

Abstract
We report the identification of a novel 12 bp deletion of the pre-mRNA splicing gene PRPF31 in a large Chinese family with autosomal dominant retinitis pigmentosa (adRP). This mutation results in the deletion of four amino acids (ΔH111K112F113I114) including H111, an amino acid residue that is highly conserved throughout evolution. The 12 bp deletion co-segregates with the disease phenotype in 19 RP patients in the family, but is not present in unaffected relatives or 100 normal individuals. Our data indicate that the novel 12 bp deletion in PRPF31 causes retinitis pigementosa in this Chinese adRP family. In contrast to the incomplete penetrance observed in most adRP families linked to chromosome band 19q13.4 (RP11), the 12 bp PRPF31 deletion identified in this study appears to show high penetrance. These data expand the spectrum of PRPF31 mutations causing adRP, and confirm the role of PRPF31 in the pathogenesis of RP.
Keywords: retinitis pigmentosa, splicing, spliceosome, snRNP, PRPF31 (PRP31), RP11, mutation, retinal degeneration and dystrophy, adRP
INTRODUCTION
Retinitis pigmentosa (RP) is a heterogeneous group of retinal dystrophies, characterized by photoreceptor cell degeneration, night blindness, a gradual loss of peripheral visual fields, and the eventual loss of central vision [Wang et al., 2001]. RP affects 1 in 4,000 persons and is responsible for visual handicap in 1.5 million individuals worldwide [Haim et al., 1992; Berson, 1993; Kumar-Singh et al., 1993]. RP exhibits high genetic heterogeneity with various inheritance modes including autosomal dominant, autosomal recessive, X-linked, and digenic forms [Dryja and Li, 1995; Inglehearn, 1998; van Soest et al., 1999; Phelan and Bok, 2000; Wang et al., 2001]. Autosomal dominant RP (adRP) and autosomal recessive RP (arRP) are each estimated to account for approximately 20% of RP cases, and X-linked RP accounts for 10% of cases [Jay, 1992; Ham, 1993]. Approximately 50% of RP cases are simplex RP, which represents either sporadic RP or arRP [Jay, 1992; Ham, 1993]. Digenic RP is rare. In this study, we genetically characterized a large Chinese family with adRP.
The genes that have been identified or cloned in adRP include HPRP3 on chromosome band 1p13-q21, RHO at 3q21-24, RDSat 6p21.1-cen, PAP1at 7p14.2, IMPDH1 at 7q31.1, RP1 at 8p11-21, RGR at 10q23, ROM1 at 11q13, NRL at 14q11.1-11.2, PRPC8 at 17p13.3, FSCN2 at 17q25, CRX at 19q13.3, and PRPF31 at 19q13.4 [Vithana et al., 2001; Wang et al., 2001; Bowne et al., 2002; Keen et al., 2002; Kennan et al., 2002]. The chromosomal location of another adRP gene was mapped by linkage analysis, locus RP17 (17q22), but the responsible gene remains to be cloned or identified [Wang et al., 2001].
The adRP gene on chromosome 19q13.4 (RP11) was identified as the PRPF31 gene that encodes a 499 amino acid protein with homology to the yeast pre-mRNA splicing factor PRP31 [Vithana et al., 2001]. PRPF31 is a component of the U4/U6 snRNP particle involved in pre-mRNA splicing [Makarova et al., 2002]. The PRPF31 gene has 14 exons and spans about 18 kb [Vithana et al., 2001]. It is expressed in many tissues including retina [Vithana et al., 2001]. In this article, we describe a novel intragenic 12-bp deletion in PRPF31 in a large Chinese family with 19 individuals affected with RP. This mutation causes the deletion of four amino acids (ΔH111K112F113I114), including amino acid H111 that is evolutionally conserved from yeast to human.
MATERIALS AND METHODS
Genomic DNA, Genotyping, and Linkage Analyses
Informed consent was obtained from the participants in accordance with guidelines established by local institutional review boards. Human genomic DNA was isolated using the DNA Isolation Kits for Mammalian Blood according to the manufacturer’s instructions (Roche Diagnostics Corporation, Indianapolis, IN). Genotyping and linkage analysis were carried out as described previously [Wang et al., 1995; Wang et al., 1996]. Lod scores were calculated for each marker by two-point linkage analysis using the linkage package 5.2 [Lathrop et al., 1985].
