

Abstract
Objective
Mutations in the cardiac sodium channel gene SCN5A are responsible for type-3 long QT disease (LQT3). The genesis of cardiac arrhythmias in LQT3 is multifaceted, and the aim of this study was to further explore mechanisms by which SCN5A mutations lead to arrhythmogenesis in vivo.
Methods
We engineered selective cardiac expression of a long QT syndrome (LQTS) mutation (N1325S) in human SCN5A and generated a transgenic mouse model, TGM(NS31).
Results
Conscious and unrestrained TGM(NS31)L12 mice demonstrated a significant prolongation of the QT-interval and a high incidence of spontaneous polymorphic ventricular tachycardia (VT) and fibrillation (VF), often resulting in sudden cardiac death (n = 52:156). Arrhythmias were suppressed by mexiletine, a sodium channel blocker for the late persistent sodium current. Action potentials (APs) from TGM(NS31)L12 ventricular myocytes exhibited early afterdepolarizations and longer 90% AP durations (APD90 = 69 ± 5.9 ms) than control (APD90 = 46.7 ± 4.8 ms). Voltage-clamp experiments in transgenic myocytes revealed a peak inward sodium current (INa) followed by a slow recovery from inactivation. After mexiletine application, transgenic ventricular APDs were shortened, and recovery from inactivation of INa was enhanced. These suggest that the N1325S transgene is responsible for the abnormal signals present in transgenic cells as well as the genesis of lethal arrhythmias in mice. Interestingly, transgenic but not wild-type myocytes displayed longer APDs with a shortening of CLs.
Conclusions
Our findings show that a new model for LQTS has been established, and we report on an arrhythmogenic mechanism that, unlike other SCN5A mutations, results in poor restitution of APD with increasing rate as a possible substrate for arrhythmogenesis.
Keywords: Cardiac sodium channel SCN5A, Long QT syndrome, Late persistent sodium current, Arrhythmia mechanisms
1. Introduction
Long QT syndrome (LQTS) is a cardiac disorder characterized by prolongation of the QT-interval on the electrocardiogram (ECG). Patients are susceptible to a specific ventricular tachycardia (VT), torsade de pointes (TdP), and ventricular fibrillation (VF), from which syncope and sudden death often precipitate. LQTS-associated mutations have been identified in six genes: KCNQ1, previously called KVLQT1, on chromosome 11p15.1 (LQT1), KCNH2 or HERG on 7q35–36 (LQT2), SCN5A on 3p21 (LQT3), ankyrin-B on 4q25–27 (LQT4), KCNE1 (LQT5) and KCNE2 (LQT6) on 21q22 [1–4].
SCN5A was the first gene identified for LQTS and encodes a member of the voltage-gated sodium channel gene family that is primarily expressed in the heart [5], but with lower expression in neuronal tissue [6]. In addition to LQTS, mutations in SCN5A are also responsible for Brugada syndrome [7] and cardiac conduction disease (CCD) [8,9]. LQTS patients with SCN5A mutations (LQT3 patients) have a higher risk of death during a cardiac event, despite a lower frequency of cardiac events when compared to LQT1 and LQT2 patients [10]. In LQT3 patients (39%), the initiation of arrhythmias is often bradycardia- or pause-induced (i.e., often occurring during sleep or rest) [11,12]. However, tachycardia-induced fatal arrhythmias have also been reported for 32% of LQT3 patients for which the mechanism is largely unclear [12].
Mutation N1325S in SCN5A, a substitution of an asparagine by a serine at position 1325 in the intracellular region between segments 4 and 5 of domain III of the cardiac sodium channel [1]. In vitro studies of N1325S expressed in either Xenopus oocytes [13] or transfected HEK-293 cells [14] have shown that the mutant channels express sodium currents that are characterized by an initial peak inward transient followed by dispersed reopenings during its inactivation phase, thus producing a late and persistent inward current [15,16]. We and others have also shown that the late current generated by mutant channels can be blocked by the sodium channel blocker, mexiletine [13,14].
Using transgenic technology to characterize the N1325S mutation in a murine model, we set out in the present study to further explore the in vivo mechanisms by which dysfunction of the cardiac sodium channel can lead to arrhythmogenesis in LQTS. Here, we report on the establishment of a new transgenic mouse model whose cardiac features are consistent with the LQTS phenotype. Prolongation of the QT-interval and ventricular action potential duration (APD), and the high mortality rates of our transgenic mice that precede VT/VF are all shown to be attributed to the N1325S SCN5A mutation. Interestingly, APD from transgenic ventricular myocytes were increasingly prolonged with increasing stimulation frequency and, as such, may reveal a non-bradycardia-related arrhythmogenic mechanism.
2. Methods
This study was approved by the Cleveland Clinic Foundation Institutional Review Board on Animal Subjects and conforms to NIH guidelines.
2.1. Generation and genotyping of transgenic mice
A 6.2-kb human SCN5A (hSCN5A) cDNA fragment containing the N1325S mutation was inserted into Clone-26 (a generous gift from J. Gulick and J. Robbins, University of Cincinnati, OH), resulting in transgenic construct pB (mMHC-hSCN5A) [17]. In pB(mMHC-hSCN5A), the hSCN5A gene was under the control of the mouse alpha-MHC (α-mMHC) promoter (5.4 kb) and followed by a human growth hormone polyadenylation signal sequence (hGH plA, 0.6 kb). A 12.4-kb fragment carrying the mMHC-α promoter, the full SCN5A coding region, and the hGH plA signal sequence was excised from pB(mMHC-hSCN5A) by digestion with Not I and injected into fertilized eggs derived from mouse strain CBA/B6. The injected zygotes were then placed back into the female foster for further development. The microinjection of pronuclei was carried out at the Cleveland Clinic Foundation Transgenic/Knockout Core Facility. The genotypes of offspring were determined by PCR and Southern blot analysis using radioactively labeled probe A in Fig. 1 and genomic DNA from tail biopsies. PCR primers were P1 (5-TGTCCGGCGCTGTCCCTGCTG-3) in combination with P2 (5-CTCATGCCCTCAAATCGTGACAGA-3) and P3 (5-GGCACCTGCTGCAACGCTCTTT-3) in combination with P4 (5-GGTGGGCACTGGAGTGGCAACTT-3).
Fig. 1.
