

Abstract
TBX5 is a T-box transcription factor that plays a critical role in organogenesis. Seven missense mutations in TBX5 have been identified in patients with Holt-Oram syndrome characterized by congenital heart defects and upper limb abnormalities. However, the functional significance and molecular pathogenic mechanisms of these mutations are not clear. In this study we describe functional defects in DNA binding, transcriptional activity, protein-protein interaction, and cellular localization of mutant TBX5 with these missense mutations (Q49K, I54T, G80R, G169R, R237Q, R237W, and S252I). Mutations G80R, R237Q, and R237W represent a group of mutations that dramatically reduce DNA-binding activity of TBX5, leading to reduced transcription activation by TBX5 and the loss of synergy in transcriptional activation between TBX5 and NKX2.5. The second group of mutations includes Q49K, I54T, G169R, and S252I, which have no or moderate effect on DNA-binding activity and the function of transcription activation of TBX5 but cause the complete loss of synergistic transcription activity between TBX5 and NKX2.5. All seven missense mutations greatly reduced the interaction of TBX5 with NKX2.5 in vivo and in vitro. Immunofluorescent staining showed that wild type TBX5 was localized completely into the nucleus, but mutants were localized in both nucleus and cytoplasm. These results demonstrate that all seven missense mutations studied here are functional mutations with a spectrum of defects ranging from decreases in DNA-binding activity and transcriptional activation to the dramatic reduction of interaction between TBX5 and NKX2.5, and loss of synergy in transcriptional activation between these two proteins, as well as impairment in the nuclear localization of TBX5. These defects are likely central to the pathogenesis of Holt-Oram syndrome.
T-box transcription factors belong to a conserved family of proteins that contribute to early cell fate determination, differentiation, and organogenesis (1). Genetic mutations in human T-box transcription factors cause a number of human congenital disorders such as Holt-Oram syndrome (TBX5) (2, 3), ulnar-mammary syndrome (TBX3) (4), X-linked cleft palate and ankyloglossia (TBX22) (5), and DiGeorge syndrome (TBX1) (6, 7, 8). T-box genes have been identified in animals from ctenophores (comb jellies) and nematodes to mammals, and may account for ~0.1% of animal genomes.
The Tbx5 gene is located on chromosome 12q24 and encodes a protein of 518 amino acids (2, 3). TBX5 contains a highly conserved DNA-binding domain, T-box domain. Tbx5 is expressed in embryonic heart and limb tissues (9–12). During embryogenesis, Tbx5 expression starts at the earliest stages of heart formation and is co-localized with genes encoding other important cardiac transcription factors, Nkx2.5 and Gata4 (10). Over-expression of Tbx5 under the control of the β-myosin heavy chain promoter in mice inhibits ventricular chamber development and loss of ventricular-specific gene expression (10). Homozygous knockout mice deficient in Tbx5 die early in embryogenesis at D9.5, and heterozygous mice showed the forelimb and congenital heart defects (13). These results indicate that Tbx5 is critical to development of the heart and skeletal structures.
Mutations in Tbx5 have been found to cause Holt-Oram syndrome, an autosomal-dominant disease characterized by upper limb malformations and congenital heart defects including atrial and ventricular septal defects or cardiac conduction disease (2, 3, 14, 15). To date, twenty-four different Tbx5 mutations (15–19) have been described in patients with Holt-Oram syndrome. Many Tbx5 mutations are nonsense, frame-shift, and splice site mutations, or large chromosomal rearrangements (translocations, deletions) that are expected to produce truncated TBX5 proteins or no TBX5 at all (e.g. nonsense-mediated mRNA decay, translocation in intron 1). Based on these findings, Tbx5 haploinsufficiency has been proposed as a mechanism underlying pathogenesis of Holt-Oram syndrome. This hypothesis is further supported by the finding that heterozygous Tbx5del/+ mice modeled Holt-Oram syndrome (13). It is interesting to note that increased Tbx5 dosage, such as a chromosome 12q2 duplication, also results in Holt-Oram syndrome (19), suggesting that Tbx5 dosage is critical to the development of the heart and limbs. However, seven missense mutations in the Tbx5 gene, which would not be expected to alter the dosage of Tbx5, have been reported in patients with Holt-Oram syndrome. It remains to be determined whether these Tbx5 missense mutations are functional mutations, and if they are, what are the molecular pathogenic mechanisms by which these mutations act?
Biochemical studies have demonstrated that TBX5 protein interacts with the homeobox transcription factor NKX2.5 to synergistically transactivate expression of target genes (13, 20). The homeodomain of NKX2.5 was shown to be necessary and sufficient for its interaction with TBX5, although the NK domain (a17 amino acid domain specific to NK-class proteins) located C-terminal to the homeodomain can significantly enhance the interaction (13, 20). For TBX5, the region including the N-terminal domain and the N-terminal portion of the T-domain (amino acids 1–90) is sufficient for its association with NKX2.5 (20).
