it takes for the peptide to reach a detector is proportional to the peptide mass. Other mass spectrometers utilise quadropoles, ion traps and Fourier transform ion cyclotron resonance (FTICR) to analyse the peptides.46 The resulting mass spectrum is converted to a list of peptide masses that is searched against the extensive genome databases, translated and trypsin digested in silico. Each protein will have a unique peptide mass fingerprint (PMF) based on its amino acid sequence, and hence the peptide masses determined by the mass spectrometer can identify the protein from the thousands of proteins in the database. An important advancement in mass spectrometry is the ability of the analyser to isolate an ion (or peptide) and subject it to further fragmentation into individual amino acids (tandem mass spectrometry, MS/MS). In this way the de novo sequence of peptides can be determined, which together with the peptide mass fingerprint can be used to positively identify the protein. Importantly, the development of specialised software algorithms that rapidly search MS data against known or predicted proteins within databases makes this process amenable to high throughput analysis.47 This is also an extremely powerful means to identify point mutations and post-translational modifications. Mass spectrometry can now be used to determine relative levels of expression without the need for prior gel separation. Isotope-coded affinity tags (ICATTM) is a high-throughput MS-based technique that facilitates direct qualitative and quantitative comparisons of complex protein mixtures.48 Samples to be compared (e.g. cancer versus normal cells) are each labelled with a heavy or light isotope, which couple to cysteine residues of the proteins, the samples are then mixed, proteins enzymatically digested, and peptides analysed by MS. Both the relative abundance of peptides from each sample, and protein identifications can be simultaneously obtained. Other labelling techniques are also available, such as O18-water labelling, global internal standard technology (GIST), and isotope tags for relative and absolute quantification (iTRAQTM), each of which have various pros and cons depending on the specific application.49 Stable isotope labelling with amino acids in cell culture (SILAC) is another quantification strategy for analysis of differential expression between two distinct cellular populations.50,51 Cells are cultured with media deficient in a natural amino acid but supplemented with a monoisotopically labelled amino acid (e.g. 12C and 13C, or 14N and 15N). Whilst a powerful technique for growing cells, only samples capable of undergoing protein synthesis in vitro are amenable.
To reduce sample complexity, MS approaches are often coupled to multi-dimensional liquid chromatography (MDLC) prior to the mass spectrometer. Most MDLC approaches utilise a strong cation exchange followed by a reverse phase separation, and the chromatography columns can be physically attached on-line to the mass spectrometer. This separates complex protein samples into numerous fractions, and the matching peptides from the two samples will have the same chromatographic properties and thus will co-elute such that peptide abundance can still be compared. Such multidimensional protein identification technology (also referred to as MudPit) is an attractive approach for analysing complex samples in a large scale manner, and has been shown to be capable of identifying up to 1484 proteins from yeast in a single experiment.52
Protein Chips
As with genomics, chip technology is beginning to be applied in the proteomics field. As proteins are so heterogeneous, a simple “one-chip for all genes” is not currently achievable as no capture molecules capable of binding all possible proteins are available. However, a variety of protein and peptide arrays have been developed to analyse a specific protein or group of proteins.53 At present a major advantage of this technology over more traditional protein separation tools is that it allows for analysis of protein-protein, protein-DNA, or protein-RNA interactions, depending on the substrate cross-linked to the chip. Affinity-based MS techniques represent a further proteomic tool. Ciphergen Biosystems, Inc. have developed the surface-enhanced laser desorption-ionisation (SELDI) Protein ChipR, which involves the affinity capture of specific subgroups of proteins based on their biochemical and/or physical properties, coupled with automated MS analysis.54 This technique is particularly useful for proteins not amenable to 2D-GE, such as low abundant, or basic proteins. In addition, analysis of serum samples is greatly enhanced with this technique as the inherent “masking” of serum proteins by high abundant albumin species on 2D-gels is greatly reduced. Indeed, the SELDI platform has been successfully used to quantify relative levels of prostate-specific membrane antigen (PSMA) from serum, and in combination with prostate-specific antigen, could discriminate between benign prostatic hyperplasias and prostate cancer patients.55 This approach is very useful for detecting marker profiles of disease, however its use in discovery research is limited due to the inherent difficulties in determining the identity of the marker polypeptides. Perhaps the most widely heralded proteomics study to date is that of Lance Liotta and Emanuel Petricoin III who used SELDI to analyse the protein patterns of serum from ovarian cancer patients.56 The pattern profiles could detect all patients with ovarian cancer in a set of 50 samples, and falsely identified just three healthy patients from 66 control samples. Of great significance, the technique worked well on patients with early stage disease, offering the prospect of earlier diagnosis which would greatly enhance the chance of successful treatment outcome. This has led to the development of a commercial test, termed OvaCheck, for diagnosis of ovarian cancer, however the clinical development of the test is still ongoing due to controversy over the validity of some of the original data (see Pitfalls in current proteome technologies section of this review). Other researchers have developed similar MALDI-based tools for protein profiling, such as magnetic, reverse-phase beads for analytical capture followed by a MALDI-MS readout.57 This method is more sensitive than surface capture on chips because spherical particles have larger combined surface areas and therefore higher binding capacity than small-diameter spots.58 Villanueva et al. have used this method to identify peptide marker profiles of various cancer types.57
A number of other protein microarray platforms are continually being developed.59–62 These include analytical arrays, for example forward phase arrays where a bait molecule (e.g. antibody) is immobilised to a solid surface and exposed to a test sample containing a mixture of proteins. Bound molecules are detected either by a secondary antibody or by direct labelling of the molecule. In a reverse phase array the sample is immobilised onto a solid surface and exposed to, for example, an antibody that is detected using a secondary antibody and signal amplification techniques.59,60 Reverse phase arrays can be designed for the detection and determination of relative levels of phosphoproteins that are important in cellular signalling pathways in cancer cells.60 In addition, functional microarray chips involving the immobilisation of purified peptides or native proteins onto suitable surfaces can also be used to study protein-protein interactions, DNA-protein interactions, or post-translational modifications and drug-target identification.59 One of the major contributions proteomics has made to the medical community is the identification of a multitude of potential drug targets. A major bottleneck in the transition of this knowledge from the bench to the bedside is the development of specific drugs to target these markers. Protein microarrays are currently being used for the high throughput screening of libraries to identify novel ligands or drugs that bind to specific bait molecules on the array.63,64
Protein chips or microarrays probably have the best potential for analysing a set of known biomarkers or the activity of proteins in specific signalling pathways. Protein arrays have been employed to measure enzyme activity of secreted and membrane proteomes of cancer cell lines,65 and are now being used to measure kinase activity via specific detection of phosphoproteins. Many diseases, in particular cancer, are characterised by alterations in certain signalling pathways and identification of the aberrant pathway in a particular patient allows for therapy to be targeted to that specific pathway. For example, epithelial ovarian cancer is often characterised by activation of the epidermal growth factor receptor (EGFR) signalling pathway, and targeted therapies including monoclonal antibodies, such as cetuximab and small molecule inhibitors such as gefitinib are either in clinical use or under clinical trial for different stages of cancer.66,67 Similarly, the c-Kit and PDGFR inhibitor, imatinib, has shown remarkable success in chronic myeloid leukaemia and gastrointestinal stromal tumours, cancers that are maintained by activation of these receptor tyrosine kinases.68 While these targeted agents are proving successful in some cases, the heterogeneous nature of some cancers results in only a subset of patients responding. Drug resistance is also a major impediment to treatment success.69 Reverse phase array technology has been utilised to profile the active molecular pathways in breast cancer and primary and metastatic ovarian cancers.70–72 Epithelial cells were microdissected from frozen tumour sections and protein lysates printed on the arrays. The slides were then probed with 26 phosphospecific antibodies to proteins known to be involved in mitogenesis including growth factor receptors, signal transducing proteins, and nuclear transcription factors, to profile the phosphoproteomic signal pathway circuitry.72 Principal component analysis identified several phosphorylated proteins that represented most of the variation between primary and metastatic tissue expression patterns. Furthermore, partition analysis then found that most of the primary and metastatic tumours could be distinguished by expression of phosphorylated c-Kit alone.72 Thus these metastatic patients are likely to benefit from the c-Kit inhibitor, imatinib. As there was substantial heterogeneity in the pattern of activated signals in other pathways, treatment could be further enhanced by combination therapy with other specific kinase inhibitors, selectively applied based on each phosphoproteomic fingerprint. Thus the use of protein arrays can move us further towards the reality of patient-tailored individualised therapy. This type of analysis could also be applied to monitor the response of patients to chemotherapy. Molecularly targeted drugs have known binding proteins and are expected to induce certain signalling pathways. The early efficacy of such treatments can be monitored during therapy to determine if the treatment is having its desired molecular effect, and hence to infer the potential success of the treatment. If a patient’s tumour is not responding in the desired manner then treatment can be changed before further progression occurs. Such powerful analyses are not possible by gene arrays.
