

Abstract
Purpose of review
Coronary artery disease, including its most severe complication myocardial infarction, is the leading cause of death; however, its genetic studies lag behind other diseases. Many advances have recently been made, however, and these are reviewed here.
Recent findings
Positional cloning based on genome-wide linkage analysis with large families identified the first non – lipid-related disease-causing gene, MEF2A (encoding a transcriptional factor), for coronary artery disease and myocardiaI infarction. The MEF2A mutations may account for up to 1.93% of the disease population; thus, genetic testing based on mutational analysis of MEF2A may soon be available for many coronary artery disease/myocardial infarction patients. Genome-wide association studies identified significant association for myocardiaI infarction with the LTA gene (encoding lymphotoxin-α), and a follow-up study found that an LTA-interacting gene, LGALS2 (encoding galectin-2), is also a susceptibility gene for myocardiaI infarction. Studies that employ genome-wide linkage scans with hundreds of small nuclear families have identified new susceptibility genes for coronary artery disease and myocardiaI infarction, including ALOX5AP (encoding 5-lipoxygenase-activating protein) associated with myocardial infarction and stroke and PDE4D (encoding phosphodiesterase 4D) for ischemic stroke.
Summary
Genetic studies provide new insights into the pathogenesis of coronary artery disease and myocardial infarction. Future studies will focus on identification of new disease-causing genes and susceptibility genes, exploration of the molecular mechanisms by which mutations cause coronary artery disease/myocardiaI infarction, and gene-specific therapies for patients.
Keywords: atherosclerosis, coronary artery disease, genetics, myocardial infarction, sudden death
Abbreviations: adCAD1 the first genetic locus for autosomal dominant form of coronary artery disease, CAD coronary artery disease, FCHL familial combined hyperlipidemia, FLAP 5-lipoxygenase-activating protein, HDL high-density lipoprotein, LDL low-density lipoprotein, LOD log base 10 of the likelihood ratio under the hypotheses of linkage and nonlinkage, LQTS long QT syndrome, MI myocardial infarction, SNP single-nucleotide polymorphism
Introduction
In the United States alone, more than 300,000 people die suddenly from a cardiac cause each year. Coronary artery disease (CAD) represents the most important cause of sudden cardiac death. More than 80% of sudden cardiac deaths are caused by atherosclerotic CAD, and the remaining 20% of cases are caused by other diseases including cardiomyopathies, left ventricular hypertrophy, long QT syndrome (LQTS), Brugada syndrome, aortic valve disease, and other cardiac disorders [1]. In the past decade, many research groups including the author’s group have contributed to many exciting advances in the genetic studies of cardiovascular disease. To date, disease-causing genes have been identified for estimated 50–70% of cases of hypertrophic cardiomyopathy, 20% of dilated cardiomyopathy, 50–75% of LQTS, 25% of Brugada syndrome, and some cases of congenital heart disease. In the case of LQTS and Brugada syndrome, commercial genetic testing is now available. Most importantly, genetics of LQTS and Brugada syndrome is leading a paradigm shift in cardiovascular medicine, known as the ‘right drug/therapy for the right patient’ or ‘personalized medicine.’ For example, LQTS patients with potassium channel KCNQ1 mutations respond to β-blockade; those with potassium channel KCNH2 mutations respond to β-blockade and increased serum potassium; sodium channel SCN5A-positive LQTS patients respond to sodium channel blockers; and symptomatic LQTS and Brugada syndrome patients (SCN5A positive) will benefit from implantation of implantable cardioverter-defibrillators [2–4].
Genetic studies of CAD and MI are lagging behind other cardiovascular disorders. The major reason for the limited success in the field of CAD/MI genetics is that CAD or MI is a complex disease, which is believed to be caused by many genetic factors, environmental factors, and interactions among these factors. Indeed, many risk factors have been identified for CAD/MI, including smoking, advanced age, male gender, diabetes mellitus, high systolic blood pressure, personal history of angina pectoris, family history of CAD or MI, high-fat diet, infectious agents, obesity, increased plasma total and low-density lipoprotein (LDL) cholesterol, increased plasma triglycerides, and decreased plasma high-density lipoprotein (HDL) cholesterol [5–11]. Among these factors, family history is one of the most significant independent risk factor for CAD/MI [5,12]. Twin studies also suggest that genetic factors contribute to the development of CAD and MI [13]. All these support the hypothesis that genetic factors contribute to the development of CAD and MI. A high death rate, late-onset characteristics of CAD and MI, and complications by phenocopy in families also present major challenges in genetic dissection of this important disease.