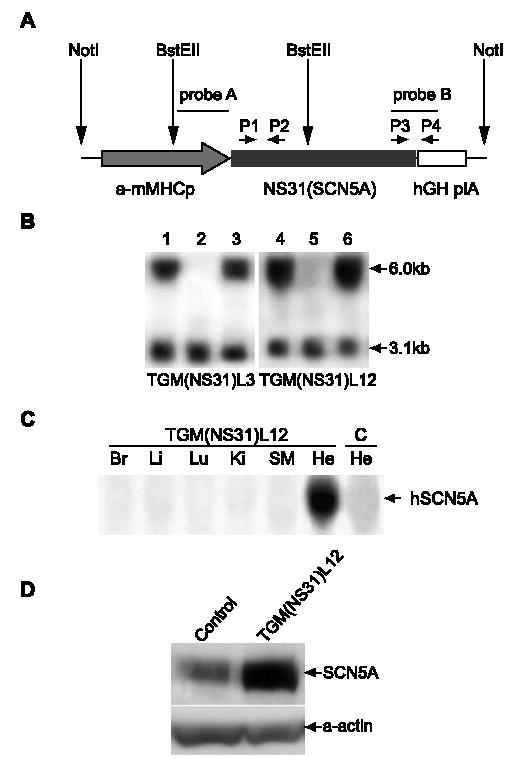
Generation and genotyping of TGM(NS31) mice. (A) Transgenic construct for the engineering of transgene, NS31(SCN5A) (human SCN5A with LQTS-associated mutation, N1325S), into the mouse genome. The Not I fragment consists of the mouse myosin heavy chain-α promoter (α-mMHCp), transgene NS31(SCN5A), and the human growth hormone poly(A) signal (hGH plA). Probe A (1.7 kb), a portion of α-mMHCp, was used as a marker for Southern blot analysis. Probe B, a junction fragment between NS31 and hGH plA, was used for Northern blot analysis to determine the expression profile of the transgene. PCR primer pairs P1–P2 and P3–P4 were used to identify transgenic mice during breeding. (B) Southern blot analysis. Mouse genomic DNA was digested with BstE II and hybridized with probe A [see (A), above]. The 6.0- and 3.1-kb fragments represents, respectively, the transgene, NS31(SCN5A), and the endogenous mouse myosin heavy chain-α gene. Two transgenic lines, TGM(NS31)L3 and TGM(NS31)L12, were generated. The following genotypes are listed: TGM(NS31)L3: Lanes 1 and 3; TGM(NS31)L12: Lanes 4 and 6; and non-transgenic mice: Lanes 2 and 5. (C) Northern blot analysis with probe B [see (A), above]. Tissues from TGM(NS31)L12 mice: Br, brain; Li, liver; Lu, lung; Ki, kidney; SM, skeletal muscle; He, heart. Heart tissue from control littermates (C–He) is also shown for comparison. (D) Western blot analysis was performed using membrane fractions of heart tissue protein extracts from TGM(NS31)L12 and control mice. A polyclonal anti-SCN5A antiserum was raised against a peptide located in the less-conserved N-terminus of SCN5A. The same filter was probed with a monoclonal anti-α-sarcomeric actin antibody (Sigma-Aldrich) in order to calibrate the amount of protein extracts loaded in each lane.
2.2. Northern and Western blot analyses and immunostaining
Northern blot analysis was performed using 20 μg of total RNA isolated from tissues using the TriZol kit (Invitrogen) and radioactively labeled probe B (Fig. 1). Western blot analysis was performed with the membrane fraction of cardiac protein extract and anti-SCN5A antiserum as the primary antibody [6]. The secondary antibody was horse-radish peroxidase-linked donkey anti-rabbit Ig (NA 934, Amersham Pharmacia Biotech). ECL Western blotting detection reagents (Amersham Pharmacia Biotech) were used to visualize the protein signals. The immunostaining of isolated myocytes was adapted from a previous study [6]. In brief, myocytes were fixed in 4% paraformaldehyde and permealized in 0.3% Triton X-100. Subsequently, slides with fixed cells were blocked with 5% BSA and incubated with the primary anti-SCN5A antibody. The secondary antibody conjugated with FITC was used to visualize the SCN5A signal and the nuclei were co-stained with DAPI.
2.3. Telemetry electrocardiogram recordings
The Data Sciences Telemetry system (TA10ETA-F20 transmitter, Data Sciences International) was used to continuously monitor and record ECGs of conscious and unrestrained mice. Mice (5–10 months; 30–35 g) were anesthetized with 0.015 ml/g Avertin (2.5%; Sigma-Aldrich) and individual transmitters were surgically implanted into the peritoneal cavity. ECG leads were implanted subcutaneously in the lead I configuration with negative and positive leads positioned on the right shoulder and chest, respectively. Non-transgenic littermates were used as controls, and all implanted mice were allowed to recover for 1 week after surgeries prior to recording. ECGs were continuously recorded and saved onto ORB disks for later analysis with the Data Sciences DATA Acquisition ECG software. Signals below 248 MHz were filtered out. The QT interval was corrected for heart rate (QTc) using Bazett’s formula [18], QTc = QT/(RR)½, or the formula proposed by Mitchell et al. [19], QTc = QT/(RR/100)½.
2.4. Ventricular myocyte isolation
Cardiac myocytes from age-matched (5–10 months) adult wild-type and transgenic mice were dispersed enzymatically. Mice were injected intraperitoneally with 0.15 ml heparin (1000 U/ml) and anesthetized with 0.65 ml/kg, i.p., Nembutal (50 mg/ml). Hearts were excised and mounted on a Langendorff apparatus for retrograde perfusion with a calcium-containing buffer solution (Solution I) of the following composition (in mM): 118, NaCl; 4.8, KCl; 2, CaCl2; 2.5 MgCl2; 1.2 KH2PO4; 11, glucose; 13.8, NaHCO3; 4.9, pyruvic acid; pH 7.2–7.4 with 95% O2:5% CO2; 37 °C. Perfusion was switched to a calcium-free Solution I with 0.03 mM EGTA (Solution II) into which 0.15 mg/ml bovine serum albumin (BSA, Sigma-Aldrich), 20 U protease (Type XXIV, Sigma-Aldrich), and 0.5 mg/ml collagenase (Type II, Worthington) was added (Solution III). Solution III was then recirculated for approximately 28 min, during which the calcium concentration was increased in a stepwise manner (with CaCl2) to a final 1.8 mM concentration. Ventricular tissue was removed, placed in normal Tyrode’s buffer (1.8 mM CaCl2), and incubated at 37 °C for 10 min. Mechanical dispersion and gentle tituration were performed and the resultant cell suspension was filtered (200-μm nylon mesh, Spectrum Laboratories) to remove undissociated tissue fragments. Final cells were collected by gravity sedimentation and resuspended in recording bath solution (see below).
2.5. Electrophysiological recordings
Only rod-shaped, quiescent cells with smooth striations were selected. Cell rupture and whole-cell configuration was achieved with fire-polished and Sylgard-coated tipped glass pipettes (Corning G86165T-4, WPI) with access resistance of 2–3 MΩ when filled with pipette solution. Using the Axopatch 1C (Axon Instruments), series resistance was 4–8 MΩ and was 30–80% electronically compensated. Action potentials (APs) were evoked by repetitive square pulses of 3 ms duration at 1.5 × stimulus threshold. Experiments were performed under constant flow conditions at 35 °C using a temperature-controlled experimental chamber (Delta T Culture Dish, Bioptechs). Myocytes were paced under current clamp conditions at cycle lengths (CLs) between 150 and 1000 ms. Runs of 12 steady-state APs at each CL were recorded with filtering at 2 kHz and sampled at 10 kHz. Data were acquired using a Pentium computer that controlled data acquisition hardware and software (pClamp 6.03+, Axon Instruments). Analysis of APs was performed by taking the average of each sweep of 12 stable and steady-state records. The average trace is illustrated in all the figures unless otherwise stated. Action potential parameters such as 90% repolarization (APD90) are summarized and listed in Tables 1 and 2.
Table 1.