Mutation Analysis
PCR-amplification of PRPF31 exons was carried out using primers listed in Table I. Mutation analysis was carried out by direct DNA sequence analysis by BigDye™ terminator cycle sequencing with an ABI-3100 Genetic Analyzer.
TABLE I.
PCR Primers for Amplification of PRPF31 Exons*
| Exon(s) | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) | Annealing temperature°C |
|---|---|---|---|
| 2, 3 | GTCGGGGCAAGTTTTTAGGG | GAGATGGGGAGGGGCACAGAGT | 64 |
| 4 | CCGAGAGGGGGTAGGGATTTAGAT | AGGCCAGTGGGGAAGGGAGAGG | 64 |
| 5 | TTAGGGCCAACCAGCAGAGTC | GAGGGGGTCCGAGAGTGAGC | 64 |
| 6, 7 | GTTCCCGAGCCTCCCCTATCTTCT | CGCTCCAGCTCCTCCTCCGACAG | 64 |
| 8 | CCGGCGGCCTGACCAACC | GGGAGGGGCCATGACGCAGTG′ | 64 |
| 9 | GCGCGGTTGCTTTGCTGTTA | ACTGCCTCCGCCTTGGTAG | 64 |
| 10, 11 | GTGGCGGTGAGGCAGCATTAGGTG | CTGGCTGGCTGTGGGGTTGAGGA | 55 |
| 12, 13 | GGGCCTGGTCGCTGA | GGGGAGGTACCTGGAGTGG | 64 |
| 14 | GGTCACAGTTGGGGCCTTCTCCTC | TACTGGGCGGTGATCTCGGTCCTG | 64 |
Exon 1 was not studied because it is outside the coding region.
Mutation analysis was also carried out using single-strand conformational polymorphism (SSCP) analysis as described previously [Zhao et al., 2001a,b]. The aberrant SSCP conformer was cut directly from dried gels, rehydrated in water, re-amplified by PCR, and sequenced.
RESULTS
We have identified a large Chinese family (kindred RPYT) with clear diagnosis of RP (Fig. 1). The inheritance pattern in kindred RPYT appears to be autosomal dominant (adRP) (Fig. 2a). Because many genetic loci have been identified in adRP, our initial genetic study of kindred RPYT was focused on linkage analysis with markers linked to known genetic loci for adRP: RHO (D3S3023, D3S1764), RDS (GATA11E02, D6S1053), NRL (D14S1280, D14S608), RP1 (D8S1110), CRX (D19S420, D19S902, D19S571), FSCN2 (D17S785, D17S928), RP9 (D7S516, D7S484), RP10 (D7S486, D7S530, D7S640), RP11 (D19S418, D19S210), RP13 (D17S849, D17S831), RP17 (D17S1868, D17S787), and RP18 (D1S252, D1S498, D1S484). Two-point lod scores varied from 0.000022 to 0.000018 with all markers tested except D19S418. Because D19S418 yielded a positive lod score of 2.8 and it is located 5 cm from the RP11 gene PRPF31, our genetic analysis of kindred RPYT was then shifted to mutation analysis of PRPF31.
Fig. 1.
