

Abstract
Hypokalemic periodic paralysis (HypoPP) is an autosomal dominant disorder which is characterized by periodic attacks of muscle weakness associated with a decrease in the serum potassium level. The skeletal muscle calcium channel α-subunit gene CACNA1S is a major disease-causing gene for HypoPP, however, only three specific HypoPP-causing mutations, Arg528His, Arg1,239His and Arg1,239Gly, have been identified in CACNA1S to date. In this study, we studied a four-generation Chinese family with HypoPP with 43 living members and 19 affected individuals. Linkage analysis showed that the causative mutation in the family is linked to the CACNA1S gene with a LOD score of 6.7. DNA sequence analysis revealed a heterozygous C to G transition at nucleotide 1,582, resulting in a novel 1,582C→G (Arg528Gly) mutation. The Arg528Gly mutation co-segregated with all affected individuals in the family, and was not present in 200 matched normal controls. The penetrance of the Arg528Gly mutation was complete in male mutation carriers, however, a reduced penetrance of 83% (10/12) was observed in female carriers. No differences were detected for age-at-onset and severity of the disease (frequency of symptomatic attacks per year) between male and female patients. Oral intake of KCl is effective in blocking the symptomatic attacks. This study identifies a novel Arg528Gly mutation in the CACNA1S gene that causes HypoPP in a Chinese family, expands the spectrum of mutations causing HypoPP, and demonstrates a gender difference in the penetrance of the disease.
Keywords: Hypokalemic periodic paralysis, Skeletal muscle calcium channel, Mutation, CACNA1S, Ion channel
Abbreviations: CACNA1S: Skeletal muscle voltage-gated calcium channel α-subunit, HyperPP: Hyperkalemic periodic paralysis, HypoPP: Hypokalemic periodic paralysis, KCNE3: Voltage-gated potassium channel β subunit gene, PCR: Polymerase chain reaction, RFLP: Restriction fragment length polymorphism, SCN4A: Skeletal muscle voltage-gated sodium channel α-subunit
Introduction
Hypokalemic periodic paralysis (HypoPP) and hyperkalemic periodic paralysis (HyperPP) are two genetic muscle disorders that are inherited in an autosomal dominant fashion, and present with clinically similar features of episodic flaccid generalized weakness [1]. An important clinical difference between the two entities is the trigger of the attacks of weakness. They can be distinguished by the changes in serum potassium levels before and during paralytic attacks. HyperPP can be provoked by oral potassium administration, whereas a low concentration of serum potassium triggers HypoPP. Co-occurrence of myotonia has been reported in HyperPP patients, but not in HypoPP patients [1].
HypoPP is the most common form of periodic paralysis in man, although it is still a rare disease with a prevalence of about 0.4 in 100,000 [2]. Familial HypoPP is a muscle disease characterized by periodic attacks of partial or total muscle weakness associated with a decrease in serum potassium. Two disease-causing genes have been identified for HypoPP, both of which encode ion channels, including the human skeletal muscle dihydropyridine-sensitive calcium channel α1-subunit gene CACNA1S [3] and the skeletal muscle sodium channel α-subunit gene SCN4A [4, 5]. A voltage-gated potassium channel β-subunit gene, KCNE3, was claimed as a HypoPP gene with the identification of one variant (Arg83His) in two families [6], but recent studies suggest that Arg83His may be a benign polymorphic variant that is not associated with HypoPP [7, 8].
The skeletal muscle calcium channel CACNA1S mediates excitation-contraction coupling. Calcium influx through CACNA1S triggers the local release of a large amount of calcium (Ca2+ sparks) from the intracellular calcium release channel, resulting in muscle contraction. CACNA1S is composed of four homologous domains (DI–DIV), each of them consisting of six transmembrane segments (S1–S6) [9]. Three specific mutations have been identified in the CACNA1S gene in families with HypoPP: Arg528His in DIIS4, and Arg1,239His and Arg123Gly in DIVS4 [3, 10–20]. These mutations are all missense mutations where an arginine residue is replaced by either a histidine or sometimes a glycine residue. These mutations are all located in the voltage sensor of the transmembrane segment of the calcium channel. Mutations in CACNA1S account for approximately 70% of HypoPP cases [12]. An Arg1,086His mutation in the loop between DIII and DIV of CACNA1S has also been linked to malignant hyperthermia [20, 21].