Action potential parameters—effects of cycle length
| Cycle length (ms)
|
|||||
|---|---|---|---|---|---|
| 1000 | 500 | 300 | 250 | 200 | |
| Control | |||||
| APD90 (ms) | 46.7±4.8 | 41.1±5.5 | 42.1±6.6 | 45.9±2.9 | |
| Em (mV) | −78.3±1.4 | −77.6±1.4 | −75.5±2.4 | −74.6±4.3 | |
| OS (mV) | 39.8±2.6 | 38.4±3.3 | 40.9±2.6 | 35.1±4.0 | |
| n | 8 | 4 | 3 | 3 | |
| TGM(NS31)L3 | |||||
| APD90 (ms) | 57±3.8 | 60±4.1 | 63±2.7* | 66±4.6* | |
| Em (mV) | −78±2.8 | −78±3.2 | −80±3.3 | −77±4.3 | |
| OS (mV) | 44±3.0 | 43±4.1 | 44±2.9 | 40±5.6 | |
| n | 5 | 5 | 5 | 5 | |
| TGM(NS31)L12 | |||||
| APD90 (ms) | 60±2.5 | 69.4±5.9* | 93.8±12.7* | ||
| Em (mV) | −76.6±3.2 | −84.5±5.4 | −84.4±5.6 | ||
| OS (mV) | 45.1±2.1 | 42.6±6.1 | 42.6±5.8 | ||
| n | 3 | 8 | 8 | ||
Resting membrane potential (Em), peak amplitude overshoot (OS), and 90% AP repolarization (APD90) are listed as AP parameters for control and transgenic ventricular myocytes under the indicated CLs. For all cells, a basal CL of 500 ms was established. For transgenic cells, values for 1000 ms CL were recorded. but for 250 and 200 ms (L3 only) CLs, values were not obtained due to their failure to maintain pacing at these CLs. Values are mean ± S.E.M. with statistical significance.
From control in the same CL group using a paired-sample t-test with P < 0.05.
Table 2.
Action potential parameters—effects of mexiletine
| (−) 10 μM Mexiletine
|
(+) 10 μM Mexiletine
|
|||||
|---|---|---|---|---|---|---|
| Em (mV) | OS (mV) | APD90 (ms) | Em (mV) | OS (mV) | APD90 (ms) | |
| Control (n=5) | −78.7±2.3 | 29.9±4.9 | 50.3±3.6 | −79.5±2.9 | 32.7±5.0 | 48.2±4.3 |
| TGM(NS31)L3 (n =5) | −77±2.9 | 40±4.1 | 60±4.9 | −78±1.4 | 39±3.3 | 55±2.9 |
| TGM(NS31)L12 (n=6) | −79.3±3.7 | 34.3±5.2 | 74.3±9.0* | −79.5±3.6 | 31.9±5.6 | 54.8±6.0 |
AP parameters are described as in Table 1 for control and transgenic ventricular myocytes in the absence and presence of 10 μM mexiletine at a pacing CL of 500 ms. Values are mean ± S.E.M for n sizes shown in parentheses.
Denotes statistical significance from control using a paired-sample t-test with P < 0.05.
Sodium currents were recorded using the ruptured, whole-cell voltage clamp configuration. For adequate voltage control, currents were recorded in low (30 mM) sodium extracellular solutions and at room temperature (20–22 °C). Before going whole-cell, capacitative transients were nulled by analog compensation. Series resistance ranged from 3 to 4 MΩ and was electronically compensated at 75–80%. Recordings were made 5 min after membrane rupture using a Pentium computer that controlled data acquisition hardware (MC 700A; Axon Instruments) and software (pClamp 9.0, Axon Instruments). Membrane currents were low-pass filtered at 10 kHz and digitized at 100 kHz, and linear leakage was subtracted online using P/2 subtraction routine. Ionic current density (pA/pF) was calculated from the ratio of current amplitude to cell capacitance. Cell capacitance was measured by integrating the area under the capacitive transient induced by a 10-mV hyperpolarizing clamp step (from −80 to −90 mV) and dividing the area by the voltage step. Persistent inward currents were measured using slow ramp stimuli ( −100 to + 50 mV at 50 mV/s from a holding potential of −100 mV) that inactivated the transient sodium current component [13]. The current–voltage relationship of the TTX-sensitive component of the ramp current was obtained by digital subtraction of records obtained before and after tetrodotoxin (TTX, 20 μmol/l) application.
2.6. Recording solutions
For AP recordings, composition of external solution was (in mM): 140, NaCl; 4, KCl; 2, CaCl2; 2, MgCl2; 10, HEPES; 10, glucose; pH 7.3 with 5N NaOH. Internal solution was (in mM): 135, KCl; 1, MgCl2; 10, EGTA; 10, HEPES; 5, glucose; pH 7.2 with 1N KOH. For sodium current measurements, external solution was (in mM): 30, NaCl; 110, CsCl; 1.8, CaCl2; 2, CdCl2; 1, MgCl2; 10, HEPES; 10, glucose; 1, 4-AP; pH 7.3 with 1N CsOH. Patch pipettes (0.9–1.4 MÙ resistance) were filled with internal solution (in mM): 10, NaCl; 130, CsCl; 5, EGTA; 10, HEPES; 10, glucose; pH 7.3.
2.7. Statistical analysis
All data are shown as mean ± S.E.M., and the Student’s t-test was performed to evaluate single-factor differences between transgenic and wild-type mice. In the experiment, to test the effect of mexiletine on VT and VF, a paired t-test was used to evaluate the difference before and after treatment in the same group. A value of P < 0.05 was considered statistically significant.
3. Results
3.1. Generation of transgenic mice with mutant human SCN5A gene (N1325S)
We generated transgenic mice with cardiac selective expression of N1325S on SCN5A under the control of the mouse cardiac-specific myosin heavy chain-α promoter (Fig. 1A). The transgenic mice were identified by PCR (data not shown) and Southern blot analyses (Fig. 1B). Two transgenic lines, TGM(NS31)L3 and L12, were established. The copy number of the transgene in TGM(NS31) was estimated by comparing the density of the band from transgenic MHC to that of endogenous MHC using Southern blot hybridization. TGM(NS31)L12 was found to carry a higher copy number of the transgene than TGM(NS31)L3 (10 vs. 1–2 copies; Fig. 1B).
Northern blot analysis (Fig. 1C) showed that the mutant human SCN5A gene was exclusively expressed at the mRNA transcript level in cardiac tissues. This was also confirmed at the protein level by Western blot analysis (Fig. 1D) using an antibody that can recognize both human and mouse SCN5A. The total amount of SCN5A protein was approximately threefold higher in TGM(NS31)L12 compared with control.
3.2. Development of LQTS, polymorphic VT and VF in transgenic mice
Continuous recording of ECGs of conscious and unrestrained mice was performed using the telemetry monitoring system. Representative traces of control and TGM (NS31)L12 mice are shown (Fig. 2A and B), and several parameters of the ECGs are summarized (Fig. 2C–F). Using the Bazett’s formula, the QT intervals corrected for heart rate were 134.4 ± 0.7 and 70.6 ± 1 ms, respectively, for TGM(NS31)L12 vs. their control littermates (Fig. 2D). Moderate prolongation of QTc was observed in TGM (NS31)L3 (89.6 ± 1.1 ms). Using Mitchell’s formula, we also found a similar increase of QTc of 91% in TGM (NS31)L12 and 23% in TGM(NS31)L3 compared with their littermate controls. Interestingly, both PR and RR intervals were shortened in transgenic mice (Fig. 2C and F), whereas QRS interval was not significantly altered (Fig. 2E).