The most frequently used method for identifying the susceptibility genes for CAD and MI has been candidate gene case-control association studies. Methodologically, this is the easiest approach by which a candidate gene is selected based on its potential involvement in CAD/MI. Single-nucleotide polymorphisms (SNPs) are identified in the gene and genotyped in a group of patients (cases) and controls. The frequencies of SNP alleles or genotypes are then analyzed. An allele or genotype is associated with the disease if its occurrence in the cases is significantly different from that in the controls. The results from the candidate gene association studies should be interpreted with caution as many of these studies are compounded by the selection bias of cases and controls, population admixture, imperfect matching of cases with controls, phenotyping errors, and small sample sizes in some studies. Numerous case-control association studies were carried out and variants in many genes have been implicated in increasing or decreasing the susceptibility to CAD or MI (for reviews, see Shen et al. [7], Wang and Pyeritz [9], and Wang and Chen [10,11]). Inconsistent results among different studies and a lack of replication in most of the numerous possible associations with CAD and MI, however, lead to debate about the true association of some genes with the disease.
Despite the pessimism discussed above, exciting discoveries have been made in the field of CAD/MI genetics in the past couple of years and are advancing the field significantly. Significant progress has been made in mapping and identifying disease-causing genes and susceptibility genes for CAD or MI using genome-wide linkage analysis with large families or hundreds of small nuclear families and genome-wide association studies. This article reviews advances of the molecular genetics of CAD and MI since 2003.
Disease-causing genes for coronary artery disease and myocardial infarction
We can classify the genes that are associated with complex human diseases such as CAD and MI into three major categories: disease-causing genes, susceptibility genes, and disease-linked genes. Disease-causing genes are the genes that are directly responsible for the pathogenesis of disease when mutated. In this case, the mutations are clearly defined or well established as the primary cause of the disease. For example, mutations in potassium channel genes KCNQ1 and KCNH2 cause LQTS [14,15] and cardiac sodium channel gene SCN5A mutations cause Brugada syndrome [16]. Disease-causing genes have great predictive values and can be used directly for genetic testing. Susceptibility genes are the genes that increase or decrease the risk of developing disease and may or may not cause the disease in the context of other genetic and environmental factors. Mutations or SNPs in these genes are present in both normal and diseased populations, but the frequencies differ in the two populations. For individuals, susceptibility genes have less predictive value for the development and prognosis of the disease. Disease-linked genes are the genes that are connected to the disease by molecular biologic, microarray, or proteomic analyses, but their relation to the disease as a cause or a consequence is not established. Some disease-linked genes may serve as biomarkers for the disease. The disease-causing genes for CAD and MI are discussed in this section, and susceptibility genes and CAD/MI – linked genes are discussed in the next two sections.
Disease-causing genes for disorders in lipid metabolism, a high risk factor for coronary artery disease
Disease-causing genes have been identified for familial hypercholesterolemia and Tangier disease, both of which increase risk of premature arteriosclerosis and CAD. Hypercholesterolemia is characterized by elevation in levels of plasma cholesterol bound to LDL, tendon xanthomas, and atheroma and can be caused by mutations in the LDL receptor (LDLR) gene [17], the ApoB-100 gene [18], the proprotein convertase subtilisin/kexin-type 9 gene (PCSK9) [19,20], the cholesterol 7-α-hydroxylase gene (CYP7A1) [21], and the ARH gene [22]. Tangier disease is a rare disorder characterized by the absence of HDL cholesterol from plasma and deposition of cholesterol esters in the reticuloendothelial system, with splenomegaly and enlargement of tonsils and lymph nodes, hepatosplenomegaly, and peripheral neuropathy [23,24]. The gene for Tangier disease has been identified as ABCA1, which encodes a member of the ATP-binding cassette transporter family involved in the efflux of lipids from peripheral cells to ApoA-I forming nascent HDL particles [23,24].
Disease-causing genes for coronary artery disease and myocardial infarction
Only one disease-causing gene, MEF2A, encoding a member of the MEF2 family of transcription factors, has been identified for primary CAD and MI without other accompanying clinical features like hypercholesterolemia and Tangier disease (Table 1) [25–31]. This discovery was selected as one of the American Heart Association’s top 10 advances for 2004 (http://www.americanheart.org/presenter.jhtml?identifier=3027694). We hypothesized that in single large families with CAD and MI, the disease behaves as a monogenic disorder and the disease-causing gene can be mapped and identified by typical positional cloning with model-based linkage analysis. To test this hypothesis, we performed a genome-wide linkage scan in a large family with 13 patients who displayed an autosomal dominant inheritance pattern of CAD and MI. The study led to the localization of the first genetic locus for autosomal dominant CAD and MI on chromosome 15q26 (adCAD1) [25]. Further molecular genetic studies established MEF2A as the gene causing CAD and MI in the family, and a 21-basepair deletion was identified in the last exon of MEF2A (exon 11) in all affected members in the family but not in the family members with normal phenotypes or in 119 individuals with normal angiograms [25]. Functional studies demonstrated that the 21-basepair deletion disrupts the transcriptional activation activity of MEF2A by a dominant-negative mechanism.