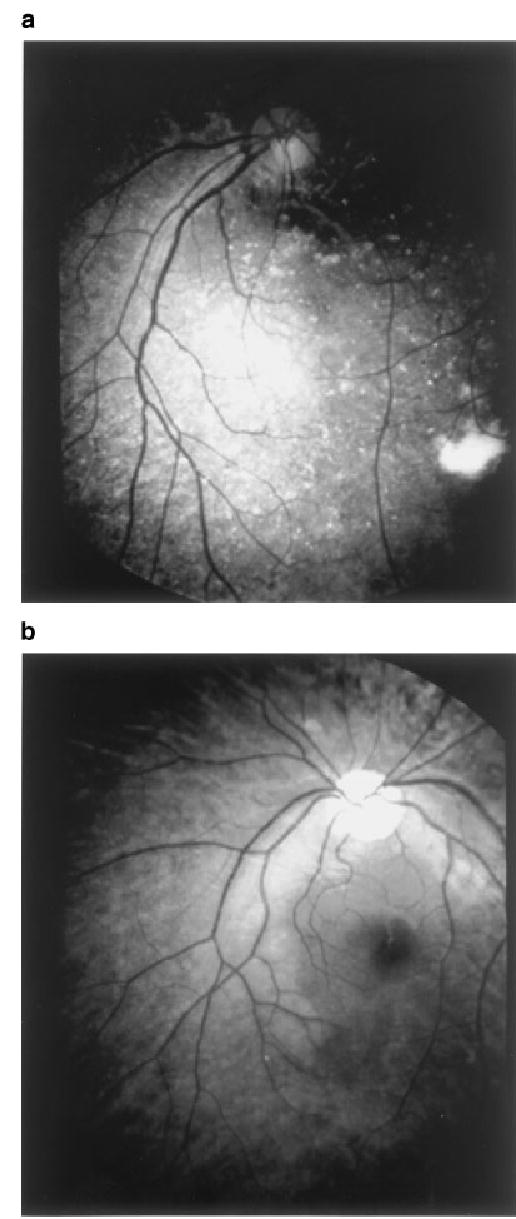
Fundus photographs of two patients with the 12-bp deletion of PRPF31. a: individual no. 7 in Figure 2a; (b): individual no. 20 in Figure 2a. Note the typical findings associated with RP: bone-spicule pigmentation, precipitates of golden particles, and dystrophy of optic nerves.
Fig. 2.
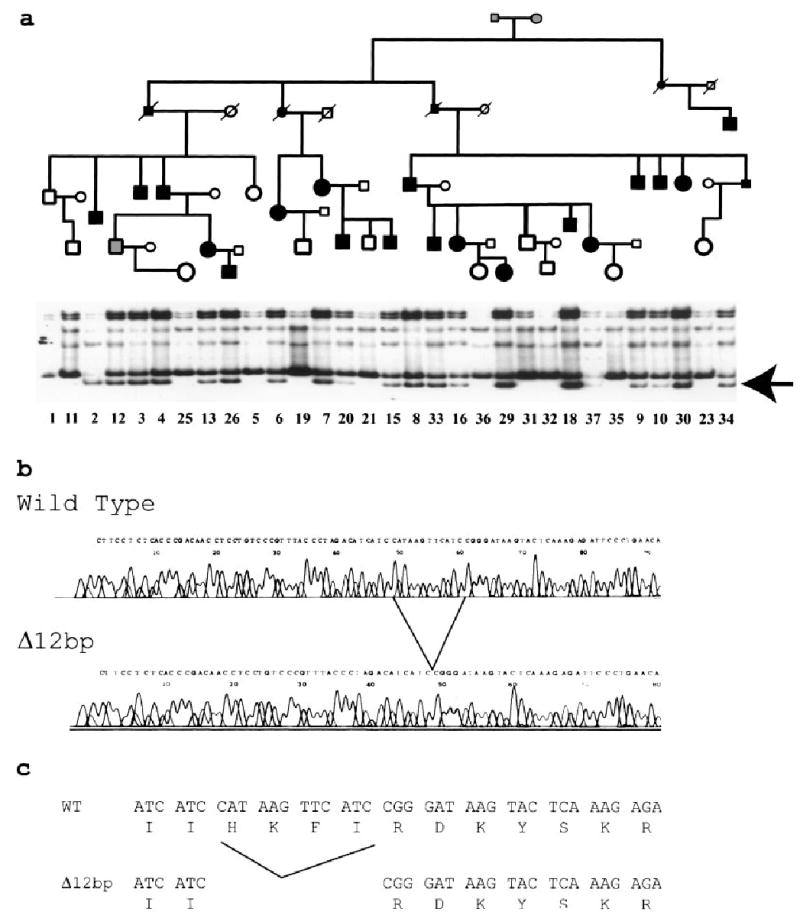
A 12-bp deletion in PRPF31 causes autosomal dominant RP in a large Chinese kindred (RPYT). a: The pedigree of RPYT is shown at the top. Normal individuals are shown as clear circles (females) or squares (males), and affected individuals are shown as solid symbols. The individual with a possible diagnosis of RP (no. 12) is shown with a gray square. The deceased individuals are shown with “/”. Results of SSCP analysis are shown below the pedigree structure. The arrow indicates the abnormal SSCP band. The numbers beneath the SSCP image are unique identification codes for individual family members. Note that only affected individuals carry the abnormal SSCP band, and all normal family members do not carry it. b: Sequence analysis of normal (wild type) and abnormal (Δ12bp) SSCP conformers. The deletion spot is indicated. c: Δ12bp leads to deletion of four amino acids of PRPF31, H111K112F113I114. Amino acid H111 is evolutionally conserved from yeast, A. thaliana, Drosophila, to humans.