Fig. 2.
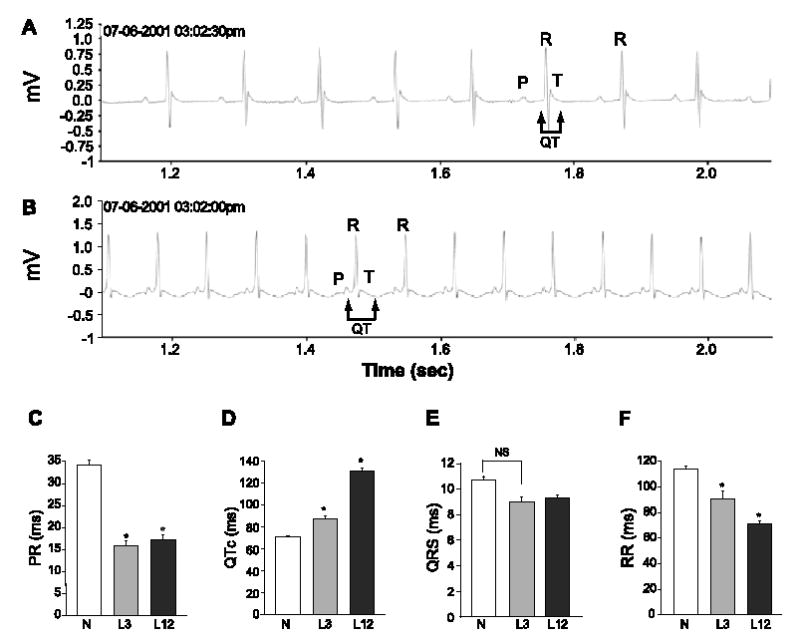
ECG recordings of non-transgenic and TGM(NS31) mice. Representative sample ECG traces from non-transgenic (A) and TGM(NS31)L12 (B) mice in 1-s duration. The P, R, T waves and the QT-interval are indicated. Differences between control (C) and transgenic (L3 and L12) mice are summarized in graphs (C–F) and QT-intervals were corrected for heart rates (QTc). Statistical significance from control ( P < 0.05) is denoted with an asterisk (*), whereas NS indicates nonsignificance.
Continuous telemetric recordings revealed a high incidence of spontaneous cardiac arrhythmias in transgenic vs. WT mice. Concurrent with these recordings was a high mortality rate amongst the transgenic mice. The mortality rate was 2:53 (3.8%) in non-transgenic mice, compared to 52:156 (33.3%) in TGM(NS31)L12 mice ( P < 0.001 compared to non-transgenic mice), and 2:43 (4.7%) in TGM(NS31)L3 mice ( P = 0.61 compared to non-transgenic mice, P < 0.001 compared to TGM(NS31)L12 mice). The majority of deaths in TGM(NS31)L12 were preceded with ECG recordings that degenerated from normal and synchronous signals to ones in which there was VF or an asynchronous signal of low amplitude that was irreversible with a duration of 5 min or greater. We defined this event as sudden cardiac death. No polymorphic VT/VF was observed in WT mice excluding the possibility that the mortalities of those mice indicated were due to cardiac arrhythmias.
Representative telemetric ECG recordings from TGM (NS31)L12 mice exhibiting spontaneous VT/VF are shown in Fig. 3A–D. Fig. 3A (top trace) is a continuous trace from one mouse showing a series of four short coupled beats (horizontal bar) followed by sudden development of VT. The monomorphic VT degenerated to polymorphic VT or TdP (middle trace) and ultimately into irreversible VF (bottom trace). Evidence for bradycardia or sinus pauses prior to the onset of cardiac arrhythmias was not observed in the telemetric recordings from TGM(NS31)L12 mice. Instead, upon closer examination of selected ECG recordings prior to the start of fatal arrhythmias, we observed premature contractions (Fig. 3B), spontaneous degeneration of normal sinus into reversible VT or VF (Fig. 3C), and possible ventricular parasystoles of varying coupling intervals[20] prior to the initiation of VT/VF (Fig. 3D; bottom trace).
Fig. 3.
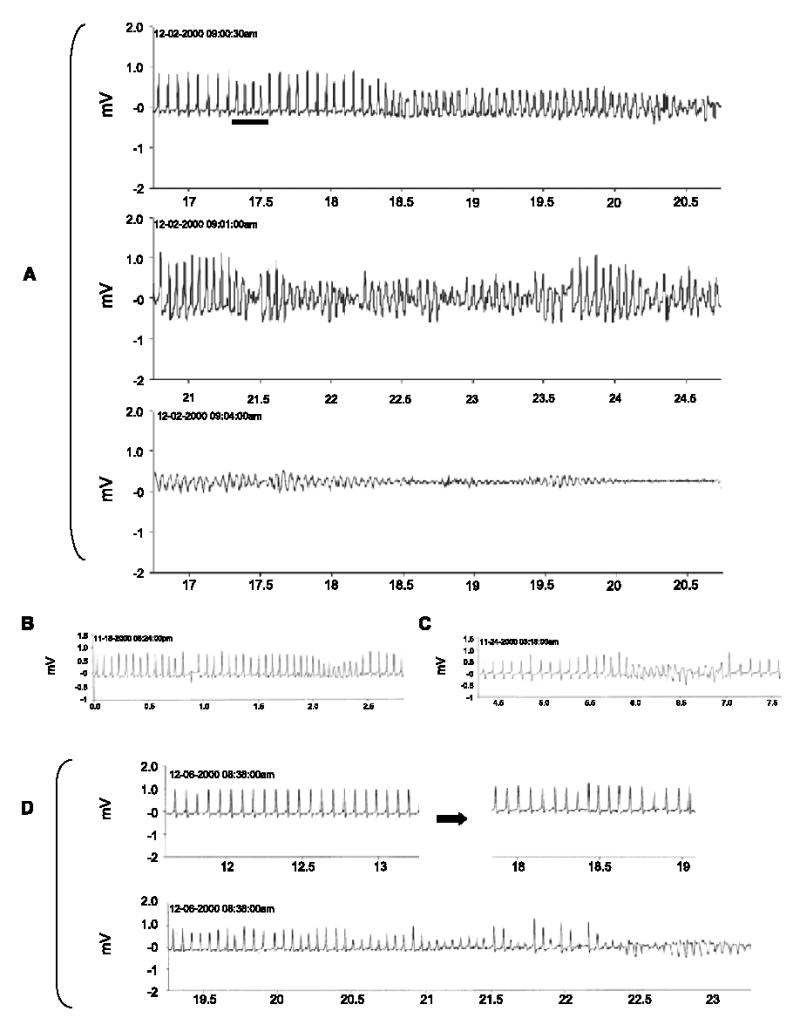
Telemetric ECG recordings illustrating the development of spontaneous cardiac arrhythmias in conscious TGM(NS31)L12 mice. Traces are represented as time frame durations in seconds with ECG signals in mV. (A) Sample traces illustrating normal sinus rhythm followed by four coupled beats (top trace; horizontal bar) and spontaneous development of monomorphic VT that degenerates into polymorphic VT (top and middle traces) and irreversible VF (bottom trace) that resulted in sudden death. (B–D) Sample traces from other TGM(NS31)L12 mice that illustrate premature contraction (B), possible ventricular parasystoles of varying coupling intervals (D; top right), and spontaneous development of reversible VT/VF (B–D) that typically preceded fatal cardiac events.