In this study, we identified and characterized a large Chinese family with HypoPP, with 43 family members (19 with a clinical diagnosis of HypoPP). We used linkage analysis to map the disease-causing gene in this family to the CACNA1S gene on chromosome 1q31-32, and further studies identified a novel mutation, 1,582C → G (Arg528-Gly), in CACNA1S. Genotype-phenotype correlation studies were carried out to characterize the clinical characteristics associated with the Arg528Gly mutation.
Materials and methods
Study subjects and isolation of genomic DNA
The study participants were identified and enrolled at Huazhong University of Science and Technology Union Hospital. Informed consent was obtained from the participants in accordance with the study protocols approved by the Ethics Committee of Huazhong University of Science and Technology. Detailed records on their medical history, physical examinations and blood potassium levels were obtained. The diagnosis of HypoPP was made on the basis of symptoms, physical signs and the blood potassium level.
Peripheral blood was collected from all participants. Genomic DNA was isolated from the whole blood with the DNA isolation kit for mammalian blood (Roche Diagnostics, Indianapolis, Ind.).
Linkage analysis
Linkage analysis was carried out as described [22–24]. Three polymorphic microsatellite markers, D1S413, D17S944 and D11S1314, were selected from the ABI PRISM Linkage Mapping Set MD10 panel for linkage analysis. These markers are closely linked to the two known HypoPP genes CACNA1S, SCN4A and the KCNE3 candiate gene. Markers were genotyped using an ABI 3100 Genetic Analyzer (Applied Biosystems, Foster City, Calif.) at Huazhong University of Science and Technology Human Genome Research Center. Genotypes were analyzed using the GeneMapper 2 Software program (Applied Biosystems). Pairwise logarithm of the odds (LOD) scores were calculated with the Linkage Package 5.2 program assuming an autosomal dominant inheritance pattern, a gene frequency of 0.001, a penetrance rate of 80%, and an allele frequency of 1/n, where n equals the number of alleles observed [22–24].
Mutation screening
Mutation screening was carried out using direct DNA sequence analysis. CACNA1S exons were PCR-amplified. PCR was performed in 25 μl of standard PCR buffer containing 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.5 μM of each primer, 1 U of Taq polymerase, and 50 ng of DNA. The amplification program was an initial 3-min denaturation at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at 56°C, 1 min at 72°C, and a final 7-min extension step at 72°C. The PCR products were extracted using the QIAquick Gel Extraction Kit (Qiagen, Valencia, Calif.), and sequenced with both forward and reverse primers. DNA sequencing analysis was performed using the BigDye Terminator Cycle Sequencing v3.1 kit and an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems).
RFLP analysis
Mutation 1,582C→G (Arg528Gly) disrupts an MspA1 I restriction site, which allowed us to perform RFLP analysis to confirm the mutation and to test whether the mutation co-segregates with the disease in the family. We PCR-amplified exon 11 containing the Arg528Gly mutation from all members of the family as well as 200 unrelated healthy Chinese individuals of Han nationality. The 364-bp PCR product was digested with 1 U of MspA1 I (New England Biolabs) at 37°C overnight. The resulting digestion products were separated on a 2.5% agarose gel.
Results
We identified a large Chinese family with 43 living individuals in four generations (Fig. 1). In the family, 19 members were clinically diagnosed as affected with HypoPP. The proband is a 53-year-old male (II-1 in Fig. 1) who experienced the first total muscle weakness attack at the age of 16 years. The attacks usually occur after excessive intake of carbohydrate-rich meals, several hours following strenuous exercise, after sudden exposure to cold, glucose perfusion, and alcohol consumption. His attacks occurred more frequently at a younger age, and became less frequent with advancing age. His blood potassium level was low and the symptoms could be relieved with potassium chloride. His serum potassium levels were 2.3 mmol/l before the attacks and 2.95 mmol/l after relief with KCl.
Fig. 1.