The target genes identified for Tbx5 include the genes for atrial natriuretic factor (ANF)1 (20) and connexin 40 (Cx40) (13, 20). Synergistic activation of both ANF and Cx40 promoters by TBX5 and NKX2.5 were demonstrated (13, 20). The promoters of ANF and Cx40 harbor binding sites for both TBX5 and NKX2.5, and these sites were shown to be capable of binding to both transcriptional factors (13, 20). Using an in vitro selection procedure with double-stranded random 26-mer oligonucleotides, a consensus sequence for TBX5 binding ((A/G)GGTGT(C/T/G)(A/G)) was identified (21). This consensus sequence is part of a half-site from the Brachyury T-box target sequence (21). TBX5 can bind to either a single site or paired non-palindromic sites in vitro (21).
In the present study, we have used a biochemical system comprising of TBX5, NKX2.5, and the ANF promoter to characterize the TBX5 missense mutations identified in Holt-Oram syndrome patients. We demonstrate that the TBX5 missense mutations are functional mutations that cause a spectrum of defects in DNA binding, transcription activation, interaction with NKX2.5, synergistic transactivation with NKX2.5, and nuclear localization. These results also provide insights into structure-function relationship of TBX5.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
The region from −270 to +1 bp upstream from the transcription start site of the ANF promoter was PCR-amplified and cloned into the pGL3-Basic vector, resulting in the ANF-Luc reporter gene. The full-length Tbx5 cDNA was cloned into the pcDNA3.1 vector, resulting in the over-expression constructs (20). Three forms of Tbx5 expression constructs were used: one with and the other without the FLAG-epitope tag as well as one construct for expressing His6-tagged TBX5. The expression constructs for hemagglutinin (HA)-tagged-NKX2.5 and GST-NKX2.5 fusion protein were as described previously (a gift from Dr. Issei Komuro) (20). The missense mutations in Tbx5 were introduced into the wild type construct by PCR-based site-directed mutagenesis and verified by DNA sequencing. The full-length Tbx5 cDNA was also cloned into the pET-28b vector, resulting in the bacterial expression construct for TBX5.
Cell Culture, Transfections, Western Blot, and Luciferase Assay
COS-7, HeLa, or NIH-3T3 cells were grown to 90% confluence in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% fetal bovine serum and transfected with LipofectAMINE 2000 and 50 ng of DNA for the expression construct, 1 μg of DNA for the reporter gene, and 50 ng of internal control plasmid pSV-β-galactosidase. Cells were harvested and lysed 48 h after transfection.
The efficiency of transfection was examined by Western blot analysis. Forty μg of total cellular lysates were separated by 12% SDS-PAGE and electro-transferred to a polyvinylidene fluoride membrane. The membrane was probed with mouse monoclonal anti-FLAG M2 antiserum (Sigma) as the primary antibody and the rabbit anti-mouse IgG horse-radish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). ECL Western blotting detection reagents (Amersham Pharmacia Biotech) were used to visualize the protein signal.
Luciferase assay was performed using a Dual-Luciferase assay kit according to the manufacturer’s instructions (Promega). β-galactosidase activity expressed from pSV-β-galactosidase was used to normalize the transfection efficiency. The experiments were repeated three times in triplicate. Data are expressed as mean ± S.E.
In Vitro Translation of TBX5 Protein and Electrophoretic Mobility Shift Assay (EMSA)
The synthetic TBX5-binding site was generated by annealing double-stranded oligonucleotides (5′-aataTCACACCTgtac-3′; 5′-gtacAGGTGTGAtatt-3′), labeled with [α-32P]dCTP and Klenow enzyme, and used in EMSA. The labeled TBX5-binding site was incubated with 2 μg of poly(dI-dC) and 4 μl of TBX5 protein synthesized using the TNT Quick Coupled Transcription/Transcription system in 20 μl of binding buffer. The TBX5-DNA complex was then separated from free DNA using 6% native polyacrylamide gels. The gels were run at 200 volts for 40 min, dried, and exposed to x-ray films.
TBX5-NKX2.5 Interactions in Vivo and in Vitro
For in vivo protein-protein interactions, HeLa cells were transiently co-transfected with expression plasmids for His-tagged TBX5 and HA-tagged NKX2.5 for 48 h. The transfected cells were harvested, washed with PBS, incubated in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 1% Nonidet P-40, a mini-protease inhibitor mixture (Roche Molecular Biochemicals)), and digested with DNase I. Total cell lysate was dialyzed with dialysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 20% glycerol, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol). The cell lysate was then mixed with 20 μl of Ni-NTA magnetic agarose beads (Qiagen) and incubated for 4–12 h at 4 °C. After extensive washing with the lysis buffer, the bound proteins were eluted with Laemmli sample buffer (Bio-Rad), separated by SDS-PAGE, and transferred onto a nitrocellulose membrane. The membrane was probed with an anti-HA antibody (Sigma), and protein signal was visualized by enhanced chemiluminescence according to the manufacturer’s instructions (Amersham Biosciences).