3.3. Suppression of VT and VF by mexiletine
Two daily treatments of 25 mg/kg, i.p., mexiletine were used to evaluate its effect on arrhythmias in telemetrically recorded TGM(NS31)L12 mice. Representative graphs are plotted as the incidence of cardiac events for each hour over a 24-h period, and the events are accumulated for the entire 30-day (Fig. 4A) and 14-day (Fig. 4B) studies. Cardiac events or arrhythmias were defined as premature contractions and mono- or polymorphic VT and VF that appeared as clusters (>0.5 s/run). As shown in Fig. 4B, the incidence of VT/VF was well suppressed by mexiletine, up to 6 h after its injection in the 14-day study. The half-life of mexiletine in rodents is approximately 2 h [21]. It was interesting to see a transient suppression of cardiac arrhythmias during the period corresponding to the administration of this sodium channel blocker, which would suggest that the mutant transgene, N1325S, is involved in the observed spontaneous developments of cardiac arrhythmias in TGM(NS31) mice.
Fig. 4.
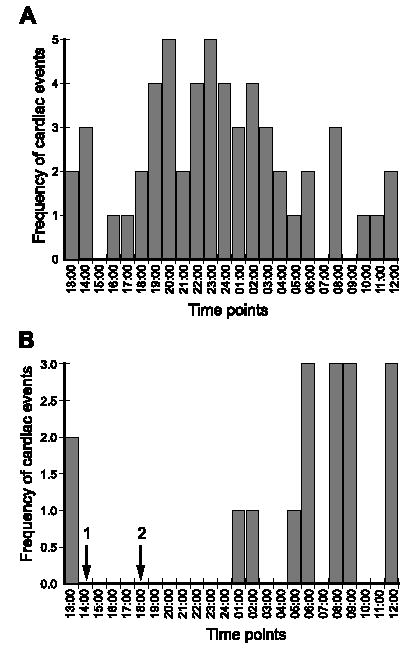
Antiarrhythmic effects of mexiletine in TGM(NS31)L12 mice. The incidence of cardiac events ( y-axis) is plotted for each hour over a 24-h period. The data are a culmination of all events for 30 (A) and 14 (B) consecutive days, the latter of which received mexiletine administrations (25 mg/kg, i.p.) at 1400 and 1800 h (arrows 1 and 2, respectively). Cardiac arrhythmias were fully suppressed immediately following and up to 6 h posttreatment with mexiletine. Since no lethal arrhythmias were detected in TGM(NS31)L3, mexiletine treatment studies were not performed.
3.4. Prolonged APD and late persistent sodium current in transgenic ventricular myocytes
Ventricular APs in isolated TGM(NS31) myocytes were analyzed to investigate the possible mechanisms of arrhythmogenesis observed in vivo. Representative ventricular APs (Fig. 5) were recorded in the ruptured whole-cell configuration at 35 °C and CLs of 150–1000 ms. Comparison of AP waveforms revealed that APD90 was increased in TGM(NS31)L12 and TGM(NS31)L3 vs. their control littermates (see also Table 1). Resting membrane potential and AP amplitude were not statistically different between mice groups (Table 1). At CL less than 300 ms, transgenic myocytes were unable to maintain pacing (Fig. 5B and C) and, during the course of recording, we regularly observed the development of EADs (Fig. 6A–C). Similarly, TGM (NS31)L3 myocytes initially followed pacing CLs of 200 ms but often failed to maintain capture and depolarized to more positive potentials (Fig. 5B). These were not observed in myocytes isolated from control littermates. In the presence of 10 μM mexiletine, APD90 from transgenic myocytes paced at 500 ms was decreased from pretreatment (Fig. 7A). Under the same conditions, APD90 in control myocytes was unaffected by mexiletine application. These results are also summarized in Table 2. Interestingly, the failure of transgenic myocytes to maintain pacing at stimulation rates less than 300 ms CL was restored following mexiletine application (Fig. 7B and C, respectively).
Fig. 5.
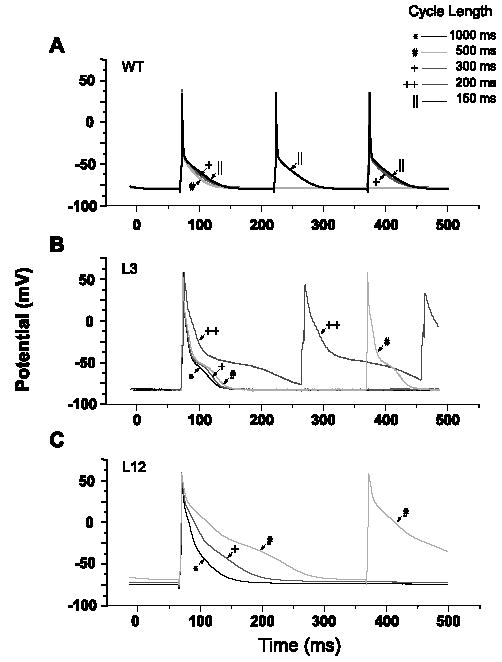
AP recordings from wild-type (WT) and transgenic (L3 and L12) ventricular myocytes at decreasing CLs. Repolarization phase for transgenic myocytes was longer than for control myocytes at all CLs; AP parameters are summarized in Table 1. The increases in the rate-dependent prolongation of APD were longer for myocytes from L12 than L3 mice. At CLs less than 300 ms, APs from L12 myocytes were unable to maintain capture and only those at CLs of 1000, 500, and 300 ms are shown. In contrast, APs from L3 mice initially maintained capture at CLs of 200 ms, but with successive sweeps, depolarized to more positive potentials and ultimately failed to uphold this stimulation frequency. Each AP trace in the figure is an average of 12 steady-state APs recorded at the specified CL; for L3, the AP recorded at a CL of 200 ms is the ninth out of 12 sweeps in the train.
Fig. 6.
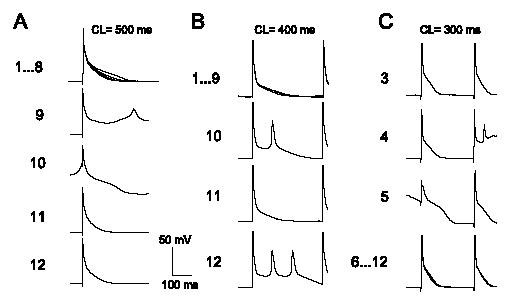
Representative trains of AP traces from TGM(NS31)L12 ventricular myocytes following pacing at 500 (A), 400 (B), and 300 ms (C) CLs with abnormal repolarization are shown. A total of 12 steady-state sweeps is associated with each train. All sweeps are superimposed, and only those traces that illustrate the sequence in the development and termination of EADs are indicated as single sweeps.
Fig. 7.