Protein microarrays still face a number of serious challenges.60 Firstly, the dynamic range of protein samples means that high abundant proteins can produce contaminating cross-reactivity, reducing the sensitivity of detection of low abundant proteins. Secondly, PCR-like direct amplification methods do not exist for proteins, and current signal amplification techniques such as biotin, peroxidases, alkaline phosphatases, fluorescent proteins, and immunoglobulins, can all cross-react with naturally occurring analytes hence increasing the background of the amplification reaction. The sensitivity of the chip is also hampered by the ability to obtain sufficient, and homogenous tissue samples. As most protein arrays utilise immunodetection, a vast number of specific antibodies must be available. Many commercial companies are beginning to accommodate this necessity, and a major initiative of the Human Proteome Organisation (HUPO) is the production and qualification of antibody libraries that will be made available to the scientific community.73,74 Of course each antibody will have unique affinity constants and may require specific conditions, thus multiplexing technology may reduce the sensitivity and/or specificity of individual antibodies due to a universal reaction.
Tissue Microarrays
Pathological assessment of tissues has been the linchpin of cancer diagnosis for the past century. With improvements in arraying technology, traditional immunohistochemical detection of protein expression in tissue sections can now be adapted to a high-throughput array format. Using immunohistochemistry on tissue microarrays, Jacquemier et al. monitored the expression of 26 selected proteins in over 1,600 cancer samples from 552 consecutive patients with early breast cancer.75 Hierarchical clustering identified relevant clusters of co-expressed proteins and clusters of tumours. The method identified a set of 21 proteins whose combined expression significantly correlated with metastasis- free survival in a learning set of 368 patients and in a validation set of 184 patients. Importantly, in a multivariate analysis the 21-protein set was the strongest independent predictor of clinical outcome, showing that protein expression profiling may be a clinically useful approach to assess breast cancer heterogeneity and prognosis.75 That this study utilised proteins of known or putative importance in breast cancer also supports the proposal that current clinical proteomics tests are best applied to a targeted subset of the proteome, rather than entire protein profiling of samples.
Imaging Mass Spectrometry
An emerging technique for discovery of protein signatures involves the identification of biomarkers by MS directly in tissue biopsies.76 Direct imaging of protein expression in normal and disease tissues has been achieved by in situ MS analysis of tissue sections. Frozen tissue is sliced and sections are applied on a MALDI plate and analysed at regular intervals. The mass spectra obtained at each interval are compared between samples, yielding a spatial distribution of individual masses across the section. Such analyses have uncovered differences in protein expression between normal and tumour tissues that may have specificity for different tumour types.77 Traditionally this technology had required substantial manual data analysis and thus was not suitable for routine clinical analysis. Recently however Schwartz et al. analysed over 100 glioma patients in a reasonably high-throughput manner.78 Application of direct tissue MALDI-MS to human brain tumours identified protein patterns that distinguished primary gliomas from normal brain tissue and one grade of gliomas from another, with high sensitivity and specificity.78 Importantly, the protein patterns described served as an independent indicator of patient survival, suggesting that this new molecular approach can provide clinically relevant information. In situ MS analysis has also been utilised on samples captured by laser capture microdissection.79 Further advancements in the data processing and analytical assessment of imaging MS is beginning to validate the utilisation of this technique in clinical practice.80
Proteomics in Cancer Research
By combining the myriad of proteome tools available to the researcher, entire proteomes can now begin to be unravelled in order to better understand the molecular basis of disease and to identify novel biomarker sets and potential drug targets. The power of this approach has been most successfully used to date in the field of cancer research.73,74 Celis and co-workers have utilised 2D-GE and MS analysis to identify differential protein expression between healthy and diseased tissue including normal urothelium vs squamous cell carcinomas (SCCs), which has defined some of the steps involved in the squamous differentiation of the bladder transitional epithelium.81 This analysis has culminated in a comprehensive 2D-gel database of bladder cancer that is publicly available.82 Similar proteome analyses have been used to identify markers of urothelial papillomas, renal cell carcinomas, breast cancer, lung cancer, ovarian cancer, and leukaemia, to name just a few (reviewed in references 81,83–85). Heterogeneity of tumour tissue, where mixtures of normal and cancerous cells co-exist, may present problems for proteomic analysis. One approach to overcome this is to use laser capture microdissection where specific cell types or tumour regions can be isolated. This technique was used in a recent study to identify proteins that distinguish between low malignant potential ovarian tumours and the invasive form.86 Since metastatic disease is often more difficult to effectively treat with chemotherapy, these new markers may prove effective targets for new drug design or predicting therapeutic response.87 Zhou et al. recently combined microdissection with the powerful technique of fluorescent 2D-DIGE to study squamous cell carcinoma of the oesophagus (ESCC), a major subtype of oesophageal carcinoma that is one of the most aggressive cancers with a dismal prognosis.88 The poor outcome for ESCC is attributed not only to the aggressive nature of the disease, but because the molecular mechanism of its progression is largely unknown, and due to the lack of adequate biomarkers for early detection and prediction of clinical behaviour. This study identified 28 proteins differentially expressed in ESCC patient cancer cells compared to adjacent normal epithelial cells.88 These proteins shed new light on the underlying mechanism of tumourigenesis in this aggressive cancer, as well as providing candidate biomarkers for earlier detection.
High-throughput MS analysis of human plasma/serum proteomes is emerging as a powerful technique for identifying distinct protein profiles in cancer patients. Proteomic patterning of serum was recently developed for the early detection of ovarian cancer.56 While at present the validity of such data is still under investigation and the specific proteins that give rise to the altered SELDI-TOF spectra are yet to be defined, it does demonstrate the utility of examining serum from patients to detect disease states. SELDI has been used to successfully discriminate serum peaks capable of distinguishing between normal, benign prostate hyperplasia, and prostate cancer patients,89 between normal and early and late stage breast cancer,56 and for prediction of chemoradiosensitivity of oesophageal cancer.90 The relative ease by which serum can be obtained from patients combined with rapid analysis is sure to see MS-based proteomics used more readily for cancer detection and progression in the future.86 In addition, LC-MS/MS MudPIT approaches have been used to identify proteins from a variety of sources,91–93 post-translational modifications,94 and quantitative expression comparisons.48,52 Global gene expression as a marker for disease classification is currently a very popular research approach. Interestingly, the use of proteomics to classify disease subtypes preceded the advent of gene arrays. In the 1980s Hanash and colleagues utilised 2D-GE to identify lineage-related protein differences in lymphoblasts from children with acute lymphoblastic leukaemia (ALL).95 Twelve polypeptides were detected that could distinguish between the major subgroups of ALL, including a new marker for common ALL and markers for cells of B and T lineages. Protein identification at the time was extremely difficult, however the new marker of ALL was later sequenced and identified as heat-shock protein 27 (HSP27).96 Additionally, a phosphorylated form of HSP27 was also identified as a marker that distinguished infant ALL from ALL in older children.97 Interestingly, 2D-GE analysis has demonstrated a correlation between increased HSP27 expression and shorter survival times for B-cell chronic lymphocytic leukaemia patients,98 suggesting that patients’ HSP27 levels may predict response to conventional chemotherapy in this disease. Research from the same laboratory has undertaken a comprehensive cell surface proteome analysis of cancer cells using 2D-GE and MS.99 Distinct patterns of expression of cell surface proteins were detected that were commonly shared amongst the different cells and distinct markers that were unique to certain cancer cell subtypes.99,100 This work represents the power of proteomic approaches for both classification of disease subtypes, identification of the origins of cancer and cancer cell surface markers.
Traditionally the combination and timing of cancer treatment has been empirical, based primarily on clinical presentation and histological features of the tumour, rather than on molecular mechanisms. While this approach has proven effective in the treatment of certain human cancers, drug resistance remains a major block in the successful treatment of this disease. Mechanisms mediating resistance, or indeed, drug sensitivity are still not well understood. Proteomic technologies have been extensively utilised for characterisation of normal and transformed cells and tissues, and the extension of this approach for the analysis of drug resistance is emerging as an exciting field.85 The Developmental Therapeutics Program of the US National Cancer Institute (NCI) has an extensive profile of 60 different cancer cell lines that are representative of different tissue origins and have been screened against over 60,000 chemical compounds. A 2D-GE database of the 60 NCI cell lines has been developed correlating changes in protein expression with drug response.101 Sinha and colleagues have performed 2D-GE analyses on numerous drug-resistant cell lines that have culminated in the identification of a number of resistance-associated protein changes.102 Further analyses of different chemoresistant cell lines continues to identify novel proteins associated with resistance, and interestingly, the 14-3-3 family of phosphoproteins are emerging as proteins that appear to be involved in resistance to many types of agents. Overexpression of 14-3-3γ and 14-3-3σ was shown to be associated with chemoresistance in malignant melanoma and pancreatic adenocarcinoma respectively.103,104 In a recent study from our laboratory on antimicrotubule resistance in leukaemia, 14-3-3τ and 14-3-3ɛ were also differentially expressed.105 We have also utilised 2D-GE and MS analysis to identify modifications to the drug target, tubulin, in antimicrotubule resistant leukaemia cells.106,107 While most of these studies have concentrated on in vitro selected cell lines, we have recently extended these analyses to a clinically relevant mouse model of drug resistant leukaemia.45 2D-DIGE was used to analyse the protein expression in ALL xenografts for which their intrinsic sensitivity to vincristine (VCR), a major component of combination chemotherapy for ALL, was known. To better understand mechanisms mediating acquired clinical drug resistance, 2D-DIGE was also utilised to examine xenografts with in vivo-derived VCR resistance. Of the 19 proteins displaying altered expression, 11 are associated with the actin cytoskeleton. A number of other proteins are associated with microtubules, showing that similar cytoskeletal proteins are altered in in vivo ALL models as were found in the in vitro cell lines.45,105 It is not only important to show that similar mechanisms are involved in in vivo animal models, but also in clinical samples obtained from patients, and we have since shown that for at least one gene, the change identified in the experimental models is reflected in non-xenografted primary samples obtained from ALL patients. γ-Actin, a major cytoskeletal protein, was down-regulated in both intrinsic and acquired drug-resistant leukaemia xenografts, and γ-actin expression was shown to be significantly lower in leukaemia cells obtained from ALL patients at relapse compared to diagnosis (Verrills et al. unpublished data). These studies provide the first evidence for a role of the actin cytoskeleton in in vivo antimicrotubule drug resistance, and highlight the power of 2D-DIGE for the discovery of drug resistance markers in relapsed leukaemia.