For in vitro protein-protein interactions, GST or GST-NKX2.5 fusion proteins were over-expressed in Escherichia coli BL21 (DE3), purified using standard protocols, and immobilized on glutathione-Sepharose beads. Aliquots (20 μl) of GST and GST-NKX2.5 beads were incubated with in vitro-translated TBX5 (described earlier) for 4 h at 4 °C. After washing with PBS buffer, bound proteins were eluted from beads with PBS containing 0.1 M glutathione and analyzed by Western blot with a monoclonal anti-His antibody (Sigma).
Immunocytochemistry
Transfected cells (NIH-3T3, COS-7, and HEK-293) were seeded on chamber slides at a density of 1 × 105 cells and incubated at 37 °C and 5% CO2 for 24–48 h. Cells were then fixed in 2% paraformaldehyde, washed in PBS, and incubated with the primary antibody (1:1000 dilution) in PBS/3% nonfat milk at 4 °C overnight. The mouse anti-FLAG M2 primary antibody recognizes the FLAG-tagged TBX5 protein. The secondary antibody, a fluorescein isothiocyanate-conjugated sheep anti-mouse IgG (1:500 dilution), was then added and incubated at room temperature for 1 h. Slides were mounted using anti-fading vectashield with DAPI and cells were viewed under a Zeiss Axioskop fluorescence microscope equipped with Photo-metrics SmartCapture. The images were analyzed with the Melanie software (Geneva Bioinformatics) for the relative distribution of the protein in the nucleus versus cytoplasm.
RESULTS
Functional Defects of TBX5 Missense Mutations in DNA Binding
Direct binding of TBX5 transcriptional factor to DNA is required for activation of its target genes including ANF encoding the atrial natriuretic factor. To identify the mechanism by which TBX5 missense mutations cause Holt-Oram syndrome, we assessed the ability of mutant TBX5 proteins to bind to its target DNA-binding site using EMSA. Seven missense mutations in TBX5 were analyzed, and they include Q49K and I54T at the N terminus, G80R, G169R, R237Q, and Q237W in the T-domain responsible for DNA binding, and S252I in the C terminus (Fig. 1). The promoter region of ANF (−252 upstream from the transcription start site) contains a TBX5-binding site, and this site was chemically synthesized as pairs of complementary single strand oligonucleotides (5′-aataTCACACCTgtac-3′; 5′-gtacAGGTGTGAtatt-3′). The synthetic site was labeled and used in EMSA with in vitro-translated TBX5 proteins. As shown in Fig. 2, the wild type TBX5 protein can bind to DNA and forms a specific DNA-protein complex. Competition experiments with unlabeled synthetic TBX5-binding site dramatically reduced the TBX5-DNA complex signal, and a synthetic DNA fragment having a sequence unrelated to the TBX5-binding site failed to form TBX5-DNA complex (data not shown). These data suggest that the observed binding of TBX5 to the synthetic binding site is specific.
Fig. 1. TBX5 missense mutations identified in patients with Holt-Oram syndrome.
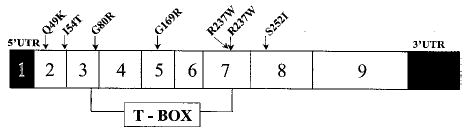
Among more than 20 TBX5 mutations, Q49K, I54T, G80R, G169R, R237Q, R237W, and S252I represent missense mutations identified in patients with Holt-Oram syndrome. TBX5 consists of 9 exons with exons 3–7 encoding the T-domain responsible for DNA binding. Note that two mutations (Q49K and I54T) are in the N terminus, four mutations (G80R, G169R, R237Q, and R237W) are within the T-box domain, and one mutation (S252I) is in the C terminus.
Fig. 2. The effect of missense mutations of TBX5 on DNA-binding activity.