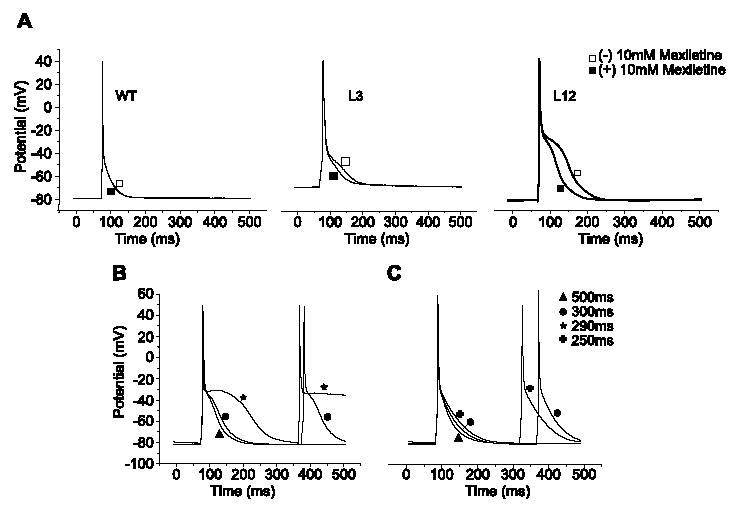
(A) APs were recorded at 500 ms CL from WT, L3, and L12 ventricular myocytes in the presence (□) and absence (▪) of 10 μM mexiletine. The sodium channel blocker shortened APD in transgenic cells with no effects in control cells. These experiments were repeated in n = 5–6 cells for each group, and the results are summarized in Table 2. (B) Failure to maintain pacing at 290 ms CL was recorded in a representative TGM(NS31)L12 myocyte in the absence of mexiletine. (C) Following 10 μM application of mexiletine, there was an improvement in repolarization and a recovery to maintain pacing up to 250 ms CL.
To determine whether a persistent non-inactivating component of INa was responsible for the differences in APDs, voltage-clamp experiments of ventricular INa were conducted in normal and transgenic myocytes (Fig. 8A). Ventricular cells were depolarized to −20 mV from a holding potential of −100 mV with a 50-ms step. INa from wild-type (WT) mice showed the characteristic pattern of an initial fast inward peak followed by a fast decaying component (inactivation) to baseline within the first 5–10 ms of the step. Currents from L3 and L12 transgenic cells showed a similar initial fast inward peak but with a subsequent slow and inactivating component well beyond 5 ms. Following the application of 10 μM mexiletine in transgenic myocytes, an increase in the rate of recovery to baseline of the slow component with no effect on peak INa amplitude was observed. Neither the peak amplitude nor the inactivating phase of INa was affected by mexiletine in non-transgenic myocytes. Our results did not show any significant differences in the maximum current densities between transgenic L12 (16.6 ± 1.1 pA/pF, n = 8), L3 (15.5 ± 2.3 pA/pF), and non-transgenic cells (15.3 ± 1.0 pA/pF, n = 6). For transgenic and non-transgenic myocytes, the average current amplitudes were 1943.8 ± 92.2 pA (L12) and 1764 ± 114.6 pA (L3) vs. 1705.0 ± 85.3 pA (WT) and for cell capacitances, 118.6 ± 4.6 pF (L12) and 114 ± 5.4 pF (L3) vs. 112.7 ± 4.8 pF (WT). Consistent with the comparison of current densities, a similar surface of sodium channel densities in both transgenic and normal cells is illustrated by the immunostaining of ventricular cells from either group (Fig. 7C).
Fig. 8.
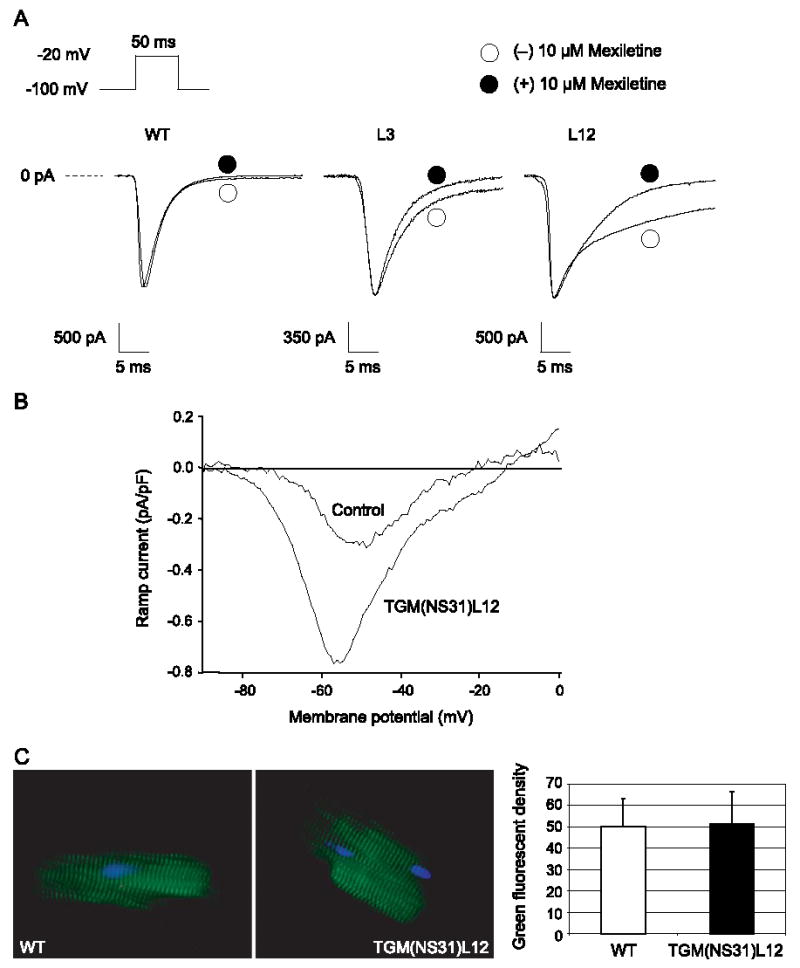
Measurments of INa. (A) Representative whole-cell INa from WT, L3, and L12 ventricular myocytes. INa was recorded with the pulse protocol shown in the inset before (○) and after (•) 10 μM mexiletine. (B) Current–voltage relationship of the late persistent sodium current from control and TGM(NS31)L12 myocytes (n = 5–6) represents the late sodium current density as the ratio of current amplitude to cell capacitance (pA/pF) over the ramp voltage range (−100 to + 50 mV). (C) Immunofluorescent staining of SCN5A (green) of isolated TGM(NS31)L12 and wild-type ventricular myocytes are shown. The nuclei were counterstained with DAPI (blue).
Additional experiments were conducted to specifically investigate the late persistent current. Late inward currents were evoked by voltage ramps that slowly depolarized the cell membrane from −100 to + 50 mV (50 mV/s). A slow depolarization inactivates the transient component of the total current leaving only the non-inactivating component. Ramp stimuli were applied before and after the addition of 20 μM TTX to sufficiently block the fast inward sodium transient. The current–voltage relationship of the TTX-sensitive component of the ramp current was obtained by digital subtraction. As shown in Fig. 8B, average currents from pooled data (5–6 TGM(NS31)L12 myocytes in each average) peaked between −60 and −40 mV. Maximum late persistent current was significantly greater in transgenic (0.81 ± 0.16 pA/pF) than in non-transgenic myocytes (0.34 ± 0.06 pA/pF).
The data on INa suggest that the increased late persistent sodium current is present and can account for the effects on AP prolongation in transgenic myocytes. The consequences for abnormal repolarization at short diastolic intervals may form the arrhythmogenic mechanisms responsible for the observed ventricular arrhythmias in this mouse model.