Single strand conformation polymorphism (SSCP) and DNA sequence analysis demonstrated a novel 12 bp deletion in exon 5 of PRPF31 (Fig. 2b). This deletion results in an in-frame deletion of four amino acids, H111K112F113I114 (Fig. 2c). The 12-bp deletion was shown to co-segregate with the RP phenotype in 18 affected members of kindred RPYT by SSCP analysis (Fig. 2a). The deletion was not observed in DNA samples from 11 unaffected members in the family (Fig. 2a) and was not present in more than 100 controls (data not shown).
DISCUSSION
We report the identification of a novel 12-bp deletion of the pre-mRNA splicing gene PRPF31 in a large Chinese family. We provide two lines of evidence that strongly suggests that the 12-bp deletion is causal. First, the 12bp deletion co-segregates with the RP phenotype in 19 patients in kindred RPYT, but not with 11 unaffected family members and 100 normal controls. Secondly, this mutation deletes four amino acids (ΔH111K112F113I114), one of which is evolutionally conserved amino acid residue H111 [Vithana et al., 2001]. The 12 bp deletion is located in exon 5 of PRPF31 in which no mutations have been identified to date. The previously identified mutations were clustered in more 3′-end exons of the gene, including exons 6, 7, 8, and 11 [Vithana et al., 2001]. Our study expands the spectrum of PRPF31 mutations causing adRP, and confirms the role of pre-mRNA splicing factor PRPF31 in the pathogenesis of RP.
The molecular mechanism underlying pathogenesis of the PRPF31 12-bp deletion in RP is not clear. PRPF31 encodes a 61 kDa pre-mRNA splicing protein that is required for U4/U6.U5 tri-snRNP (small nuclear ribo-nucleoprotein) formation [Makarova et al., 2002]. The tri-snRNP formation starts with the association between the U4 and U6 snRNPs through extensive RNA-RNA base-pairing, which is followed by the association of the U4/U6 snRNP complex with U5 snRNP [Staley and Guthrie, 1998]. The PRPF31 protein appears to tether U4/U6 to U5 snRNP to form the tri-snRNP complex [Makarova et al., 2002]. The tri-snRNP formation is a critical step in the assembly of the catalytically active spliceosome. Lack of the PRPF31 protein blocks tri-snRNP formation and pre-mRNA splicing [Makarova et al., 2002]. It is interesting to note that two other newly-identified adRP genes also encode proteins involved in pre-mRNA splicing: PRPC8 at 17p13.3 (RP13) coding for PRP8 (a core component of the U5 snRNP) [McKie et al., 2001], and HPRP3 at 1p13-q21 (RP18) for PRP3 (a component of the U4/U6) [Chakarova et al., 2002]. These data together suggest that disruptions in tri-SNP formation and function contribute to the pathogenesis of adRP. Thus, we speculate that the 12-bp deletion of the PRPF31 gene identified in this study may disrupt the association of the U4/U6 snRNP complex to U5-snRNP, leading to defective pre-mRNA splicing. It is puzzling as why these splicing defects cause a defect in the vision system only. Deery et al. [2002] recently found that two missense mutations (A194E, A216P) in PRPF31 may affect splicing by impeding the translocation of PRPF31 into the nucleus. The splicing defect of A216P was further demonstrated by the finding that PRPF31 with A216P failed to fully complement functional deficiency in a temperature-sensitive, PRP31p-deficient yeast strain at the high restrictive temperature (higher growth rate, high demand for splicing) [Deery et al., 2002]. It was argued that rod photoreceptors may have a high demand for splicing of important molecules such as opsin mRNA, and subtle defects in splicing due to loss-of-function of PRPF31 may lead to a disease in this system (RP).