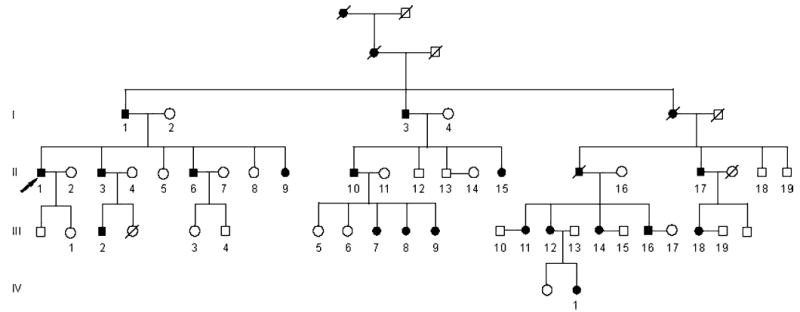
Pedigree structure of a large Chinese family affected with HypoPP. Affected males and females are indicated by filled squares and circles, respectively. Normal individuals are shown as empty symbols. Deceased individuals are indicated by slashes (/). The proband is indicated by an arrow
Similar clinical features were detected in the other 18 affected family members. The age of onset and the frequency of HypoPP attacks are shown in Table 1 for each affected individual in the family. The frequency and severity of attacks and the age of onset vary from patient to patient. The age of onset ranged from 3 to 23 years in females and from 16 to 20 years in males (Table 1). The attacks became less frequent after 40 years of age and disappeared after 60. The frequency of attacks varied from patient to patient, ranging from six times to 300 times per year (Table 1). The duration of the attacks was from 2 h to 2 days. Attacks commonly started in the second half of the night; awakening the patient who was unable to move, and the symptoms could be relieved only with the intake of potassium chloride salt. No significant differences of age-at-onset and the frequency of attacks were identified between males and females (Table 2).
Table 1.
Age at onset and the frequency of attacks for nine male and ten female HypoPP patients in the Chinese HypoPP family with the Arg528Gly mutation of CACNA1S
| Age at onset (male) | Age at onset (female) | Frequency of attacks per year (male) | Frequency of attacks per year (female) |
|---|---|---|---|
| 16 | 20 | 6 | 17 |
| 17 | 14 | 24 | 8 |
| 20 | 20 | 24 | 7 |
| 17 | 23 | 8 | 18 |
| 17 | 20 | 8 | 20 |
| 18 | 17 | 20 | 20 |
| 16 | 14 | 300 | 20 |
| 18 | 3 | 22 | 300 |
| 18 | 21 | 23 | 20 |
| 17 | 21 |
Table 2.
Comparison of key clinical features of HypoPP in male and female patients in the Chinese family with the Arg528Gly mutation of CACNA1S. A P value of <0.05 was considered significant
| Male | Female | P value | |
|---|---|---|---|
| Penetrance | 1.00 (9/9) | 0.8333 (10/12) | – |
| Mean age at onset (years) | 15.22 ± 9.12 | 16.99 ± 17.11 | 0.94 |
| Mean frequency of attacks per year | 48.33 ± 94.67 | 45.00 ± 89.76 | 0.78 |
To identify the molecular basis of HypoPP in this family, we carried out linkage analysis for two genes identified as being involved in HypoPP, CACNA1S on chromosome 1q31–32 and SCN4A on 17q23.2–25.3, and also the KCNE3 candidate gene on 11q13–24. Detection of multiple obligate recombinants (affected individuals carrying the normal allele) excluded SCN4A and KCNE3 as the causative gene in the Chinese HypoPP family. Co-segregation of marker D1S413 close to CACNA1S with all affected individuals in the family and a LOD score of 6.70 between the marker and disease suggest that the disease gene in the family is the CACNA1S gene.
Direct sequence analysis of DNA from the proband revealed a heterozygous C to G transition at nucleotide 1,582 of CACNA1S, which results in a substitution of a highly conserved, positively charged amino acid arginine with a neutral glycine residue at codon 528, 1,582C→G (Arg528-Gly) (Fig. 2a). Mutation Arg528Gly is located in the trans-membrane segment S4 of domain II, a critical region serving as the voltage sensor of the channel and regulating the activation of the calcium channel. Amino acid residue R528 is evolutionally conserved from C. elegans to humans (Fig. 2b).