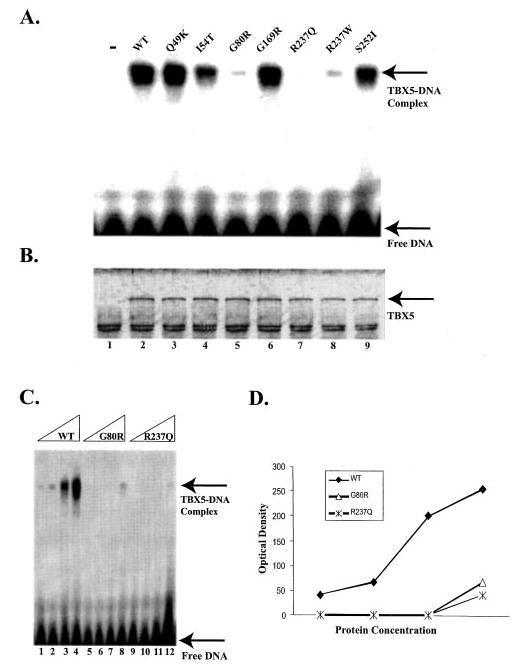
The wild type and mutant TBX5 proteins were synthesized in vitro using the TNT-coupled transcription/translation system. The TBX5-binding site is a synthetic double-stranded DNA fragment corresponding to the region from −257 to −242 bp upstream from the ANF transcriptional start site (5′-aataTCACACCTgtac-3′). A, EMSA. Lane “−,” no TBX5 protein; WT, wild type TBX5; Q49K, I54T, G80R, G169R, R237Q, R237W, and S252I, TBX5 with individual mis-sense mutations. B, a silver-stained protein gel showing the approximately equal level of synthesis of wild type and various mutant TBX5 proteins used in EMSA. Lane 1, no TBX5 expression plasmid DNA; lane 2, wild type TBX5; lanes 3–9 represent Q49K, I54T, G80R, G169R, R237Q, R237W, and S252I mutant TBX5, respectively. C, EMSA with various amounts of wild type TBX5 and mutants G80R and R237Q. D, the amount of the TBX5-DNA complex in panel C was plotted against TBX5 concentrations. The fitted slope for specific binding of wild type TBX5 (WT) and mutants G80R and R237Q is 79.5, 19.5, and 11.7, respectively.
Three mutations in the T-domain, including G80R, R237Q, and R237W, dramatically reduced the binding ability of TBX5 to DNA (Fig. 2A). The relative DNA-binding abilities of wild type and mutant G80R and R237Q TBX5 were determined using various concentrations of proteins. As shown in Fig. 2, C and D, both mutant proteins have much lower affinity to DNA than wild type TBX5. Mutations I54T and S252I had moderate effect on DNA binding of TBX5. In contrast, there was no significant difference in DNA binding between the wild type TBX5 and mutants Q49K and G169R (Fig. 2A). A silver-stained protein gel showed that both wild type and mutant TBX5 proteins were successfully synthesized at approximately equal amounts (Fig. 2B). Our results suggest that a functional defect in DNA binding is the molecular mechanism for some TBX5 mutations (G80R, R237Q, and R237W), but not a universal mechanism for all mutations.
Functional Defects of TBX5 Missense Mutations in Transcriptional Activation
TBX5 alone can activate transcription of its target genes, but in the presence of NKX2.5, synergy in transcriptional activation by these two proteins was observed (13, 20). We assessed the functional effect of TBX5 mutations on transcriptional activation using a reporter gene (ANF-Luc, Fig. 3A) in which the ANF promoter (region from −270 to +1 bp) was fused to the luciferase gene. The ANF-Luc reporter gene was co-transfected with wild type or various mutant TBX5 expression constructs into COS-7 cells. Transcriptional activity was examined and expressed as relative luciferase units. As shown in Fig. 3B, expression of wild type TBX5 activated transcription of the ANF promoter. Three mutations, including G80R, R237Q, and R237W, reduced transcription activation by TBX5, consistent with our data that these three mutations reduced DNA binding of TBX5. In contrast, TBX5 mutations Q49K, I54T, G169R, and S252I had little effect on transcriptional activation by TBX5 (Fig. 3B). Western blot analysis showed that both wild type and all mutant TBX5 proteins were successfully expressed in transfected cells (Fig. 3C).
Fig. 3. The effect of missense mutations of TBX5 on transcription activation activity in the presence or absence of NKX2.5.
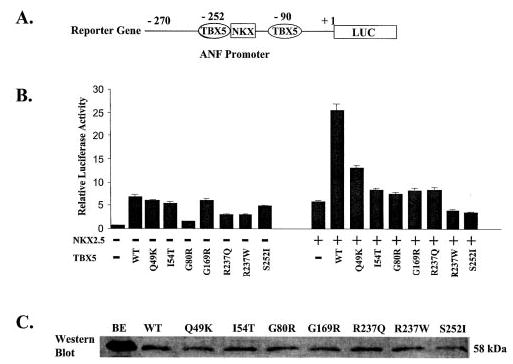
A, the reporter gene used for transcriptional activation assay. The promoter region, from −270 to +1 bp upstream from the transcriptional start site, of ANF was fused to the luciferase gene (LUC). B, transcriptional activation assay for all seven missense mutations of TBX5 in the absence (left block) and presence (right block) of NKX2.5. Transcriptional activity is shown as relative luciferase activity on the y-axis. The transcriptional activity for the vector only was set arbitrarily to 1. WT, wild type. Note that all missense mutations of TBX5 abolished synergistic transcription activation of the ANF promoter between TBX5 and NKX2.5. C, Western blot analysis to determine whether mutant TBX5 were successfully expressed in transfected COS-7 cells. BE, purified wild type TBX5 control from a bacterial over-expression system; WT, lane with lysate from COS-7 cells containing expressed FLAG-tagged wild type TBX5; Q49K, I54T, G80R, G169R, R237Q, R237W, and S252I, lanes with lysates from COS-7 cells containing expressed TBX5 proteins with corresponding mutations.