One caveat to these powerful proteomic approaches is that they can identify a large number of candidate proteins that require validation as therapeutic targets. This process would be greatly facilitated if differential proteomics can be combined with specific functional proteomics profiling. Such an approach would allow one to pinpoint proteins involved in both drug response and drug resistance, and in turn, identification of such targets would allow them to be used for the development of specific inhibitors. By identifying proteins involved in the response of leukaemia cells to antimicrotubule agents, together with protein expression changes in leukaemia cells resistant to antimicrotubule agents, we were able to restrict the number of potential targets to ten proteins by identifying which are altered in both drug response and drug resistance.105 These proteins highlight known and novel pathways involved in the mechanism of action of microtubule-targeted anticancer drugs, and are potential targets for improved therapy of drug resistant cancer. This was a valuable approach to study resistance mechanisms that could be used in the investigation of a broad range of anti-cancer agents, and can lead to the identification of novel markers of relapsed cancer.
Proteomics is a powerful tool for analysis of post-translational modifications (PTMs), not possible by analysis at the gene level. Phosphorylation is a dynamic PTM that regulates the function of many proteins, and is intimately involved in cellular signalling pathways. Using a proteomic approach, Nishio et al. identified marked differences in the phosphorylation status of specific nuclear proteins between drug sensitive, and cis-diamminedichloroplatinum (II)-resistant cell lines.108 Interestingly, using more traditional techniques, no difference in protein kinase C activity or total protein phosphatase activity, nor total cellular phosphorylation was detected, in this case highlighting the power of proteomics over traditional approaches. In addition, dynamic changes in phosphorylation in signal transduction pathways can now be profiled using protein microarrays and has been applied to the study of breast and ovarian cancer.70–72 Post-translational modification of drug targets can also affect the efficacy of treatment. The extent of glutamylation for tubulin proteins has been determined by tandem MS.109 The level of tubulin glutamylation modulates the binding of microtubule-associated proteins,110,111 and thus may affect the stability of microtubules and hence the action of antimicrotubule drugs. Tyrosination-detyrosination is another PTM of tubulin, and Western blotting analysis has shown that tyrosinated tubulin is increased in paclitaxel-resistant breast cancer cells.112 MS has also been used to study polyglycylation of tubulin.113 These powerful proteomic approaches could be used to analyse antimicrotubule resistant cells, and may lead to the identification of novel changes in tubulin isotype expression and PTMs associated with the resistance phenotype.
Autoantibodies may be useful biomarkers of some diseases, in particular cancer.114–117 Le Naour et al. utilised 2D-GE of cultured cells and tumour and non-tumour tissues, followed by immunoblotting with patient serum, to identify a distinct repertoire of autoantibodies associated with hepatocellular carcinoma that may have utility in early diagnosis among high risk subjects.118 Importantly, using a proteomics approach to identify autoantibodies allows for detection of immune responses to post-translationally modified proteins. For example, a study on lung cancer identified autoantibodies to a glycosylated form of annexin I and II in 60% of patients with lung adenocarcinoma and from 33% of patients with squamous cell lung carcinoma, but from no normal controls.119
Proteomics for Other Diseases
While the power of proteomics has been used extensively in cancer research, the same approaches can also be applied to the myriad of other human diseases. Infectious diseases remain a leading cause of death worldwide. The sequencing of many pathogenic genomes now allows for substantial proteomic analyses of such pathogens. For example, a comparative proteomic study of the malaria parasite, Plasmodium falciparum, has led to the identification of new potential drug and vaccine targets.120,121 Pseudomonas aeruginosa is an opportunistic pathogen for humans, causing infections in cystic fibrosis and burns patients, as well as other immuno-compromised individuals.122 Nouwens et al. identified numerous potential drug targets by performing comprehensive 2D-gel based proteomic analysis of the membrane fractions of P. aeruginosa clinical isolates.123,124 Drug resistance is also a major problem for the treatment of infectious diseases, and identification of mechanisms of resistance and biomarkers specific to the resistant strains are required if treatments are to be effective. Tuberculosis remains one of the world’s most serious infectious diseases, claiming millions of lives every year.125 2D-GE and MS were used to identify proteins secreted by common clinical isolates of Mycobacterium tuberculosis. Two of these proteins have shown potential as serodiagnostic antigens.126 Clearly proteins that are secreted into the serum of infected patients are strong candidates for simple kit-based serum screening tests. Various other genomics and proteomics studies have also uncovered new vaccine candidates currently in clinical trials for tuberculosis.127 Severe acute respiratory syndrome (SARS) is a major priority area for current research. Proteomic analysis of sera from SARS patients has identified a potential protein marker, truncated α1-antitrypsin, which was consistently increased in SARS patients compared to healthy controls, that could be used as a specific diagnostic or potential drug target.128 Similar studies using SELDI technology have identified other potential biomarkers for the early diagnosis of SARS patients.129–131 One potential caveat of these studies is that the control cases were either healthy persons or patients with non-SARS infections, with the degree of similarity of symptoms between the SARS and control groups not considered. For effective diagnosis of infectious disease, differentiation is most often required to differentiate disease carrying patients from those patients presenting with similar symptoms, rather than healthy individuals. Indeed, it was recently shown that biomarkers identified by comparing SARS versus normal patients may not be clinically useful.132 In a recent study Pang et al. took this into account and identified a number of serum biomarkers present in SARS patients compared to non-SARS patients presenting with similar symptoms, that were correlated with clinical and/or biological variables.132 This powerful study suggests that serum proteomic fingerprints could be used to identify SARS cases early during onset, and could enable the correct treatment to be administered to each patient group by differentiating similarly presenting SARS and non-SARS patients.
Protein microarrays have been used in the clinical analysis of proteins that induce an antibody response in autoimmune disorders.133 Microarrays, generated by attaching hundreds of proteins and peptides to the surface of derivatised glass slides, were incubated with patient serum and fluorescent labels were used to detect autoantibody binding to specific proteins known to be associated with autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematosus.133 Many years of research have culminated in the identification of numerous biomarkers associated with cardiovascular disease. As with most diseases, the use of a single biomarker has limited applicability in cardiovascular disease, and a recent review compiled a list of 177 potential biomarkers for the different forms of this disease.1 Such a list is a powerful start to developing multiplexed panels for candidate-based proteome analysis. Proteomics is also being applied to diseases such as asthma,134–136 Alzheimers disease,137 dermatological disorders,138 rheumatoid arthritis139 and cystic fibrosis140 to name just a few.