Fig. 2.
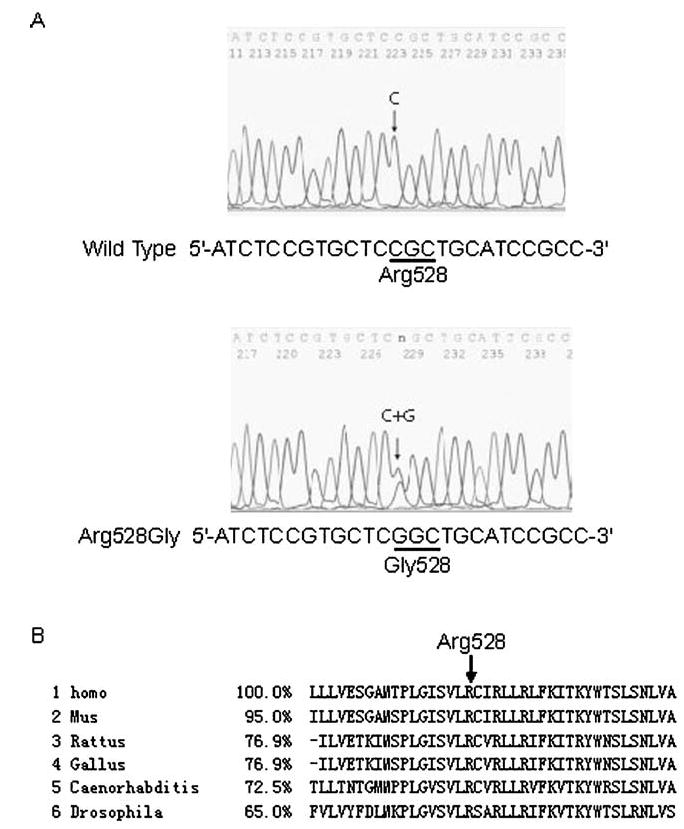
Identification of a novel mutation 1,582C →G (Arg528-Gly) of the CACNA1S gene in the Chinese family with Hy-poPP. a DNA sequences for a normal family member (wild type) and the proband III-1 (Arg528Gly) are shown. The nucleotide residue where the mutation occurs is indicated by an arrow. The C to G change at codon 528 results in the substitution of an arginine residue by a glycine residue in DIIS4 (GenBank accession NM_000069). b The alignment of amino acids around Arg528 revealed evolutionary conservation of this residue from the C. elegans and fly (Drosophila) to the humans (Homo). The percentage of identity to the human gene is shown. Homo Homo sapiens, Mus Mus musculus, Rattus Rattus norvegicus, Gallus Gallus gallus, Caenorhabditis Caenorhabditis elegans, Drosophila Drosophila melanogaster
Detection of mutation Arg528Gly was further confirmed by RFLP analysis showing the presence of both the wild type allele (252 and 112 bp bands) and the mutant allele (364 bp band) (proband III-1, Fig. 3). RFLP results showed that all affected members in the family carried the Arg528-Gly mutation, but the mutation was not present in 200 normal controls (Fig. 3). All normal individuals except two young female individuals, III-1 and III-3, in the family do not carry the Arg528Gly mutation (Fig. 3). Identification of two normal individuals (III-1 and III-3) who carry the Arg528Gly mutation suggests reduced penetrance of the mutation in the female patient population.
Fig. 3.