We then investigated whether the missense mutations in TBX5 could disrupt the synergistic transactivation between TBX5 and NKX2.5. As shown in Fig. 3B, either wild type TBX5 or NKX2.5 alone activated expression of the ANF promoter, but co-transfection of TBX5 and NKX2.5 into COS-7 cells showed synergistic activation of the ANF promoter as reported previously (12, 16). However, all seven missense mutations of TBX5 did not produce such synergistic effect with NKX2.5 (Fig. 3B).
Defects of TBX5 Missense Mutations in TBX5 and NKX2.5 Interaction
Because all seven TBX5 missense mutations caused loss of synergistic activation of transcription by TBX5 and NKX2.5, we determined whether these mutations affect the physical interaction between these two transcriptional factors. We used both in vivo and in vitro protein-protein interaction assays. For the in vivo assay, His-tagged TBX5 and NKX2.5 tagged with HA epitope at the C terminus were co-expressed in HeLa cells. Western blot analysis showed that both TBX5 and NKX2.5 were successfully expressed in the same cells (Fig. 4A). The protein mixture was then incubated with Ni-NTA beads (which bind His-tagged TBX5 or His-TBX5-NKX2.5-HA complex). The protein or protein-complex bound to Ni-NTA beads was eluted and analyzed by Western blot with an anti-HA antibody to detect the presence of NKX2.5. As expected, interaction between wild type TBX5 and NKX2.5 was observed (Fig. 4A, lane 2). Faint signal (very weak interaction) was observed for TBX5 proteins with mutation Q49K or G169R. No interactions were found for mutants I54T, G80R, R237Q, R237W, or S252I. These results suggest that seven missense mutations studies here dramatically reduce the interaction between TBX5 and NKX2.5.
Fig. 4. The effect of missense mutations of TBX5 on the interaction between TBX5 and NKX2.5 in vivo (A) and in vitro (B).
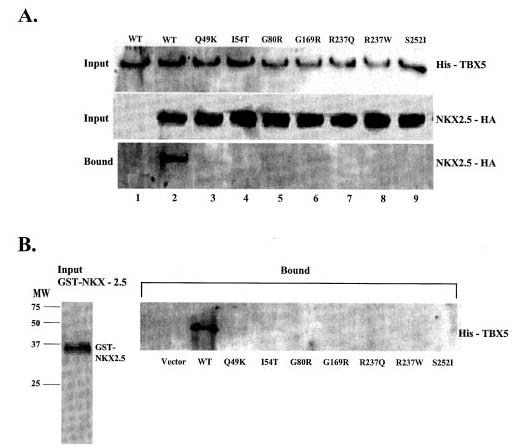
A, HeLa cells were transiently transfected with expression plasmids for His-TBX5 and NKX2.5-HA. Total cell lysates containing His-tagged TBX5 and/or HA-tagged NKX2.5 were incubated with Ni-NTA beads, separated by 12% SDS-PAGE, and analyzed by Western blot with an anti-His antibody (upper panel) or with an anti-HA monoclonal antibody (middle panel). Proteins bound to Ni-NTA beads were washed with washing buffer, eluted, and fractionated by 12% SDS-PAGE and analyzed by Western blot with anti-mouse HA for NKX2.5. Lane 1, wild type (WT) TBX5 without co-transfection of NKX2.5; lane 2, wild type TBX5 with co-transfection of NKX2.5; lanes 2–9, TBX5 with Q49K, I54T, G80R, G169R, R237Q, R237W, and S252I co-transfected with NKX2.5-HA, respectively. B, GST-NKX2.5 fusion protein and in vitro-translated His-tagged TBX5 were incubated with glutathione-Sepharose 4B beads, and proteins bound to the beads were washed with PBS, eluted, fractionated by 12% SDS-PAGE, and analyzed by Western blot with an anti-His antibody for detecting TBX5. Approximately equal amounts of wild type and various mutant TBX5 proteins were used in the GST pull-down assay as shown in the legend to Fig. 2B.
We also used GST pull-down assay to determine the interaction of various TBX5 mutants with NKX2.5. GST-NKX2.5 fusion protein was purified, immobilized to glutathione-Sepharose 4B beads, and mixed with in vitro-translated TBX5. We separated TBX5 bound to GST-NKX2.5 and analyzed it using Western blot. Wild type TBX5, but none of the mutant TBX5, showed direct physical interaction with NKX2.5 (Fig. 4B). These results further confirm the in vivo data that missense mutations severely disrupt the direct interaction between TBX5 and NKX2.5.