Table 1.
Major disease-causing and susceptibility genes for patients with coronary artery disease and myocardial infarction
| Chromosomal location | Gene (function) | Phenotype | Study | |
|---|---|---|---|---|
| Disease-causing genses | ||||
| adCAD1 (autosomal dominant CAD locus 1) | 15q26 | MEF2A (transcription factor in endothelium of coronary arteries) | CAD, MI | [25,26••] |
| adCAD2 | ? | ? | ? | ? |
| Susceptibility genes | ||||
| 1 | 1p34–36 | ? | MI | [27] |
| 2 | 2q21.1–22 | ? | CAD | [28] |
| 3 | 2q36–37 | ? | Acute coronary syndrome | [29] |
| 4 | 3q13 | ? | CAD | [30] |
| 5 | 5q12 | PDE4D (phosphodiesterase 4D, cAMP signaling, inflammation) | Ischemic stroke | [31,32] |
| 6 | 6p21 | LTA (lymphotoxin-α, cytokine, inflammation, immune response) | MI | [33] |
| 7 | 13q12–13 | ALOX5AP (FLAP, generating leukotrienes, inflammatory mediators) | MI, stroke | [34••] |
| 8 | 14q | ? | MI | [35] |
| 9 | 16p13-ter | ? | CAD | [36] |
| 10 | 22q13.1 | LGALS2 (galectin-2, regulates secretion of LTA; inflammation) | MI | [37••] |
| 11 | Xq23–26 | ? | CAD | [28] |
CAD, coronary artery disease; LTA, lymphotoxin-α; MI, myocardial infarction; PDE4D, phosphodiesterase 4D.
In a follow-up study, we performed mutation analysis in MEF2A with 207 independent CAD/MI patients and 191 controls with normal angiograms. Three new MEF2A mutations were identified in four patients, all in exon 7, but no mutations were found in the normal population [26••]. Different from the 21-basepair deletion identified in the earlier report, the three new mutations suppress transcriptional activation activity of MEF2A by a loss-of-function mechanism and are associated with a less severe form of CAD. The results suggest that up to 1.93% of all CAD/MI patients may carry a mutation in MEF2A, which makes genetic testing for heart disease a step closer to reality. It is important to point out, however, that the current study is still a small study with just over 200 patients, and more studies with large samples are needed to validate the frequency of MEF2A mutations in the CAD/MI population.
The significance of identification of MEF2A as the first disease-causing gene for CAD and MI is twofold. First, it makes genetic testing possible for many individuals with a very high risk for CAD and MI. Aggressive lifestyle modifications and pharmacologic strategies may be used to delay or prevent the development of MI in the gene carriers. Second, it provides a molecular mechanism for the pathogenesis of CAD and MI. The results indicate that an early step in the development of CAD and MI may involve deregulation of specific transcriptional programs in the endothelium of coronary arteries. Our immunostaining study with an antibody for MEF2A revealed that the MEF2A protein is highly expressed in the endothelium [25]. The endothelium plays a protective role for coronary arteries and prevents the arteries from damage by blood elements such as platelets and monocytes. Inflammation mediated by adhesion of monocytes to the endothelium is considered to be critical to the pathogenesis of CAD, and defective or abnormally developed vascular endothelium will be more susceptible to inflammation and the formation of an atherosclerotic plaque, which may result in thrombosis, MI, and sudden death [25].
Our views of the pathophysiology of CAD and MI have evolved over time (Fig. 1). In 1970s, the breakthrough discovery that mutations in the LDL receptor cause hyper-cholesterolemia established a firm connection between lipids and CAD, and the disease was then considered as a disease of lipid metabolism. In the 1980s, a frequent observation of restenosis in the clinical setting prompted the view that CAD/MI is a disease of smooth muscle cell proliferation. In the 1990s, identification of inflammatory cells and other inflammatory molecules in the plaques from human patients led to the prevailing view that CAD/MI is a disease of inflammation and immune responses. The recent discovery of MEF2A as a disease-causing gene for CAD and MI and its high expression in the endothelium lead us to hypothesize that an early trigger for the pathogenesis of CAD and MI may be dysfunction or abnormal development of the endothelium, which increases susceptibility of the coronary arteries to inflammation, leading to the development of CAD and MI. It is important to note that the last view remains a hypothesis that needs further validation with more molecular biologic and genetic studies.