The chromosome 19q13.4 linked RP (RP11) is thought to be a frequent cause of adRP [Al Maghtheh et al., 1996]. It is noteworthy that mutations at the RP11 locus show “all or none” form of incomplete penetrance, where gene carriers display either fully symptomatic phenotype or completely asymptomatic phenotype [Al Maghtheh et al., 1994, 1996; Evans et al., 1995; McGee et al., 1997]. Interestingly, the 12 bp deletion of PRPF31 identified in this study shows high penetrance (Fig. 2a). The cause of the high penetrance phenotype associate with the 12 bp deletion is unknown. One possible explanation is that the 12 bp deletion may be a highly severe mutation, and reduces the splicing activity in the photoreceptors below the threshold level that is sufficient for the development of RP.
Acknowledgments
We thank Donald Kikta for technical help. Qing Wang is an outstanding Young Investigator of the China Natural Science Foundation. This work was partly supported by two China National Natural Science Foundation Awards (to QW and KZ). QW is also supported by NIH grants R01 HL65630 and R01 HL66251, and a Doris Duke Innovationin Clinical Research Award.
Footnotes
Lejin Wang, Michael Ribaudo, and Kanxing Zhao have contributed equally to this work.
Grant sponsor: China National Natural Science Foundation (to QW and KZ); Grant sponsor: NIH (to QW); Grant numbers: R01 HL65630, R01 HL66251.
References
- Al Maghtheh M, Inglehearn CF, Keen TJ, Evans K, Moore AT, Jay M, Bird AC, Bhattacharya SS. Identification of a sixth locus for autosomal dominant retinitis pigmentosa on chromosome 19. Hum Mol Genet. 1994;3:351–354. doi: 10.1093/hmg/3.2.351. [DOI] [PubMed] [Google Scholar]
- Al Maghtheh M, Vithana E, Tarttelin E, Jay M, Evans K, Moore T, Bhattacharya S, Inglehearn CF. Evidence for a major retinitis pigmentosa locus on 19q13.4 (RP11) and association with a unique bimodal expressivity phenotype. Am J Hum Genet. 1996;59:864–871. [PMC free article] [PubMed] [Google Scholar]
- Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34:1659–1676. [PubMed] [Google Scholar]
- Bowne SJ, Sullivan LS, Blanton SH, Cepko CL, Blackshaw S, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, Papaioannou MG, Inglehearn CF, Keen TJ, Willis C, Moore AT, Rosenberg T, Webster AR, Bird AC, Gal A, Hunt D, Vithana EN, Bhattacharya SS. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- Deery EC, Vithana EN, Newbold RJ, Gallon VA, Bhattacharya SS, Warren MJ, Hunt DM, Wilkie SE. Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum Mol Genet. 2002;11:3209–3219. doi: 10.1093/hmg/11.25.3209. [DOI] [PubMed] [Google Scholar]
- Dryja TP, Li T. Molecular genetics of retinitis pigmentosa. Hum Mol Genet. 1995;4:1739–1743. doi: 10.1093/hmg/4.suppl_1.1739. [DOI] [PubMed] [Google Scholar]
- Evans K, Al Maghtheh M, Fitzke FW, Moore AT, Jay M, Inglehearn CF, Arden GB, Bird AC. Bimodal expressivity in dominant retinitis pigmentosa genetically linked to chromosome 19q. Br J Ophthalmol. 1995;79:841–846. doi: 10.1136/bjo.79.9.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim M, Holm NV, Rosenberg T. Prevalence of retinitis pigmentosa and allied disorders in Denmark. I main results. Acta Ophthalmol (Copenh) 1992;70:178–186. doi: 10.1111/j.1755-3768.1992.tb04121.x. [DOI] [PubMed] [Google Scholar]
- Ham M. Retinitis pigmentosa: Problems associated with genetic classification. Clin Genet. 1993;44:62–70. doi: 10.1111/j.1399-0004.1993.tb03848.x. [DOI] [PubMed] [Google Scholar]
- Inglehearn CF. Molecular genetics of human retinal dystrophies. Eye. 1998;12:571–579. doi: 10.1038/eye.1998.147. [DOI] [PubMed] [Google Scholar]