Proteomics for Prediction of Clinical Outcome
Cancer is an extremely complex disease, with a multitude of molecular aberrations resulting in huge variability in clinical behaviour. As such, traditional analysis of one, or even a few biological parameters, has proved to be insufficient for accurate prediction of disease outcome. Advancements in gene expression profiling are beginning to allow for correlations of clinical data with genome-wide expression.4 Similar correlations were recently demonstrated using proteomic profiles.98 Importantly, by identifying functional components, i.e. the proteins, as precise prognostic markers, novel drug targets and chemotherapy strategies specifically designed for individual patients is a foreseeable outcome in the near future. Correlating the protein expression profiles by 2D-GE with clinical staging of 24 B-cell chronic lymphocytic leukaemia patients (B-CLL), allowed for the identification of 20 proteins with characteristic expression in the three patient distributions studied. Among those, HSP27 was over-expressed in patients with shorter survival times. Down-regulation of thioredoxin peroxidase 2 and protein disulfide isomerase also correlated to decreased survival times.98 Identification of these proteins is of particular prognostic value in B-CLL patients, but with further analysis may also be useful targets for improved therapy. Similar studies in other cancer types are now emerging. Prediction of outcome is also possible via other proteomics methods. As described above, a powerful application of proteomics is the identification of protein activity in specific pathways known to be involved in a disease process, or in response to treatment, and phosphorylation-specific arrays allow the simultaneous detection of pathway activation. Another approach is the use of tissue microarrays which require much less sample than 2D-gels, and as described above can be extremely useful for analysing known sets of protein markers.75 Access to sufficient clinical material can sometimes preclude large-scale proteomic analyses of clinical samples. For the discovery phase of proteomics projects the use of animal models can be a powerful means to overcome this. Moreover, using such models, investigations into in vivo drug response and drug resistance can be conducted in controlled experimental conditions.45
Many researchers are also investigating the use of blood or urine proteomes as these samples are much more readily available. Indeed, serum profiling has been shown to predict response to the cyclooxygenase-2 inhibitor, celecoxib, for cancer prevention and treatment.141 Celecoxib was shown to be efficacious in cancer prevention of patients with the inherited autosomal dominant condition, familial adenomatous polyposis (FAP), however there was a large heterogeneity in patients’ responses.142 Using SELDI TOF MS serum proteomic profiles from patients on this FAP/celecoxib clinical trial, Xiao et al. identified expression changes of several markers that were modulated after treatment with celecoxib, and identified one marker as a strong discriminator between response and non-response.141 SELDI-TOF MS serum profiling has also been used for prediction of liver fibrosis and cirrhosis in chronic hepatitis B infection,143 and, as described above, has been used to predict treatment outcome in patients with SARS.132
Proteomics can also be a powerful tool for identification of disease causing genes. In a seminal study using 2D-GE and affinity chromatography of autopsy brain tissue from late-infantile neuronal ceroid lipofuscinosis (LINCL) patients, a fatal neurodegenerative disease whose defective gene had remained elusive, Sleat et al. identified a protein absent in LINCL patients compared to normals as a pepstatin-insensitive lysosomal peptidase, termed CLN2.144 Importantly, sequence analysis identified mutations in this gene in LINCL patients.144 Future analysis of the myriad of proteomic markers identified in other diseases is certain to reveal further disease causing genes.
Pitfalls of Current Proteomics Technologies
While proteomics has proven extremely useful for discovery based research, its routine use in the clinic is currently hampered by a number of factors. The international Human Proteome Organisation (HUPO) (http://www.hupo.org) was launched in 2001 to start to overcome some of these pitfalls. HUPO’s mission is (i) to consolidate national and regional proteome organisations into a worldwide organisation (ii) to spread proteomics technologies and disseminate knowledge pertaining to the human proteome and model organisms by engaging in scientific and educational activities and (iii) to assist in the coordination of public proteome initiatives aimed at characterising specific tissue and cell proteomes.73 An extremely important aspect of HUPO is to provide standardisation of techniques particularly once proteome analyses become routine use in the clinical setting. HUPO’s Proteomics Standards Initiative is developing standards for data generation and presentation, including standardised formats for databases of all proteomics measurements.
Sample collection procedures must be carefully considered in any clinical laboratory. Specimen manipulation, be it sample collection, pipetting, and diluting, contributes to pre-analytical variables.145 Using MALDI-TOF MS, Marshall et al. demonstrated that changes in serum protein profiles can be generated simply by the amount of time between sample draw and analysis.146 Another source of variation to be considered is biological inter- and intra-patient variability.147 As with any biochemical analysis, protein expression between people resulting from factors such as age, gender, or race contributes to between-subject variation. Within patient variation can also occur based on time of day, fasting state, or age. Testing protein changes due to such variation requires good experimental design, a solid understanding of the limitations of proteomic technologies, and the proper use of statistics.145
In biomarker research, samples are usually collected from multiple sites and randomly divided into discovery (training) sets and validation (testing) sets. Differences in sample collection, handling or storage, and profiling techniques, may influence the protein profile obtained from a given sample.148,149 To obtain meaningful results a sufficiently large number of collection sites and sample populations must be employed to best represent the target population of interest.145 As mentioned previously, the use of SELDI to diagnose ovarian cancer at the earliest stages of the disease was heralded as a major advancement in cancer research.56 However, since the publication of this paper, several reports have emerged that question the validity of the data.150,151 The problems were only brought to light because the original authors made their data publicly available on the internet. The subsequent studies reanalysed the data and found numerous differences in the protein patterns that discriminated between the cancer patients and healthy controls, however they were concerned that the differences looked more like experimental artefacts rather than real biological differences.150,151 The original group have since refined its methods in a new paper152 and make the point that their 2002 Lancet paper56 still shows the feasibility of the approach, and the results prove that there are indeed low molecular weight molecules that can discriminate between the disease states.153 In addition, other groups have examined their data and support the original conclusions.154 This experience is testimony to the fact that relevant data must be made publicly available and must be able to be validated in independent laboratories. As this is one of the major initiatives of HUPO, more public scrutiny is sure to improve the validity of proteomics tests. OvaCheck, the commercial test based on the SELDI patterns described earlier is still being validated,56 however the company says it has validated the test and ensure it will work for women under many different testing conditions. If the company releases its primary data leading to this conclusion such that other researchers can examine the data, and allows researchers to send blinded test samples to determine if the test gives accurate results, the scientific community is then likely to regain confidence in the test.
Plasma, or its close cognate serum, is the primary biochemically useful clinical specimen. However plasma contains the largest and deepest human proteome making it the most difficult sample to work with in proteomics. The enormous depth of the plasma proteome comes from the huge dynamic range of protein concentrations within it. Approximately half of the total protein content in plasma comes from albumin (~55 mg/mL), and a further 10 proteins together make up 90% of the total. On the other hand, low abundance proteins such as cytokines are normally present at 1–5 pg/mL. Indeed, there may be even more proteins at lower concentrations that we have not yet discovered. Thus there are at least 10 orders of magnitude difference between the highest and lowest abundant proteins in plasma. Currently any one proteomics technique can only analyse proteins within 3–4 orders of magnitude, and mostly at the higher concentration end of the spectrum. The removal of high abundant proteins from plasma or serum is thus a prerequisite for conducting more detailed proteomic studies of low abundant proteins, and has been the subject of intense recent investigations (reviewed in reference 145). The most powerful method to date is the simultaneous removal of the 12 most abundant blood proteins by immunological means,155 and reasonably high-throughput columns are now commercially available to achieve this. However, it is also important to note that many potentially important biomarkers may also be lost in this process due to non-specific binding or the co-removal of peptides/proteins intrinsically bound to the high abundant carrier proteins. In particular albumin removal has been shown to result in significant loss of cytokines.156
Another precluding factor for the widespread use of proteomics in the clinical laboratory is the cost. Most proteomics technologies use complex instrumentation, critical computing power, and expensive consumables. At this stage, expertise of operators is still required, particularly for high-throughput analysis of MS data. It has been suggested that this technology could be most reliable and cost-effective if it is offered through large clinical 4 laboratories that are experienced in MS technology.157 While the cost of analyses at this stage may still preclude routine testing, prices are sure to reduce once bulk samples are being analysed. Furthermore, the technology is still a major tool in the discovery armoury, and identification of specific protein markers for disease are better developed into a cheaper and more user-friendly test, such as a protein or tissue microarray, or the ‘tried and tested’ ELISA. Indeed, the technology of ELISAs has improved in recent years and multiplexed assays to detect a combination of proteins are now possible.158
Future Perspectives
Medical research is poised to benefit greatly from the increasing use of proteomics, with the potential to develop better diagnostic and prognostic tests, to identify potential new therapeutic targets, and to head towards individualised patient therapy (Figure 3). Although proteomics has proved its promise for biomarker discovery, further work is still required to enhance the performance and reproducibility of established proteomics tools before they can be routinely used in the clinical laboratory. Issues regarding pre-analytical variables, analytical variability and biological variation must be tackled. The efforts of many researchers, together with the HUPO consortium, are now starting to address many of these issues, such that the future remains extremely bright for the widespread use of clinical proteomics. As current standard proteomics technologies require skilled operators and expensive equipment, their use in routine clinical laboratories is not yet feasible. Thus it is likely that the major input from standard proteomics studies will continue to be the identification of a particular set of biological markers for a specific disease, and more traditional biochemical and immunological tests will then be developed based on these discoveries. However the emerging multiplexing technology of protein chips and arrays should further enhance the throughput of such analytical tests in the near future.
Figure 3.
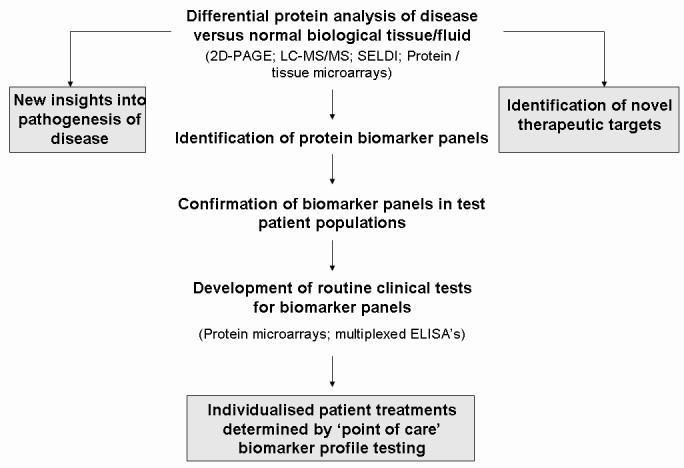
The promise of proteomics. With current technologies, proteomics has the power to greatly enhance our understanding of the molecular basis of disease and to identify novel drug targets. Recent advancements allow for identification of protein biomarkers that will be used for individualising patient care in the near future.