4. Discussion
The main clinical features of LQTS typically include (i) prolongation of the QTc on the ECG, (ii) development of VT and VF, and (iii) syncope and sudden cardiac death. In this study, we have established a new transgenic mouse model for LQTS by expressing mutation N1325S in SCN5A in the mouse genome. Our TGM(NS31) mice showed prolongation of the QTc and high incidences of spontaneous VT and VF followed by sudden cardiac death. Therefore, we conclude that these mice can serve as an animal model for dissecting the molecular mechanisms that underlie the pathogenesis of LQTS associated with SCN5A mutations.
In vitro electrophysiological studies on N1325S and other LQT3 mutations in HEK293 cells or Xenopus oocytes have reported that mutant sodium channels exhibit a peak inward transient followed by a late persistent current, the latter of which is attributed to late reopenings of the sodium channel [13–16]. Mexiletine has been shown to be a selective blocker of these delayed reopenings at concentrations that do not affect the peak sodium current [13,14]. In this study, similar findings were found for mutation N1325S when it was expressed in cardiac cells. The results from ventricular myocytes isolated from transgenic mice showed a higher current density of the late persistent sodium current than in control myocytes and that this was blocked by mexiletine (Fig. 8). Action potential recordings in TGM(NS31) vs. control myocytes further revealed that ventricular APD was prolonged, which may reflect the prolonged QTc detected on ECGs. According to our in vivo studies, we observed a reduction in arrhythmia incidence in TGM(NS31) mice along with a shortening of ventricular APD after mexiletine administration which would suggest that the LQTS phenotype identified in these transgenic mice is modulated by the late persistent sodium current generated by the N1325S transgene.
Conditions whereby cardiac arrhythmias result from include (1) disturbance in impulse conduction (e.g., reentry), (2) disturbance in impulse formation, (3) or both. Triggered activity, as one form of abnormal impulse formation, can arise from EADs or DADs and EAD-induced triggered activity has been increasingly recognized as the substrate for the pathogenesis of arrhythmias in LQTS [22–24]. A recent study involving ΔKPQ knock-in mice has attempted to elucidate the triggering mechanisms that lead to the development of EADs associated with a gain-of-function sodium channel mutation [25]. Myocytes from mice with the ΔKPQ deletion in SCN5A gene were shown to develop EADs at prolonged stimulation rates (>1 Hz.) Bradycardia and/or sinus pauses are recognized as arrhythmogenic triggers (mechanisms or events that lead to development of arrhythmias) in LQT3 patients [12]. However, this is but a single “instigator” out of other arrhythmogenic triggers that has been documented for LQT3 patients. Based on genotype–phenotype correlation studies by Schwartz et al. [12], the existence of a heterogenous group of LQT3 patients was summarized for which the arrhythmogenic trigger is multifaceted: triggers include bradycardia (during sleep or rest, 39%), non-bradycardia (exercise and emotion, 32%), and unknown triggers (29%).
Unlike the report on ΔKPQ mice [25] (n = 2:36; 5%), our transgenic mice exhibited a higher incidence of fatal cardiac events (n = 52:156; 33%). These were characterized as frequent and spontaneous development of lethal arrhythmias (VT/VF) from which many were telemetrically recorded as preceding the degeneration of the ECG and, likely, the indication of sudden cardiac death. Moreover, the arrhythmias occurred while our mice were conscious, unrestrained, and without any invasive electrical pacing—conditions which we suggest approximate physiological parameters for a given in vivo arrhythmia model. It is unclear why TGM(NS31)L12 mice displays a more severe phenotype than ΔKPQ mice [25], namely, spontaneous and lethal VF. This aspect was somehow “masked” in ΔKPQ mice. Notably, we found equivalent densities of SCN5A protein on the cell surface (Fig. 8C) between the control and transgenic groups despite a higher expression in the latter group. It is likely that tight regulations exists in the number of the channels for a given myocyte surface. The severity of the phenotype may reflect overexpression of the SCN5A mutant relative to WT SCN5A, a proportionality that favors the former, while maintaining a “constant” population of SCN5A proteins for a given cell. Presumably, the proportionality of mutant vs. WT SCN5A channels on the myocytes surface is higher in TGM(NS31)L12 than in ΔKPQ mice, thereby resulting in a more severe phenotype. The same reason may also explain why the phenotype in TGM(NS31)L12 is more severe than TGM(NS31)L3.
Interestingly, no evidence of a bradycardic pause prior to the onset of cardiac arrhythmias was detected in our transgenic mice. As in the clinical form, multiple arrhythmogenic triggers may be associated with the LQT3 phenotype. As such, our mouse model for LQTS may make it practical to study the development of EADs and fatal arrhythmias that are not preceded by pauses or bradycardia. In an attempt to offer an explanation for adrenergic- or exercised-induced arrhythmias in LQT3 patients, the report on ΔKPQ mice [25] also showed that abrupt accelerations in rate resulted in a paradoxical (albeit, transient) lengthening of the AP, which favored the development of EADs. This may serve as a partial explanation for LQT3 patients with abrupt emotions or physical exertion, but it may not sufficiently apply as the trigger in cases where the rate is progressively increased and maintained.
Unexpectedly, expression of N1325S mutation in our transgenic mice resulted in abnormally high resting heart rates, and the reason for this is unclear. Additionally, APD in TGM(NS31)L3 and L12 ventricular myocytes was augmented as the diastolic interval was decreased (Fig. 5). This positive rate-dependent prolongation of ventricular APD could potentially increase the risk and vulnerability for induction of premature contractions and cardiac arrhythmias, and it is a likely “trigger” for arrhythmogenesis in TGM(NS31) mice. Evidence for this increased vulnerability was shown in isolated ventricular myocytes with EAD development (Fig. 6A–C) and a failure to maintain pacing when CL was reduced below 300 ms (Fig. 7B). It should be noted that when CL was increased to 1000 ms (Fig. 5; Table 1), the stability of transgenic AP waveforms was maintained and no development of premature afterdepolarizations was observed. In addition, CLs less than 200 ms (or 500–600 beats/min) are a close approximation to the physiological heart rates of mice. Interestingly, telemetric ECG data on arrhythmia incidence of TGM(NS31)L12 mice was most prevalent between 1700 and 900 h (Fig. 4). During the life cycle of rodents, this is often when they are most active, and this may explain the high frequency of cardiac events during this time. The observed rate-dependent prolongation of ventricular APD is intriguing from the perspective of a non-bradycardic mechanism, but the basis for this requires further investigation.
Based on our results, the coupling of APD prolongation and development of EADs with increasing stimulation rate does suggest that poor restitution of APD during shorter CLs may be a trigger for arrhythmogenesis in our transgenic mice. We also do not discount the role of increased dispersion of repolarization as an arrhythmogenic mechanism since mexiletine was shown to aid in the recovery of repolarization and possibly reduce dispersion following higher stimulation rates.