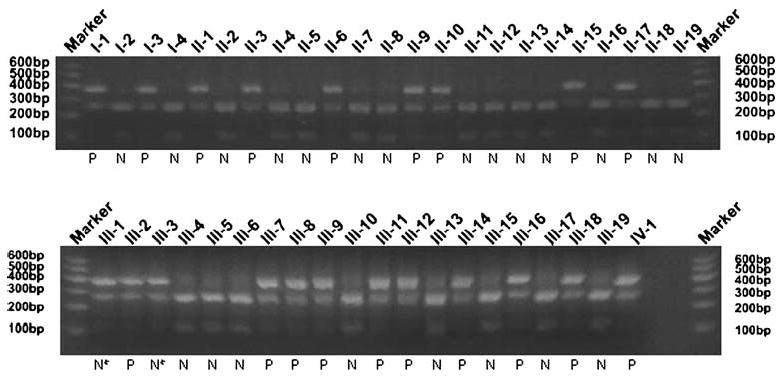
Mutation Arg528Gly of the CACNA1S gene co-segregates with HypoPP in the family. Marker 100-bp molecular weight ladder/ marker (Invitrogen), N normal phenotype, P affected phenotype. The sample lanes are labeled with the unique identification numbers for each individual in the Chinese family with HypoPP as in Fig. 1, e.g. I-1 is for individual 1 at generation I in Fig. 1. The Arg528Gly mutation abolishes an MspA1 I site. The wild type PCR product can be cut by MspA1 I, yielding two shorter DNA fragments of 252 and 112 bp. Normal family members display two 252 and 112 bp bands except for the two female individuals III-1 and III-3 (marked with N*) who show reduced penetrance. The PCR fragment containing mutation Arg528Gly cannot be cut by the enzyme, resulting in only one DNA fragment of 364 bp. All affected individuals in the family are heterozygous for the Arg528Gly mutation (three fragments: 364, 252, and 112 bp). the PCR primers used were: 528-1 forward, 5′-AAGGCTGGGAGTCAGGAGAAG-3′ and 528-1 reverse, 5′-ATGAGATGGGTGTAGACCAGAC-3′
Discussion
In this study, we present a novel mutation, 1,582C→G (Arg528Gly), in the skeletal muscle calcium channel gene, CACNA1S, that causes the autosomal dominant muscle disorder HypoPP in a large Chinese family. Although CACNA1S mutations are considered to be responsible for the majority of HypoPP cases (70%), only three disease-causing mutations have been identified, Arg528His, Arg1, 239His and Arg1,239Gly. Previously, the arginine residue at codon 1,239 was found to be frequently mutated to either a histidine or glycine residue, but only a mutation to a histidine residue from the arginine at codon 528 was detected. With the identification of the Arg528Gly mutation, our results suggest that Arg528 can also be mutated to a glycine residue. Furthermore, since both Arg528His and Arg528Gly mutations can be evidenced by the loss of the MspA1 I restriction site, both mutations should be considered when screening HypoPP patients using RFLP analysis with this enzyme.
Mutation Arg528Gly co-segregates with the disease in a large family with 19 HypoPP patients, and does not occur in 200 normal controls. The arginine residue at codon 528 is highly conserved from C. elegans to humans, and is located in the critical voltage-sensor domain of the calcium channel. Together with the finding that mutation Arg528His at the same location also causes HypoPP, these results strongly support the conclusion that mutation Arg528Gly is a disease-causing mutation.
The penetrance rate is 100% (9/9) in the male patients and 83.3% (10/12) in the female patients (Table 2). Recently, a Japanese boy with HypoPP was found to carry the Arg528His mutation in CACNA1S, but his mother and grandmother are clinically normal even though they are mutation carriers [16]. All these results suggest that there is a gender difference for the mutation penetrance of HypoPP. The molecular mechanism for the gender difference in the penetrance of this disease is unknown.
The functional effect of the Arg528Gly mutation on the skeletal calcium channel is not known. We speculate that the mutation may act similarly to the Arg528His mutation and lead to reduction of the L-type calcium current amplitude or density and affect the voltage dependence of channel activation [25, 26]. These effects are expected to result in a lesser calcium influx inside cells, and a failure of generation of Ca2+ sparks and muscle contraction, resulting in HypoPP. However, it is equally possible that the Arg528 Gly mutation may act differently from the Arg528His mutation. Future electrophysiological characterization of the Arg528Gly mutation will test these hypotheses and provide insights into the mechanism underlying the pathogenesis of HypoPP.

Junguo Yang received his medical degree from the Chinese Union University, P. R. China. He is presently a Professor of Medicine at Huazhong University of Science and Technology Human Genome Research Center and the Institute of Cardiovascular Disease, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, P. R. China. His research interests include the molecular genetics of cardiovascular disease.