Defects in Nuclear Localization by TBX5 Missense Mutations
TBX5 is a transcription factor, and it is expected to be localized in the nucleus. We hypothesized that missense mutations in TBX5 may cause conformational changes of the protein and result in protein trafficking defects. Such defects will prevent TBX5 from exerting its function as a transcription factor. To test this hypothesis, we expressed wild type and mutant TBX5 proteins tagged with a FLAG-epitope into NIH-3T3 cells and studied cellular localization of TBX5 by immunofluorescence staining. The relative amount of the nuclear and cytoplasmic protein was quantified and shown in Fig. 5B. As expected, wild type TBX5 was localized completely into the nucleus (Fig. 5, WT, green signal). Mutant Q49K TBX5 exhibited a minor defect in trafficking with most of the protein molecules in the nucleus and few molecules in the cytoplasm (the protruded bud) (Fig. 5A). Mutation R237Q caused the most severe defect in TBX5 trafficking with most of the signals in the cytoplasm and few in the nucleus (Fig. 5A). The rest of mutations (I54T, G80R, G169R, R237W, and S252I) had moderate effect on nuclear localization of TBX5 with immunofluorescence signals found in both the nucleus and cytoplasm (Fig. 5A). Similar results were obtained with HEK-293 or COS-7 cells (data not show).
Fig. 5. Immunostaining of NIH-3T3 cells expressing human TBX5.
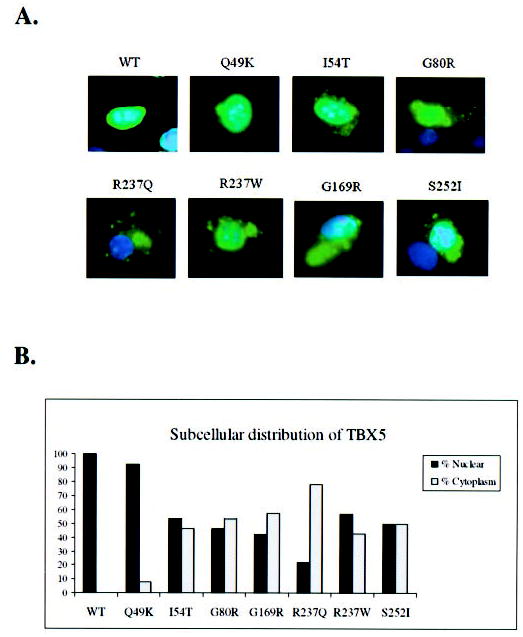
A, WT, cell transfected with the FLAG-tagged wild type TBX5 construct; Q49K, I54T, G80R, R237Q, R237W, G169R, and S252I represent cells over-expressing various mutant TBX5 proteins. Transfected cells were immunostained with anti-FLAG (green) for TBX5 and the nucleus was stained with DAPI (blue). No detectable immunofluorescence staining was observed in non-transfected cells. Note that wild type TBX5 is completely localized into the nucleus, whereas TBX5 proteins with various missense mutations are distributed in both the nucleus and cytoplasm. B, percentage of nuclear versus cytoplasm distribution of TBX5 for wild type (WT) and mutant (Q49K, I54T, G80R, G169R, R237Q, R237W, and S252I). The data were based on the images in panel A.
DISCUSSION
This study provides evidence that the seven TBX5 missense mutations identified in Holt-Oram syndrome patients are functional mutations with a spectrum of defects. We evaluated the functional effect of these missense mutations on DNA binding and transcriptional activation by TBX5 as well as synergistic activation by the interaction between TBX5 and NKX2.5. Based on our results, TBX5 missense mutations can be divided into two different functional groups. The first group consists of mutations G80R, R237W, and R237Q, which exert their effect mainly by disrupting the DNA-binding function of TBX5. Reduced DNA binding could lead to observed reduction in transcription activation and loss of synergy between TBX5 and NKX2.5 in transactivation of target genes. The second group consists of mutations Q49K, I54T, G169R, and S252I, which had no or moderate effect on DNA binding. This group of mutations did not affect the transcriptional activation by TBX5, but they caused dramatic reduction in the interaction between TBX5 and NKX2.5 and the complete loss of synergy between TBX5 and NKX2.5 in transcriptional activation.