A balance of inward and outward currents typically orchestrates the generation of a cardiac action potential. In the case of the late persistent inward sodium current, that balance is shifted in favor of inward currents and, perhaps, influences the recovery from inactivation and reactivation of other inward currents (e.g., ICa,L, INa – Ca). Coupled with a decreasing diastolic interval, there would be an abnormal handling of net increases in inward currents in TGM(NS31) ventricular myocytes that would lead to higher incidences of abnormal repolarizations. According to Nagatomo et al. [26], the late persistent sodium current generated by the ΔKPQ mutation decreases with increasing stimulation rates because of accumulation of inactivation of the late current. Whether this holds true for the N1325S mutation remains to be determined. The rate-dependent prolongation of APD observed in this study does imply that inward currents exceed outward currents during repolarization. The question is whether there exists an abnormal handling and/or signaling of intracellular ions, namely, calcium, that is secondary to the late sodium current, and whether this abnormality is accentuated with increasing depolarization rates. This is a likely possibility since intracellular calcium and its involvement in arrhythmogenesis is well documented.
It is also likely that influences associated from the expression of the N1325S mutation may affect slow inward currents (e.g., ICa,L) that are critical in the control and/or maintenance of the sinoatrial conduction system, and that this would be reminiscent of the abnormally high resting heart rates that was present in our transgenic mice. Alternatively, this gain-of-function mutation in our transgenic mice may have modified the cardiac conduction system, i.e., enhanced conduction, since impulse propagation in the heart depends on the magnitude of the depolarizing current (usually INa) for which INa current densities in the conduction system are often larger than in any other cardiac tissue. This would be reminiscent of the abnormally high resting heart rates and shorter PR intervals that were present in our transgenic mice. Notably, cardiac conduction abnormalities (e.g., Brugada syndrome [7] and CCD [8,9]) have been linked to genetic defects on SCN5A.
In conclusion, our findings on the N1325S mutation offer a new mouse model for the study of LQT3. Genotype–phenotype observations in the LQTS clinical population suggest that the mechanisms that initiate arrhythmias are gene specific and that life-threatening arrhythmias occur under specific circumstances for a given genotype [15]. A closer inspection into these observations reveals that the specificity of an arrhythmogenic trigger for a given LQT genotype is not absolute, i.e., for a given LQT genotype, there may be different triggering events. The triggering event in TGM (NS31) mice was found to be non-bradycardic while still retaining features identifiable as LQT3. Based on these findings, it is intriguing to consider that functional expression of gain-of-function sodium channel mutations may give rise to LQT3 phenotypes with unlikely arrhythmogenic triggers.
5. Limitations
There is a continuing debate as to the relevance of mouse models to elucidate human arrhythmic disorders and caution should be considered when extrapolating these results to the human form of LQTS. Although L3 has one to two copies of the transgene and is more comparable to the human genotype in LQTS patients, this line showed mild phenotypic changes (i.e., long QTc with a mortality rate that is comparable to control). Despite some similarities in phenotype to clinical LQT3, TGM(NS31)L12 has disadvantages for a direct relationship to LQT3 because of the high copy number of the transgene. Nevertheless, the TGM(NS31)L12 model may be important to investigate some of the mechanisms underlying the arrhythmias, and caution should be considered in its relationship to LQT3.
Acknowledgments
The authors thank M. Lamorgese and L. Castel, and the CCF Lerner Research Institute Transgenic/Knockout Core Facility for their technical assistance, D. Kikta, Y. Cheng, D. Janigro, and Y. Zhang for their discussion and comments on the manuscript. QW wishes to thank Dr. J. W. Pollard and his colleagues for their help and discussion during the 5th Annual Mouse Developmental Genetics Course at Albert Einstein College of Medicine. XLT was supported by an AHA-Ohio-Affiliate Postdoctoral Fellowship. This work was supported by the Cleveland Clinic Foundation Seed Grant (QW) and an NIH Grant R01 66251 (QW).
References
- 1.Wang Q, Shen J, Li Z, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603–7. doi: 10.1093/hmg/4.9.1603. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–11. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q, Pyeritz RE, Seidman CE, et al. Genetic studies of myocardial and vascular disease. In: Topol EJ, editor. Textbook of cardiovascular medicine. 2nd ed. Philadelphia: Lippincott William & Wilkins; 2002. pp. 1967–90. [Google Scholar]
- 4.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–9. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 5.Gellens ME, George ALJ, Chen LQ, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89:554–8. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu L, Nishiyama K, Hollyfield JG, et al. Localization of Nav1.5 sodium channel protein in the mouse brain. NeuroReport. 2002;13:2547–51. doi: 10.1097/01.wnr.0000052322.62862.a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 8.Schott JJ, Alshinawi C, Kyndt F, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20–1. doi: 10.1038/12618. [DOI] [PubMed] [Google Scholar]
- 9.Tan HL, Bink-Boelkens MT, Bezzina CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–7. doi: 10.1038/35059090. [DOI] [PubMed] [Google Scholar]
- 10.Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long-QT syndrome. International long-QT syndrome registry research group. N Engl J Med. 1998;339:960–5. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–6. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 13.Dumaine R, Wang Q, Keating MT, et al. Multiple mechanisms of Na+ channel-linked long-QT syndrome. Circ Res. 1996;78:916–24. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 14.Wang DW, Yazawa K, Makita N, et al. Pharmacological targeting of long QT mutant sodium channels. J Clin Invest. 1997;99:1714–20. doi: 10.1172/JCI119335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett PB, Yazawa K, Makita N, et al. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–5. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 16.Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999;400:566–9. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]
- 17.Gulick J, Subramaniam A, Neumann J, et al. Isolation and characterization of the mouse cardiac myosin heavy chain genes. J Biol Chem. 1991;266:9180–5. [PubMed] [Google Scholar]
- 18.Bazett HC. An analysis of the time relations of electrocardiograms. Heart. 1920;7:353–70. [Google Scholar]
- 19.Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 1998;274:H747–51. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- 20.Castellanos AS, Moleiro N, Myerburg RJ. Parasystole. In: Zipes DaJ J, editor. Cardiac electrophysiology: from cell to bedside. 4th ed. Philadelphia: Saunders; 2000. pp. 690–5. [Google Scholar]
- 21.Igwemezie LN, Beatch GN, McErlane KM, et al. Mexiletine’s anti-fibrillatory actions are limited by the occurrence of convulsions in conscious animals. Eur J Pharmacol. 1992;210:271–7. doi: 10.1016/0014-2999(92)90415-z. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu W, Ohe T, Kurita T, et al. Epinephrine-induced ventricular premature complexes due to early afterdepolarizations and effects of verapamil and propranolol in a patient with congenital long QT syndrome. J Cardiovasc Electrophysiol. 1994;5:438–44. doi: 10.1111/j.1540-8167.1994.tb01183.x. [DOI] [PubMed] [Google Scholar]
- 23.Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;17:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Kurita T, Ohem T, Shimizu W, et al. Early afterdepolarization-like activity in patients with class IA induced long QT syndrome and torsades de pointes. Pacing Clin Electrophysiol. 1997;20:695–705. doi: 10.1111/j.1540-8159.1997.tb03888.x. [DOI] [PubMed] [Google Scholar]
- 25.Nuyens D, Stengl M, Dugarmaa S, et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–7. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- 26.Nagatomo T, January CT, Ye B, et al. Rate-dependent QT shortening mechanism for the LQT3 ΔKPQ mutant. Cardiovasc Res. 2002;54:624–9. doi: 10.1016/s0008-6363(02)00265-1. [DOI] [PubMed] [Google Scholar]