Qiufen Wang received her Ph.D. from Tongji Medical University, P. R. China. She is presently an Associate Professor of Medicine at Huazhong University of Science and Technology Human Genome Research Center and the Institute of Cardiovascular Disease, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, P. R. China. Her research interests include the molecular genetics and the immune mechanisms of cardiovascular disease.
Acknowledgments
We are grateful to the family members. This study would be impossible without their enthusiastic participation. This work was supported by two grants from the Chinese Ministry of Science and Technology National High Technology "863” Programs of China No. 2002BA711A07 (one to Q.W. and the other to J.Y.; Professor Yan Shen is the Chief Scientist on the overall project).
Footnotes
Qiufen Wang and Mugen Liu contributed equally to this work
References
- 1.Lehmann-Horn F, Engel AG, Ricker K, Rüdel R. The periodic paralyses and paramyotonia congenita. In: Engel AG, Franzini-Armstrong C, editors. Myology. McGraw-Hill; New York: 1994. pp. 1304–1334. [Google Scholar]
- 2.Kantola IM, Tarssanen LT. Familial hypokalaemic periodic paralysis in Finland. J Neurol Neurosurg Psychiatry. 1992;55:322–324. doi: 10.1136/jnnp.55.4.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ptacek LJ, Tawil R, Griggs RC, Engel AG, Layzer RB, Kwiecinski H, McManis PG, Santiago L, Moore M, Fouad G. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. 1994;77:863–868. doi: 10.1016/0092-8674(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 4.Ptacek LJ, George AL, Jr, Griggs RC, Tawil R, Kallen RG, Barchi RL, Robertson M, Leppert MF. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. 1991;67:1021–1027. doi: 10.1016/0092-8674(91)90374-8. [DOI] [PubMed] [Google Scholar]
- 5.Ptacek LJ, George AL, Jr, Barchi RL, Griggs RC, Riggs JE, Robertson M, Leppert MF. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron. 1992;8:891–897. doi: 10.1016/0896-6273(92)90203-p. [DOI] [PubMed] [Google Scholar]
- 6.Abbott GW, Butler MH, Bendahhou S, Dalakas MC, Ptacek LJ, Goldstein SA. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell. 2001;104:217–231. doi: 10.1016/s0092-8674(01)00207-0. [DOI] [PubMed] [Google Scholar]
- 7.Jurkat-Rott K, Lehmann-Horn F. Periodic paralysis mutation MiRP2-R83H in controls: interpretations and general recommendation. Neurology. 2004;62:1012–1015. doi: 10.1212/01.wnl.0000119392.29624.88. [DOI] [PubMed] [Google Scholar]
- 8.Sternberg D, Tabti N, Fournier E, Hainque B, Fontaine B. Lack of association of the potassium channel-associated peptide MiRP2-R83H variant with periodic paralysis. Neurology. 2003;61:857–859. doi: 10.1212/01.wnl.0000082392.66713.e3. [DOI] [PubMed] [Google Scholar]
- 9.Hogan K, Gregg RG, Powers PA. The structure of the gene encoding the human skeletal muscle alpha 1 subunit of the dihydropyridine-sensitive L-type calcium channel (CACN-L1A3) Genomics. 1996;31:392–394. doi: 10.1006/geno.1996.0066. [DOI] [PubMed] [Google Scholar]
- 10.Caciotti A, Morrone A, Domenici R, Donati MA, Zammarchi E. Severe prognosis in a large family with hypokalemic periodic paralysis. Muscle Nerve. 2003;27:165–169. doi: 10.1002/mus.10298. [DOI] [PubMed] [Google Scholar]
- 11.Elbaz A, Vale-Santos J, Jurkat-Rott K, Lapie P, Ophoff RA, Bady B, Links TP, Piussan C, Vila A, Monnier N. Hypokalemic periodic paralysis and the dihydropyridine receptor (CACN-L1A3): genotype/phenotype correlations for two predominant mutations and evidence for the absence of a founder effect in 16 Caucasian families. Am J Hum Genet. 1995;56:374–380. [PMC free article] [PubMed] [Google Scholar]