Acknowledgements
Nicole M. Verrills is supported by a National Health and Medical Research Council (NHMRC) Peter Doherty (Biomedical) Fellowship, and grants from the Hunter Medical Research Institute (HMRI).
Footnotes
Competing Interests: None declared.
References
- 1.Anderson L. Candidate-based proteomics in the search for biomarkers of cardiovascular disease. J Physiol. 2005;563:23–60. doi: 10.1113/jphysiol.2004.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rai A J, Zhang Z, Rosenzweig J, et al. Proteomic approaches to tumor marker discovery. Arch Pathol Lab Med. 2002;126:1518–26. doi: 10.5858/2002-126-1518-PATTMD. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Z, Bast RC, Jr, Yu Y, et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 2004;64:5882–90. doi: 10.1158/0008-5472.CAN-04-0746. [DOI] [PubMed] [Google Scholar]
- 4.Weinstein JN, Myers TG, O'Connor PM, et al. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–9. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 5.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 6.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–48. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 8.Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–26. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 9.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 10.Esteva FJ, Valero V, Booser D, et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:1800–8. doi: 10.1200/JCO.2002.07.058. [DOI] [PubMed] [Google Scholar]
- 11.van't Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 12.van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 13.Ross ME, Zhou X, Song G, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102:2951–9. doi: 10.1182/blood-2003-01-0338. [DOI] [PubMed] [Google Scholar]
- 14.Yeoh EJ, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 15.Haslinger C, Schweifer N, Stilgenbauer S, et al. Microarray gene expression profiling of B-cell chronic lymphocytic leukemia subgroups defined by genomic aberrations and VH mutation status. J Clin Oncol. 2004;22:3937–49. doi: 10.1200/JCO.2004.12.133. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Jie S, Zou P, Zou G. CDNA microarray analysis of chronic myeloid leukemia. Int J Hematol. 2002;75:388–93. doi: 10.1007/BF02982130. [DOI] [PubMed] [Google Scholar]
- 17.Lindvall C, Furge K, Bjorkholm M, et al. Combined genetic and transcriptional profiling of acute myeloid leukemia with normal and complex karyotypes. Haematologica. 2004;89:1072–81. [PubMed] [Google Scholar]
- 18.Ross ME, Mahfouz R, Onciu M, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–87. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- 19.Cheok MH, Yang W, Pui CH, et al. Treatment-specific changes in gene expression discriminate in vivo drug response in human leukemia cells. Nat Genet. 2003;34:85–90. doi: 10.1038/ng1151. [DOI] [PubMed] [Google Scholar]
- 20.Holleman A, Cheok MH, den Boer ML, et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med. 2004;351:533–42. doi: 10.1056/NEJMoa033513. [DOI] [PubMed] [Google Scholar]
- 21.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–30. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong YK, Lo YM. Diagnostic developments involving cell-free (circulating) nucleic acids. Clin Chim Acta. 2006;363:187–96. doi: 10.1016/j.cccn.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 23.Taback B, Hoon DS. Circulating nucleic acids in plasma and serum: past, present and future. Curr Opin Mol Ther. 2004;6:273–8. [PubMed] [Google Scholar]
- 24.O'Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–21. [PMC free article] [PubMed] [Google Scholar]
- 25.Bjellqvist B, Ek K, Righetti PG, et al. Isoelectric focusing in immobilized pH gradients: principle, methodology and some applications. J Biochem Biophys Methods. 1982;6:317–39. doi: 10.1016/0165-022x(82)90013-6. [DOI] [PubMed] [Google Scholar]
- 26.Scherl A, Coute Y, Deon C, et al. Functional proteomic analysis of human nucleolus. Mol Biol Cell. 2002;13:4100–9. doi: 10.1091/mbc.E02-05-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cordwell SJ, Nouwens AS, Verrills NM, Basseal DJ, Walsh BJ. Subproteomics based upon protein cellular location and relative solubilities in conjunction with composite two-dimensional electrophoresis gels. Electrophoresis. 2000;21:1094–103. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1094::AID-ELPS1094>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 28.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 29.Hoffmann P, Ji H, Moritz RL, et al. Continuous free-flow electrophoresis separation of cytosolic proteins from the human colon carcinoma cell line LIM 1215: a non two-dimensional gel electrophoresis-based proteome analysis strategy. Proteomics. 2001;1:807–18. doi: 10.1002/1615-9861(200107)1:7<807::AID-PROT807>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 30.Gorg A, Weiss W, Dunn MJ. Current two-dimensional electrophoresis technology for proteomics. Proteomics. 2004;4:3665–85. doi: 10.1002/pmic.200401031. [DOI] [PubMed] [Google Scholar]
- 31.Tonge R, Shaw J. Middleton B, et al. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1:377–96. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 32.Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–7. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 33.Von Eggeling F, Gawriljuk A, Fiedler W, et al. Fluorescent dual colour 2D-protein gel electrophoresis for rapid detection of differences in protein pattern with standard image analysis software. Int J Mol Med. 2001;8:373–7. doi: 10.3892/ijmm.8.4.373. [DOI] [PubMed] [Google Scholar]
- 34.Alban A, David SO, Bjorkesten L, et al. A novel experimental design for comparative two-dimensional gel analysis: two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics. 2003;3:36–44. doi: 10.1002/pmic.200390006. [DOI] [PubMed] [Google Scholar]
- 35.Friedman DB, Hill S, Keller JW, et al. Proteome analysis of human colon cancer by two-dimensional difference gel electrophoresis and mass spectrometry. Proteomics. 2004;4:793–811. doi: 10.1002/pmic.200300635. [DOI] [PubMed] [Google Scholar]
- 36.Somiari RI, Sullivan A, Russell S, et al. High-throughput proteomic analysis of human infiltrating ductal carcinoma of the breast. Proteomics. 2003;3:1863–73. doi: 10.1002/pmic.200300560. [DOI] [PubMed] [Google Scholar]
- 37.Zhou G, Li H, DeCamp D, et al. 2D differential in-gel electrophoresis for the identification of esophageal scans cell cancer-specific protein markers. Mol Cell Proteomics. 2002;1:117–24. doi: 10.1074/mcp.m100015-mcp200. [DOI] [PubMed] [Google Scholar]
- 38.Wang D, Jensen R, Gendeh G, Williams K, Pallavicini MG. Proteome and transcriptome analysis of retinoic acid-induced differentiation of human acute promyelocytic leukemia cells, NB4. J Proteome Res. 2004;3:627–35. doi: 10.1021/pr049976r. [DOI] [PubMed] [Google Scholar]
- 39.Seike M, Kondo T, Fujii K, et al. Proteomic signature of human cancer cells. Proteomics. 2004;4:2776–88. doi: 10.1002/pmic.200300795. [DOI] [PubMed] [Google Scholar]
- 40.Gharbi S, Gaffney P, Yang A, et al. Evaluation of two-dimensional differential gel electrophoresis for proteomic expression analysis of a model breast cancer cell system. Mol Cell Proteomics. 2002;1:91–8. doi: 10.1074/mcp.t100007-mcp200. [DOI] [PubMed] [Google Scholar]
- 41.Lee JR, Baxter TM, Yamaguchi H, Wang TC, Goldenring JR, Anderson MG. Differential protein analysis of spasomolytic polypeptide expressing metaplasia using laser capture microdissection and two-dimensional difference gel electrophoresis. Appl Immunohistochem Mol Morphol. 2003;11:188–93. doi: 10.1097/00129039-200306000-00018. [DOI] [PubMed] [Google Scholar]
- 42.Knowles MR, Cervino S, Skynner HA, et al. Multiplex proteomic analysis by two-dimensional differential in-gel electrophoresis. Proteomics. 2003;3:1162–71. doi: 10.1002/pmic.200300437. [DOI] [PubMed] [Google Scholar]
- 43.Kernec F, Unlu M, Labeikovsky W, Minden JS, Koretsky AP. Changes in the mitochondrial proteome from mouse hearts deficient in creatine kinase. Physiol Genomics. 2001;6:117–28. doi: 10.1152/physiolgenomics.2001.6.2.117. [DOI] [PubMed] [Google Scholar]
- 44.Evans CA, Tonge R, Blinco D, et al. Comparative proteomics of primitive hematopoietic cell populations reveals differences in expression of proteins regulating motility. Blood. 2004;103:3751–9. doi: 10.1182/blood-2003-09-3294. [DOI] [PubMed] [Google Scholar]