Our study represents the most comprehensive analysis of all missense mutations identified in TBX5 to date. Two previous studies reported partial characterization of a few missense mutations in TBX5. Ghosh et al. (21) reported analysis of the effect of four point mutations, G80R, G169R, R237Q and S252I, on DNA binding to an in vitro selected fragment with two half TBX5-binding sites oriented tail-tail using a truncated form of TBX5 (amino acids 1–279). These authors, however, did not assay the effect of these mutations on transcription activation. Mutations G80R and R237Q were found to eliminate DNA binding of TBX5, whereas mutations G169R and S252I retained DNA-binding activity (21). Our DNA binding analysis with the full-length TBX5 and a native TBX5-binding site in the ANF promoter (Fig. 2) yielded similar results on mutations G80R, G169R, R237Q, and S252I. Hiroi et al. (20) analyzed the effect of mutations G80R and R237Q on transcription activity of the ANF promoter but did not examine their effect on DNA binding. Mutation G80R caused significant reduction of transcriptional activation activity of TBX5 and loss of synergistic activation with NKX2.5. Surprisingly, mutation R237Q, which eliminated DNA binding, had minor effects on transcriptional activation by TBX5 or synergistic activation by TBX5 and NKX2.5 (20) In our study, similar results were obtained for mutation G80R; however, we found much more dramatic effect by mutation R237Q, which is consistent the DNA binding data on this mutation. Moreover, we analyzed the effects of seven missense mutations on the direct interaction and synergy in transactivation between TBX5 and NKX2.5. Our study provides further definition of molecular mechanisms of TBX5 mutant protein dysfunctions.
Among seven missense mutations studied, only the three mutations in the T-domain, G80R, R237W, and R237Q, dramatically reduced binding of TBX5 to the DNA target, further confirming that the T-domain is the DNA-binding domain. It is surprising that TBX5 with mutation G169R, also at a highly conserved position in the T-domain, retained nearly full binding activity, suggesting that not all amino acid residues in the T-domain are responsible for DNA binding. Based on the x-ray crystallographic data from the T-domain of Xenopus laevis T protein (22), both Gly-80 and Arg-237 are located immediately next to the critical amino acid residues for DNA binding, whereas Gly-169 is five residues to the left and four residues to the right of the critical DNA-binding residues. The different spatial positioning in relation to critical DNA-binding residues may explain the difference of binding activity to DNA for mutations at Gly-80 and Arg-237 from that for mutation G169R.
Deletion studies implicated the N terminus of TBX5 and the N-terminal part of the T-domain (amino acids 1–90) as the binding site for NKX2.5 (21). Our results that mutations Q49K, I54T, and G80R disrupt the physical interaction and synergistic transcriptional activity between TBX5 and NKX2.5 further support that the N terminus of TBX5 is involved in its interaction with NKX2.5. Our data also implicate other regions, including residues Gly-169, Arg-237, and Ser-252, as critical residues involved in the interaction and synergistic transcription activation between TBX5 and NKX2.5. Future structure-function relationship studies and x-ray crystallographic studies with the full TBX5 and NKX2.5 will define all critical residues involved in their interaction.
Immunostaining of cells transfected with FLAG-tagged TBX5 showed that TBX5 proteins with the missense mutations were localized in both the nucleus and cytoplasm. The molecular mechanism for such trafficking defects is unknown. We speculate that TBX5 missense mutations cause improper folding of TBX5, which will prolong the time required for TBX5 to be transported into the nucleus. This slow trafficking of mutant TBX5 proteins leads to the observation of mutant TBX5 in both the nucleus and cytoplasm. Delayed entry of TBX5 molecules into the nuclei will prevent these molecules from executing their transcriptional activation function. These results demonstrate impaired nuclear localization of mutant TBX5 as a molecular mechanism for TBX5 missense mutations. Similar protein trafficking defects have been reported for other cardiovascular diseases including long QT syndrome (23) and Brugada syndrome (24).
In summary, we have shown that the missense mutations in TBX5 investigated in this study are functional mutations that cause a spectrum of biochemical and cellular defects. Our results indicate three distinct molecular mechanisms underlying the pathogenesis of Holt-Oram syndrome. First, some TBX5 mutations cause a dramatic decrease in its DNA-binding activity, which leads to reduced transcription activation by TBX5 and the loss of synergy between TBX5 and NKX2.5. Second, some TBX5 mutations do not affect the DNA binding and transcriptional activation functions of TBX5 but cause the complete loss of synergy between TBX5 and NKX2.5 in transcription activation due to dramatically reduced interaction between TBX5 and NKX2.5. Third, impaired nuclear localization is a common defect shared by all missense mutations studied here. All these mechanisms are likely central to the pathogenesis of Holt-Oram syndrome. Our data are consistent with the hypothesis that the missense mutations in TBX5 act by a loss-of-function mechanism.
Acknowledgments
We thank Dr. Rajkumar Kadaba for help and Dr. Issei Komuro for the generous gift of expression plasmids.
Footnotes
This study was supported by a Fourjay Foundation Cardiovascular Research grant (to Q. W.) and National Institutes of Health Grants R01 HL65630 and R01 HL66251 (to Q. W.).