- Jay M. On the heredity of retinitis pigmentosa. Br J Ophthalmol. 1992;66:405–416. doi: 10.1136/bjo.66.7.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen TJ, Hims MM, McKie AB, Moore AT, Doran RM, Mackey DA, Mansfield DC, Mueller RF, Bhattacharya SS, Bird AC, Markham AF, Inglehearn CF. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur J Hum Genet. 2002;10:245–249. doi: 10.1038/sj.ejhg.5200797. [DOI] [PubMed] [Google Scholar]
- Kennan A, Aherne A, Palfi A, Humphries M, McKee A, Stitt A, Simpson DA, Demtroder K, Orntoft T, Ayuso C, Kenna PF, Farrar GJ, Humphries P. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho(−/−) mice. Hum Mol Genet. 2002;11:547–557. doi: 10.1093/hmg/11.5.547. [DOI] [PubMed] [Google Scholar]
- Kumar-Singh R, Farrar GJ, Mansergh F, Kenna P, Bhattacharya S, Gal A, Humphries P. Exclusion of the involvement of all known retinitis pigmentosa loci in the disease present in a family of Irish origin provides evidence for a sixth autosomal dominant locus (RP8) Hum Mol Genet. 1993;2:875–878. doi: 10.1093/hmg/2.7.875. [DOI] [PubMed] [Google Scholar]
- Lathrop GM, Lalouel JM, Julier C, Ott J. Multilocus linkage analysis in humans: Detection of linkage and estimation of recombination. Am J Hum Genet. 1985;37:482–498. [PMC free article] [PubMed] [Google Scholar]
- Makarova OV, Makarov EM, Liu S, Vornlocher HP, Luhrmann R. Protein 61K, encoded by a gene (PRPF31) linked to autosomal dominant retinitis pigmentosa, is required for U4/U6center dotU5 tri-snRNP formation and pre-mRNA splicing. EMBO J. 2002;21:1148–1157. doi: 10.1093/emboj/21.5.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee TL, Devoto M, Ott J, Berson EL, Dryja TP. Evidence that the penetrance of mutations at the RP11 locus causing dominant retinitis pigmentosa is influenced by a gene linked to the homologous RP11 allele. Am J Hum Genet. 1997;61:1059–1066. doi: 10.1086/301614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, Lith-Verhoeven JJ, Greenberg J, Ramesar RS, Hoyng CB, Cremers FP, Mackey DA, Bhattacharya SS, Bird AC, Markham AF, Inglehearn CF. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum Mol Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- Phelan JK, Bok D. A brief review of retinitis pigmentosa and the identified retinitis pigmentosa genes. Mol Vis. 2000;6:116–124. [PubMed] [Google Scholar]
- Staley JP, Guthrie C. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- van Soest S, Westerveld A, de Jong PT, Bleeker-Wagemakers EM, Bergen AA. Retinitis pigmentosa: Defined from a molecular point of view. Surv Ophthalmol. 1999;43:321–334. doi: 10.1016/s0039-6257(98)00046-0. [DOI] [PubMed] [Google Scholar]
- Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, Al Maghtheh M, Ebenezer ND, Willis C, Moore AT, Bird AC, Hunt DM, Bhattacharya SS. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11) Mol Cell. 2001;8:375–381. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang Q, Chen Q, Zhao K, Wang L, Wang L, Traboulsi EI. Update on the molecular genetics of retinitis pigmentosa. Ophthalmic Genet. 2001;22:133–154. doi: 10.1076/opge.22.3.133.2224. [DOI] [PubMed] [Google Scholar]
- Zhao K, Wang L, Wang L, Wang L, Zhang Q, Wang Q. Novel deletion of the RPGR gene in a Chinese family with X-linked retinitis pigmentosa. Ophthalmic Genet. 2001a;22:187–194. doi: 10.1076/opge.22.3.187.2221. [DOI] [PubMed] [Google Scholar]
- Zhao K, Xiong S, Wang L, Wang L, Cui Y, Wang Q. Novel rhodopsin mutation in a Chinese family with autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2001b;22:155–162. doi: 10.1076/opge.22.3.155.2225. [DOI] [PubMed] [Google Scholar]