- 45.Verrills NM, Liem NL, Liaw TY, Hood BD, Lock RB, Kavallaris M. Proteomic analysis reveals a novel role for the actin cytoskeleton in vincristine resistant childhood leukemia - An in vivo study. Proteomics. 2006;6:1681–94. doi: 10.1002/pmic.200500417. [DOI] [PubMed] [Google Scholar]
- 46.Lim MS, Elenitoba-Johnson KS. Proteomics in pathology research. Lab Invest. 2004;84:1227–44. doi: 10.1038/labinvest.3700167. [DOI] [PubMed] [Google Scholar]
- 47.Yates JR., 3rd Mass spectrometry and the age of the proteome. J Mass Spectrom. 1998;33:1–19. doi: 10.1002/(SICI)1096-9888(199801)33:1<1::AID-JMS624>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 48.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 49.Schneider LV, Hall MP. Stable isotope methods for high-precision proteomics. Drug Discov Today. 2005;10:353–63. doi: 10.1016/S1359-6446(05)03381-7. [DOI] [PubMed] [Google Scholar]
- 50.Ong SE, Kratchmarova I, Mann M. Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC) J Proteome Res. 2003;2:173–81. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 51.Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 52.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 53.Fey SJ, Larsen PM. 2D or not 2D. Two-dimensional gel electrophoresis. Curr Opin Chem Biol. 2001;5:26–33. doi: 10.1016/s1367-5931(00)00167-8. [DOI] [PubMed] [Google Scholar]
- 54.Merchant M, Weinberger SR. Recent advancements in surface-enhanced laser desorption/ionization-time of flight-mass spectrometry. Electrophoresis. 2000;21:1164–77. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1164::AID-ELPS1164>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 55.Xiao Z, Adam BL, Cazares LH, et al. Quantitation of serum prostate-specific membrane antigen by a novel protein biochip immunoassay discriminates benign from malignant prostate disease. Cancer Res. 2001;61:6029–33. [PubMed] [Google Scholar]
- 56.Petricoin EF, Ardekani AM, Hitt BA, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–7. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 57.Villanueva J, Shaffer DR, Philip J, et al. Differential exoprotease activities confer tumor-specific serum peptidome patterns. J Clin Invest. 2006;116:271–84. doi: 10.1172/JCI26022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinberger SR, Dalmasso EA, Fung ET. Current achievements using ProteinChip Array technology. Curr Opin Chem Biol. 2002;6:86–91. doi: 10.1016/s1367-5931(01)00282-4. [DOI] [PubMed] [Google Scholar]
- 59.Phizicky E, Bastiaens PI, Zhu H, Snyder M, Fields S. Protein analysis on a proteomic scale. Nature. 2003;422:208–15. doi: 10.1038/nature01512. [DOI] [PubMed] [Google Scholar]
- 60.Liotta LA, Espina V, Mehta AI, et al. Protein microarrays: meeting analytical challenges for clinical applications. Cancer Cell. 2003;3:317–25. doi: 10.1016/s1535-6108(03)00086-2. [DOI] [PubMed] [Google Scholar]
- 61.Zhu H, Snyder M. Protein arrays and microarrays. Curr Opin Chem Biol. 2001;5:40–5. doi: 10.1016/s1367-5931(00)00170-8. [DOI] [PubMed] [Google Scholar]
- 62.Kodadek T. Protein microarrays: prospects and problems. Chem Biol. 2001;8:105–15. doi: 10.1016/s1074-5521(00)90067-x. [DOI] [PubMed] [Google Scholar]
- 63.Greenbaum D, Baruch A, Hayrapetian L, et al. Chemical approaches for functionally probing the proteome. Mol Cell Proteomics. 2002;1:60–8. doi: 10.1074/mcp.t100003-mcp200. [DOI] [PubMed] [Google Scholar]
- 64.Wilson DS, Nock S. Recent developments in protein microarray technology. Angew Chem Int Ed Engl. 2003;42:494–500. doi: 10.1002/anie.200390150. [DOI] [PubMed] [Google Scholar]
- 65.Jessani N, Liu Y, Humphrey M, Cravatt BF. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc Natl Acad Sci U S A. 2002;99:10335–40. doi: 10.1073/pnas.162187599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003;21:2787–99. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- 67.Sewell JM, Macleod KG, Ritchie A, Smyth JF, Langdon SP. Targeting the EGF receptor in ovarian cancer with the tyrosine kinase inhibitor ZD 1839 (“Iressa”) Br J Cancer. 2002;86:456–62. doi: 10.1038/sj.bjc.6600058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peggs K. Imatinib mesylate--gold standards and silver linings. Clin Exp Med. 2004;4:1–9. doi: 10.1007/s10238-004-0032-5. [DOI] [PubMed] [Google Scholar]
- 69.Chen LL, Sabripour M, Andtbacka RH, et al. Imatinib resistance in gastrointestinal stromal tumors. Curr Oncol Rep. 2005;7:293–9. doi: 10.1007/s11912-005-0053-6. [DOI] [PubMed] [Google Scholar]
- 70.Petricoin EF, 3rd, Bichsel VE, Calvert VS, et al. Mapping molecular networks using proteomics: a vision for patient-tailored combination therapy. J Clin Oncol. 2005;23:3614–21. doi: 10.1200/JCO.2005.02.509. [DOI] [PubMed] [Google Scholar]
- 71.Wulfkuhle JD, Aquino JA, Calvert VS, et al. Signal pathway profiling of ovarian cancer from human tissue specimens using reverse-phase protein microarrays. Proteomics. 2003;3:2085–90. doi: 10.1002/pmic.200300591. [DOI] [PubMed] [Google Scholar]
- 72.Sheehan KM, Calvert VS, Kay EW, et al. Use of reverse phase protein microarrays and reference standard development for molecular network analysis of metastatic ovarian carcinoma. Mol Cell Proteomics. 2005;4:346–55. doi: 10.1074/mcp.T500003-MCP200. [DOI] [PubMed] [Google Scholar]
- 73.Hanash S. Disease proteomics. Nature. 2003;422:226–32. doi: 10.1038/nature01514. [DOI] [PubMed] [Google Scholar]
- 74.Tyers M, Mann M. From genomics to proteomics. Nature. 2003;422:193–7. doi: 10.1038/nature01510. [DOI] [PubMed] [Google Scholar]
- 75.Jacquemier J, Ginestier C, Rougemont J, et al. Protein expression profiling identifies subclasses of breast cancer and predicts prognosis. Cancer Res. 2005;65:767–79. [PubMed] [Google Scholar]
- 76.Caprioli RM. Deciphering protein molecular signatures in cancer tissues to aid in diagnosis, prognosis, and therapy. Cancer Res. 2005;65:10642–5. doi: 10.1158/0008-5472.CAN-04-3581. [DOI] [PubMed] [Google Scholar]
- 77.Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues. Nat Med. 2001;7:493–6. doi: 10.1038/86573. [DOI] [PubMed] [Google Scholar]
- 78.Schwartz SA, Weil RJ, Thompson RC, et al. Proteomic-based prognosis of brain tumor patients using direct-tissue matrix-assisted laser desorption ionization mass spectrometry. Cancer Res. 2005;65:7674–81. doi: 10.1158/0008-5472.CAN-04-3016. [DOI] [PubMed] [Google Scholar]
- 79.Xu BJ, Caprioli RM, Sanders ME, Jensen RA. Direct analysis of laser capture microdissected cells by MALDI mass spectrometry. J Am Soc Mass Spectrom. 2002;13:1292–7. doi: 10.1016/S1044-0305(02)00644-X. [DOI] [PubMed] [Google Scholar]
- 80.Maddalo G, Petrucci F, Iezzi M, et al. Analytical assessment of MALDI-TOF Imaging Mass Spectrometry on thin histological samples. An insight in proteome investigation. Clin Chim Acta. 2005;357:210–8. doi: 10.1016/j.cccn.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 81.Celis JE, Gromov P. Proteomics in translational cancer research: Toward an integrated approach. Cancer Cell. 2003;3:9–15. doi: 10.1016/s1535-6108(02)00242-8. [DOI] [PubMed] [Google Scholar]
- 82.Celis JE, Ostergaard M, Jensen NA, Gromova I, Rasmussen HH, Gromov P. Human and mouse proteomic databases: novel resources in the protein universe. FEBS Lett. 1998;430:64–72. doi: 10.1016/s0014-5793(98)00527-4. [DOI] [PubMed] [Google Scholar]
- 83.Alaiya AA, Franzen B, Auer G, Linder S. Cancer proteomics: from identification of novel markers to creation of artificial learning models for tumor classification. Electrophoresis. 2000;21:1210–7. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1210::AID-ELPS1210>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 84.Srinivas PR, Srivastava S, Hanash S, Wright GL., Jr Proteomics in early detection of cancer. Clin Chem. 2001;47:1901–11. [PubMed] [Google Scholar]
- 85.Verrills NM, Kavallaris M. Drug resistance mechanisms in cancer cells: a proteomics perspective. Curr Opin Mol Ther. 2003;5:258–65. [PubMed] [Google Scholar]
- 86.Jones MB, Krutzsch H, Shu H, et al. Proteomic analysis and identification of new biomarkers and therapeutic targets for invasive ovarian cancer. Proteomics. 2002;2:76–84. [PubMed] [Google Scholar]
- 87.Celis JE, Celis P, Palsdottir H, et al. Proteomic strategies to reveal tumor heterogeneity among urothelial papillomas. Mol Cell Proteomics. 2002;1:269–79. doi: 10.1074/mcp.m100031-mcp200. [DOI] [PubMed] [Google Scholar]
- 88.Zhou G, Li H, Gong Y, et al. Proteomic analysis of global alteration of protein expression in squamous cell carcinoma of the esophagus. Proteomics. 2005;5:3814–21. doi: 10.1002/pmic.200401230. [DOI] [PubMed] [Google Scholar]