- 12.Fouad G, Dalakas M, Servidei S, Mendell JR, Van den Bergh P, Angelini C, Alderson K, Griggs RC, Tawil R, Gregg R, Hogan K, Powers PA, Weinberg N, Malonee W, Ptacek LJ. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromuscul Disord. 1997;7:33–38. doi: 10.1016/s0960-8966(96)00401-4. [DOI] [PubMed] [Google Scholar]
- 13.Grosson CL, Esteban J, Kenna-Yasek D, Gusella JF, Brown RH., Jr Hypokalemic periodic paralysis mutations: confirmation of mutation and analysis of founder effect. Neuromuscul Disord. 1996;6:27–31. doi: 10.1016/0960-8966(95)00018-6. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda Y, Watanabe M, Shoji M. Mutation analysis of the CACNL1A3 gene in Japanese hypokalemic periodic paralysis families. Nippon Rinsho. 1997;55:3247–3252. [PubMed] [Google Scholar]
- 15.Jurkat-Rott K, Lehmann-Horn F, Elbaz A, Heine R, Gregg RG, Hogan K, Powers PA, Lapie P, Vale-Santos JE, Weissenbach J. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet. 1994;3:1415–1419. doi: 10.1093/hmg/3.8.1415. [DOI] [PubMed] [Google Scholar]
- 16.Kawamura S, Ikeda Y, Tomita K, Watanabe N, Seki K. A family of hypokalemic periodic paralysis with CACNA1S gene mutation showing incomplete penetrance in women. Intern Med. 2004;43:218–222. doi: 10.2169/internalmedicine.43.218. [DOI] [PubMed] [Google Scholar]
- 17.Kim SH, Kim UK, Chae JJ, Kim DJ, Oh HY, Kim BJ, Lee CC. Identification of mutations including de novo mutations in Korean patients with hypokalaemic periodic paralysis. Nephrol Dial Transplant. 2001;16:939–944. doi: 10.1093/ndt/16.5.939. [DOI] [PubMed] [Google Scholar]
- 18.Kusumi M, Kumada H, Adachi Y, Nakashima K. Muscle weakness in a Japanese family of Arg1239His mutation hypokalemic periodic paralysis. Psychiatry Clin Neurosci. 2001;55:539–541. doi: 10.1046/j.1440-1819.2001.00902.x. [DOI] [PubMed] [Google Scholar]
- 19.Sillen A, Sorensen T, Kantola I, Friis ML, Gustavson KH, Wadelius C. Identification of mutations in the CACNL1A3 gene in 13 families of Scandinavian origin having hypokalemic periodic paralysis and evidence of a founder effect in Danish families. Am J Med Genet. 1997;69:102–106. doi: 10.1002/(sici)1096-8628(19970303)69:1<102::aid-ajmg20>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 20.Stewart SL, Hogan K, Rosenberg H, Fletcher JE. Identification of the Arg1086His mutation in the alpha subunit of the voltage-dependent calcium channel (CACNA1S) in a North American family with malignant hyperthermia. Clin Genet. 2001;59:178–184. doi: 10.1034/j.1399-0004.2001.590306.x. [DOI] [PubMed] [Google Scholar]
- 21.Monnier N, Procaccio V, Stieglitz P, Lunardi J. Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet. 1997;60:1316–1325. doi: 10.1086/515454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Fan C, Topol SE, Topol EJ, Wang Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science. 2003;302:1578–1581. doi: 10.1126/science.1088477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 25.Morrill JA, Brown RH, Jr, Cannon SC. Gating of the L-type Ca channel in human skeletal myotubes: an activation defect caused by the hypokalemic periodic paralysis mutation R528H. J Neurosci. 1998;18:10320–10334. doi: 10.1523/JNEUROSCI.18-24-10320.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrill JA, Cannon SC. Effects of mutations causing hypokalaemic periodic paralysis on the skeletal muscle L-type Ca2+ channel expressed in Xenopus laevis oocytes. J Physiol. 1999;520:321–336. doi: 10.1111/j.1469-7793.1999.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]