The abbreviations used are: ANF, the atrial natriuretic factor gene; WT, wild type; Luc, luciferase; EMSA, electrophoretic mobility shift assay; HA, hemagglutinin; GST, glutathione S-transferase; PBS, phosphate-buffered saline; Ni-NTA, nickel-nitrilotriacetic acid; DAPI, 4′,6-diamidino-2-phenylindole.
References
- 1.Papaioannou VE. Int Rev Cytol. 2001;207:1–70. doi: 10.1016/s0074-7696(01)07002-4. [DOI] [PubMed] [Google Scholar]
- 2.Basson CT, Bachinsky DR, Lin RC. Nat Genet. 1997;15:30–35. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- 3.Li QY, Newbury-Ecob RA, Terrett JA. Nat Genet. 1997;15:21–29. doi: 10.1038/ng0197-21. [DOI] [PubMed] [Google Scholar]
- 4.Bamshad M, Aoki VT, Adkison MG, Warren WS, Bartness TJ. Nat Genet. 1997;16:311–315. [Google Scholar]
- 5.Braybrook C, Doudney K, Marcano AC, Arnason A, Bjornsson A, Patton MA, Goodfellow PJ, Moore GE, Stanier P. Nat Gene. 2001;29:179–183. doi: 10.1038/ng730. [DOI] [PubMed] [Google Scholar]
- 6.Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 7.Jerome LA, Papaioannou VE. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 8.Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 9.Sepulveda JL, Vlahopoulos S, Iyer D, Belaguli N, Schwartz RJ. J Biol Chem. 2002;277:25775–25782. doi: 10.1074/jbc.M203122200. [DOI] [PubMed] [Google Scholar]
- 10.Liberatore CM, Searcy-Schrick RD, Yutzey KE. Dev Biol. 2000;223:169–180. doi: 10.1006/dbio.2000.9748. [DOI] [PubMed] [Google Scholar]
- 11.Hatcher CJ, Kim MS, Mah CS. Dev Biol. 2001;230:177–188. doi: 10.1006/dbio.2000.0134. [DOI] [PubMed] [Google Scholar]
- 12.Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, Seidman CE. Dev Biol. 1999;211:100–108. doi: 10.1006/dbio.1999.9298. [DOI] [PubMed] [Google Scholar]
- 13.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. Cell. 2001;106:709–721. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 14.Holt M, Oram S. Br Heart J. 1960;22:236. doi: 10.1136/hrt.22.2.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newbury-Ecob RA, Leanage R, Raeburn JA, Young ID. J Med Genet. 1996;33:300–307. doi: 10.1136/jmg.33.4.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basson CT, Huang T, Lin RC, Bachinsky DR, Weremowicz S, Vaglio A, Bruzzone R, Quadrelli R, Lerone M, Romeo G, Silengo M, Pereira A, Krieger J, Mesquita SF, Kamisago M, Morton CC, Pierpont ME, Muller CW, Seidman JG, Seidman CE. Proc Natl Acad Sci U S A. 1999;96:2919–2924. doi: 10.1073/pnas.96.6.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cross SJ, Ching YH, Li QY, Armstrong-Buisseret L, Spranger S, Lyonnet S, Bonnet D, Penttinen M, Jonveaux P, Leheup B, Mortier G, van Ravenswaaij C, Gardiner CA. J Med Genet. 2000;37:785–787. doi: 10.1136/jmg.37.10.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Hu D, Xia J, Yang Y, Ying B, Hu J, Zhou X. Am J Med Genet. 2000;92:237–240. doi: 10.1002/(sici)1096-8628(20000605)92:4<237::aid-ajmg2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 19.Vaughan CJ, Basson CT. Am J Med Genet. 2000;97:304–309. doi: 10.1002/1096-8628(200024)97:4<304::aid-ajmg1281>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I. Nat Genet. 2001;28:276–280. doi: 10.1038/90123. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh TK, Packham EA, Bonser AJ, Robinson TE, Cross SJ, Brook JD. Hum Mol Genet. 2001;10:1983–1994. doi: 10.1093/hmg/10.18.1983. [DOI] [PubMed] [Google Scholar]
- 22.Muller CW, Hermann BG. Nature. 1997;389:884–888. doi: 10.1038/39929. [DOI] [PubMed] [Google Scholar]
- 23.Bianchi L, Shen Z, Dennis AT, Priori SG, Napolitano C, Ronchetti E, Bryskin R, Schwartz PJ, Brown AM. Hum Mol Genet. 1999;8:1499–1507. doi: 10.1093/hmg/8.8.1499. [DOI] [PubMed] [Google Scholar]
- 24.Baroudi G, Acharfi S, Larouche C, Chahine M. Circ Res. 2000;90:e11–e16. [PubMed] [Google Scholar]