- 89.Wright ME, Han DK, Aebersold R. Mass spectrometry-based expression profiling of clinical prostate cancer. Mol Cell Proteomics. 2005;4:545–54. doi: 10.1074/mcp.R500008-MCP200. [DOI] [PubMed] [Google Scholar]
- 90.Hayashida Y, Honda K, Osaka Y, et al. Possible prediction of chemoradiosensitivity of esophageal cancer by serum protein profiling. Clin Cancer Res. 2005;11:8042–7. doi: 10.1158/1078-0432.CCR-05-0656. [DOI] [PubMed] [Google Scholar]
- 91.Wu CC, MacCoss MJ, Howell KE, Yates JR., 3rd A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–8. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 92.Florens L, Washburn MP, Raine JD, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520–6. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- 93.Schirmer EC, Florens L, Guan T, Yates JR, 3rd, Gerace L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 2003;301:1380–2. doi: 10.1126/science.1088176. [DOI] [PubMed] [Google Scholar]
- 94.MacCoss MJ, McDonald WH, Saraf A, et al. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci U S A. 2002;99:7900–5. doi: 10.1073/pnas.122231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hanash SM, Baier LJ, McCurry L, Schwartz SA. Lineage-related polypeptide markers in acute lympho-blastic leukemia detected by two-dimensional gel electrophoresis. Proc Natl Acad Sci U S A. 1986;83:807–11. doi: 10.1073/pnas.83.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Strahler JR, Kuick R, Eckerskorn C, et al. Identification of two related markers for common acute lymphoblastic leukemia as heat shock proteins. J Clin Invest. 1990;85:200–7. doi: 10.1172/JCI114413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hanash SM, Kuick R, Strahler J, et al. Identification of a cellular polypeptide that distinguishes between acute lymphoblastic leukemia in infants and in older children. Blood. 1989;73:527–32. [PubMed] [Google Scholar]
- 98.Voss T, Ahorn H, Haberl P, Dohner H, Wilgenbus K. Correlation of clinical data with proteomics profiles in 24 patients with B-cell chronic lymphocytic leukemia. Int J Cancer. 2001;91:180–6. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1037>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 99.Shin BK, Wang H, Yim AM, et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278:7607–16. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 100.Adam PJ, Boyd R, Tyson KL, et al. Comprehensive proteomic analysis of breast cancer cell membranes reveals unique proteins with potential roles in clinical cancer. J Biol Chem. 2003;278:6482–9. doi: 10.1074/jbc.M210184200. [DOI] [PubMed] [Google Scholar]
- 101.Myers TG, Anderson NL, Waltham M, et al. A protein expression database for the molecular pharmacology of cancer. Electrophoresis. 1997;18:647–53. doi: 10.1002/elps.1150180351. [DOI] [PubMed] [Google Scholar]
- 102.Hutter G, Sinha P. Proteomics for studying cancer cells and the development of chemoresistance. Proteomics. 2001;1:1233–48. doi: 10.1002/1615-9861(200110)1:10<1233::AID-PROT1233>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 103.Sinha P, Kohl S, Fischer J, et al. Identification of novel proteins associated with the development of chemo- resistance in malignant melanoma using two- dimensional electrophoresis. Electrophoresis. 2000;21:3048–57. doi: 10.1002/1522-2683(20000801)21:14<3048::AID-ELPS3048>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 104.Sinha P, Hutter G, Kottgen E, Dietel M, Schadendorf D, Lage H. Increased expression of epidermal fatty acid binding protein, cofilin, and 14-3-3-sigma (stratifin) detected by two-dimensional gel electrophoresis, mass spectrometry and microsequencing of drug-resistant human adenocarcinoma of the pancreas. Electrophoresis. 1999;20:2952–60. doi: 10.1002/(SICI)1522-2683(19991001)20:14<2952::AID-ELPS2952>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 105.Verrills NM, Walsh BJ, Cobon GS, Hains PG, Kavallaris M. Proteome analysis of vinca alkaloid response and resistance in acute lymphoblastic leukemia reveals novel cytoskeletal alterations. J Biol Chem. 2003;278:45082–93. doi: 10.1074/jbc.M303378200. [DOI] [PubMed] [Google Scholar]
- 106.Kavallaris M, Tait AS, Walsh BJ, et al. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res. 2001;61:5803–9. [PubMed] [Google Scholar]
- 107.Verrills NM, Flemming CL, Liu M, et al. Microtubule alterations and mutations induced by desoxyepothilone B: implications for drug-target interactions. Chem Biol. 2003;10:597–607. doi: 10.1016/s1074-5521(03)00141-8. [DOI] [PubMed] [Google Scholar]
- 108.Nishio K, Sugimoto Y, Kasahara K, et al. Increased phosphorylation of nuclear phosphoproteins in human lung-cancer cells resistant to cisdiamminedichloro-platinum (II) Int J Cancer. 1992;50:438–42. doi: 10.1002/ijc.2910500319. [DOI] [PubMed] [Google Scholar]
- 109.Rao S, Aberg F, Nieves E, Band Horwitz S, Orr G. A. Identification by mass spectrometry of a new alpha-tubulin isotype expressed in human breast and lung carcinoma cell lines. Biochemistry. 2001;40:2096–103. doi: 10.1021/bi002323d. [DOI] [PubMed] [Google Scholar]
- 110.Larcher JC, Boucher D, Lazereg S, Gros F, Denoulet P. Interaction of kinesin motor domains with alpha- and beta-tubulin subunits at a tau-independent binding site. Regulation by polyglutamylation. J Biol Chem. 1996;271:22117–24. doi: 10.1074/jbc.271.36.22117. [DOI] [PubMed] [Google Scholar]
- 111.Boucher D, Larcher JC, Gros F, Denoulet P. Polyglutamylation of tubulin as a progressive regulator of in vitro interactions between the microtubule-associated protein Tau and tubulin. Biochemistry. 1994;33:12471–7. doi: 10.1021/bi00207a014. [DOI] [PubMed] [Google Scholar]
- 112.Banerjee A. Increased levels of tyrosinated alpha-, beta(III)-, and beta(IV)-tubulin isotypes in paclitaxel-resistant MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2002;293:598–601. doi: 10.1016/S0006-291X(02)00269-3. [DOI] [PubMed] [Google Scholar]
- 113.Banerjee A. Coordination of posttranslational modifications of bovine brain alpha-tubulin. Polyglycylation of delta2 tubulin. J Biol Chem. 2002;277:46140–4. doi: 10.1074/jbc.M208065200. [DOI] [PubMed] [Google Scholar]
- 114.Stockert E, Jager E, Chen YT, et al. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med. 1998;187:1349–54. doi: 10.1084/jem.187.8.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Boon T, Old LJ. Cancer Tumor antigens. Curr Opin Immunol. 1997;9:681–3. doi: 10.1016/s0952-7915(97)80049-0. [DOI] [PubMed] [Google Scholar]
- 116.Soussi T. p53 Antibodies in the sera of patients with various types of cancer: a review. Cancer Res. 2000;60:1777–88. [PubMed] [Google Scholar]
- 117.Old LJ, Chen YT. New paths in human cancer serology. J Exp Med. 1998;187:1163–7. doi: 10.1084/jem.187.8.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Le Naour F, Brichory F, Misek DE, Brechot C, Hanash SM, Beretta L. A distinct repertoire of autoantibodies in hepatocellular carcinoma identified by proteomic analysis. Mol Cell Proteomics. 2002;1:197–203. doi: 10.1074/mcp.m100029-mcp200. [DOI] [PubMed] [Google Scholar]
- 119.Brichory FM, Misek DE, Yim AM, et al. An immune response manifested by the common occurrence of annexins I and II autoantibodies and high circulating levels of IL-6 in lung cancer. Proc Natl Acad Sci U S A. 2001;98:9824–9. doi: 10.1073/pnas.171320598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lasonder E, Ishihama Y, Andersen JS, et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002;419:537–42. doi: 10.1038/nature01111. [DOI] [PubMed] [Google Scholar]
- 121.Nilsson CL. Bacterial proteomics and vaccine development. Am J Pharmacogenomics. 2002;2:59–65. doi: 10.2165/00129785-200202010-00005. [DOI] [PubMed] [Google Scholar]
- 122.Lyczak JB, Cannon CL, Pier GB. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2000;2:1051–60. doi: 10.1016/s1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 123.Nouwens AS, Willcox MD, Walsh BJ, Cordwell SJ. Proteomic comparison of membrane and extracellular proteins from invasive (PAO1) and cytotoxic (6206) strains of Pseudomonas aeruginosa. Proteomics. 2002;2:1325–46. doi: 10.1002/1615-9861(200209)2:9<1325::AID-PROT1325>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 124.Nouwens AS, Cordwell SJ, Larsen MR, et al. Complementing genomics with proteomics: the membrane subproteome of Pseudomonas aeruginosa PAO1. Electrophoresis. 2000;21:3797–809. doi: 10.1002/1522-2683(200011)21:17<3797::AID-ELPS3797>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 125.Anon. Global Tuberculosis Control, WHO Report 2002. World Health Organisation 2002;181.
- 126.Bahk YY, Kim SA, Kim JS, et al. Antigens secreted from Mycobacterium tuberculosis: identification by proteomics approach and test for diagnostic marker. Proteomics. 2004;4:3299–307. doi: 10.1002/pmic.200400980. [DOI] [PubMed] [Google Scholar]
- 127.Doherty T M. Real world TB vaccines: clinical trials in TB-endemic regions. Vaccine. 2005;23:2109–14. doi: 10.1016/j.vaccine.2005.01.060. [DOI] [PubMed] [Google Scholar]
- 128.Ren Y, He QY, Fan J, et al. The use of proteomics in the discovery of serum biomarkers from patients with severe acute respiratory syndrome. Proteomics. 2004;4:3477–84. doi: 10.1002/pmic.200400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yip TT, Chan JW, Cho WC, et al. Protein chip array profiling analysis in patients with severe acute respiratory syndrome identified serum amyloid a protein as a biomarker potentially useful in monitoring the extent of pneumonia. Clin Chem. 2005;51:47–55. doi: 10.1373/clinchem.2004.031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Poon TC, Chan KC, Ng PC, et al. Serial analysis of plasma proteomic signatures in pediatric patients with severe acute respiratory syndrome and correlation with viral load. Clin Chem. 2004;50:1452–5. doi: 10.1373/clinchem.2004.035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kang X, Xu Y, Wu X, et al. Proteomic fingerprints for potential application to early diagnosis of severe acute respiratory syndrome. Clin Chem. 2005;51:56–64. doi: 10.1373/clinchem.2004.032458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pang RT, Poon TC, Chan KC, et al. Serum proteomic fingerprints of adult patients with severe acute respiratory syndrome. Clin Chem. 2006;52:421–9. doi: 10.1373/clinchem.2005.061689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Robinson WH, DiGennaro C, Hueber W, et al. Auto-antigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 134.Chung YW, Oh HY, Kim JY, Kim JH, Kim IY. Allergen-induced proteolytic cleavage of annexin-1 and activation of cytosolic phospholipase A2 in the lungs of a mouse model of asthma. Proteomics. 2004;4:3328–34. doi: 10.1002/pmic.200400895. [DOI] [PubMed] [Google Scholar]
- 135.Yeo S, Roh GS, Kim DH, et al. Quantitative profiling of plasma peptides in asthmatic mice using liquid chromatography and mass spectrometry. Proteomics. 2004;4:3308–17. doi: 10.1002/pmic.200400918. [DOI] [PubMed] [Google Scholar]
- 136.Roh GS, Shin Y, Seo SW, et al. Proteome analysis of differential protein expression in allergen-induced asthmatic mice lung after dexamethasone treatment. Proteomics. 2004;4:3318–27. doi: 10.1002/pmic.200400930. [DOI] [PubMed] [Google Scholar]
- 137.Shin SJ, Lee SE, Boo JH, et al. Profiling proteins related to amyloid deposited brain of Tg2576 mice. Proteomics. 2004;4:3359–68. doi: 10.1002/pmic.200400961. [DOI] [PubMed] [Google Scholar]
- 138.Park YD, Kim SY, Jang HS, et al. Towards a proteomic analysis of atopic dermatitis: a two-dimensional-polyacrylamide gel electrophoresis/mass spectrometric analysis of cultured patient-derived fibroblasts. Proteomics. 2004;4:3446–55. doi: 10.1002/pmic.200400998. [DOI] [PubMed] [Google Scholar]
- 139.Ali M, Manolios N. Proteomics in rheumatology: a new direction for old diseases. Semin Arthritis Rheum. 2005;35:67–76. doi: 10.1016/j.semarthrit.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 140.Sloane AJ, Lindner RA, Prasad SS, et al. Proteomic analysis of sputum from adults and children with cystic fibrosis and from control subjects. Am J Respir Crit Care Med. 2005;172:1416–26. doi: 10.1164/rccm.200409-1215OC. [DOI] [PubMed] [Google Scholar]
- 141.Xiao Z, Luke BT, Izmirlian G, et al. Serum proteomic profiles suggest celecoxib-modulated targets and response predictors. Cancer Res. 2004;64:2904–9. doi: 10.1158/0008-5472.can-03-3754. [DOI] [PubMed] [Google Scholar]
- 142.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–52. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 143.Poon TC, Hui AY, Chan HL, et al. Prediction of liver fibrosis and cirrhosis in chronic hepatitis B infection by serum proteomic fingerprinting: a pilot study. Clin Chem. 2005;51:328–35. doi: 10.1373/clinchem.2004.041764. [DOI] [PubMed] [Google Scholar]
- 144.Sleat DE, Donnelly RJ, Lackland H, et al. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–5. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- 145.Colantonio DA, Chan DW. The clinical application of proteomics. Clin Chim Acta. 2005;357:151–8. doi: 10.1016/j.cccn.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 146.Marshall J, Kupchak P, Zhu W, et al. Processing of serum proteins underlies the mass spectral fingerprinting of myocardial infarction. J Proteome Res. 2003;2:361–72. doi: 10.1021/pr030003l. [DOI] [PubMed] [Google Scholar]
- 147.Petersen PH, Sandberg S, Fraser CG, Goldschmidt H. Influence of index of individuality on false positives in repeated sampling from healthy individuals. Clin Chem Lab Med. 2001;39:160–5. doi: 10.1515/CCLM.2001.027. [DOI] [PubMed] [Google Scholar]
- 148.White CN, Chan DW, Zhang Z. Bioinformatics strategies for proteomic profiling. Clin Biochem. 2004;37:636–41. doi: 10.1016/j.clinbiochem.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 149.Drake SK, Bowen RA, Remaley AT, Hortin GL. Potential interferences from blood collection tubes in mass spectrometric analyses of serum polypeptides. Clin Chem. 2004;50:2398–401. doi: 10.1373/clinchem.2004.040303. [DOI] [PubMed] [Google Scholar]
- 150.Sorace JM, Zhan M. A data review and re-assessment of ovarian cancer serum proteomic profiling. BMC Bioinformatics. 2003;4:24. doi: 10.1186/1471-2105-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Baggerly KA, Morris JS, Coombes KR. Reproducibility of SELDI-TOF protein patterns in serum: comparing datasets from different experiments. Bioinformatics. 2004;20:777–85. doi: 10.1093/bioinformatics/btg484. [DOI] [PubMed] [Google Scholar]
- 152.Conrads TP, Fusaro VA, Ross S, et al. High-resolution serum proteomic features for ovarian cancer detection. Endocr Relat Cancer. 2004;11:163–78. doi: 10.1677/erc.0.0110163. [DOI] [PubMed] [Google Scholar]
- 153.Check E. Proteomics and cancer: running before we can walk? Nature. 2004;429:496–7. doi: 10.1038/429496a. [DOI] [PubMed] [Google Scholar]
- 154.Zhu W, Wang X, Ma Y, Rao M, Glimm J, Kovach JS. Detection of cancer-specific markers amid massive mass spectral data. Proc Natl Acad Sci U S A. 2003;100:14666–71. doi: 10.1073/pnas.2532248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huang L, Harvie G, Feitelson JS, et al. Immunoaffinity separation of plasma proteins by IgY microbeads: meeting the needs of proteomic sample preparation and analysis. Proteomics. 2005;5:3314–28. doi: 10.1002/pmic.200401277. [DOI] [PubMed] [Google Scholar]
- 156.Granger J, Siddiqui J, Copeland S, Remick D. Albumin depletion of human plasma also removes low abundance proteins including the cytokines. Proteomics. 2005;5:4713–8. doi: 10.1002/pmic.200401331. [DOI] [PubMed] [Google Scholar]
- 157.Petricoin E, 3rd, Liotta LA. Counterpoint: The vision for a new diagnostic paradigm. Clin Chem. 2003;49:1276–8. doi: 10.1373/49.8.1276. [DOI] [PubMed] [Google Scholar]
- 158.Nielsen UB, Geierstanger BH. Multiplexed sandwich assays in microarray format. J Immunol Methods. 2004;290:107–20. doi: 10.1016/j.jim.2004.04.012. [DOI] [PubMed] [Google Scholar]