Figure 1. Evolution of our views of the pathophysiology of atherosclerosis (coronary artery disease and myocardial infarction).
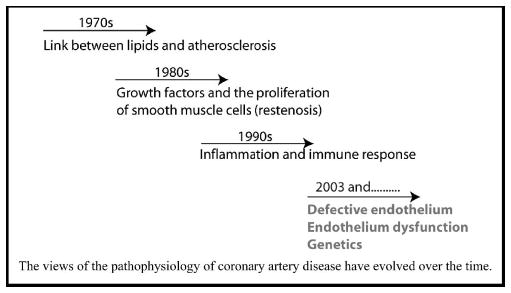
Views of the pathophysiology of coronary artery disease have evolved over time.
Our laboratory has recently studied several other large families with CAD and MI. Linkage analysis with these families excluded MEF2A as the disease-causing genes in these specific families (data not shown), which suggests that CAD/MI is genetically heterogeneous and more disease-causing genes (adCAD2 and others; Table 1) are present and await to be mapped and identified.
Susceptibility genes for coronary artery disease and myocardial infarction
As discussed above, numerous case-control studies have been carried out to identify susceptibility genes for CAD and MI. Genetic variants or SNPs in many candidate genes with physiologic relevance to CAD and MI have been found to be associated with increased or decreased risks for CAD and MI. Numerous possible susceptibility genes have been identified for CAD and MI [7,10]. Many of these studies need to be further validated or replicated as false-positive results can be generated easily in a case-control association study due to selection bias of cases and controls and population admixture. This article reviews only susceptibility genes identified by genome-wide association studies and by positional cloning based on genetic linkage analysis with small nuclear families.
Transcriptional factor gene USF1 and familial combined hyperlipidemia
Familial combined hyperlipidemia (FCHL) is present in about 20% of patients with CAD and is characterized by elevated serum total cholesterol or triglycerides. A major susceptibility locus for FCHL was mapped to chromosome 1q21–23 in a Finnish population [38]. Recently, two synonymous SNPs in the USF1 gene were found to be significantly associated with FCHL (P = 0.00002) [39•]. USF1 encodes a transcriptional factor belonging to the basic helix-loop-helix leucine zipper family and regulates genes involved in glucose and lipid metabolism, including ABCA1 and apolipoproteins CIII, AII, and E [39•]. The characteristics of the downstream genes regulated by USF1 make it an attractive gene for the pathogenesis of FCHL; however, it remains to be determined whether the two SNPs associated with FCHL are the true causative variants, a common problem with association studies.
Cytokine gene lymphotoxin-α and myocardial infarction
Development of a high-density SNP map covering the entire human genome makes it possible to identify susceptibility genes for complex disease such as CAD and MI using genome-wide association studies. For a genome-wide association study, 50,000–100,000 SNPs are estimated to be sufficient to provide genome coverage, and a P value of <5 × 10−7 was proposed to be a cutoff value for achieving significance [40]. The first genome-wide case-control association study for CAD and MI was carried out using 92 788 gene-based SNPs with 94 Japanese patients with MI. Positive SNPs with a nominal significance P value of 0.01 were then genotyped in 1133 MI cases and 1006 controls [33]. Three SNPs in the LTA gene (exon 1 10G/A, intron 1 252A/G, exon 3 p.Thr26Asn) were found to be significantly associated with a high risk of MI when they were homozygous (odds ratio = 1.69–1.78; P = 2.2 × 10−5 to 3.3 × 10−6) [33]. Lymphotoxin-α is a cytokine that mediates immune responses and inflammation. The functional effects of the three LTA SNPs associated with MI were examined with some biologic assays. SNP 252A/G in intron 1, but not SNP 10G/A in exon 1, increased expression of LTA by 1.5-fold. SNP p.Thr26Asn in exon 3 increased expression of adhesion molecules and cytokines including vascular cell adhesion molecule-1, intercellular adhesion molecule-1, tumor necrosis factor, interleukin-1A, interleukin-1B, and selectin E by twofold [33]. The results from functional studies strengthen the conclusion that LTA variants increase susceptibility to MI.
Although the P value of 3.3 × 10−6 from the Japanese study does not reach the proposed cutoff value for significance for a genome-wide association study, it is one of the most impressive P values that was ever achieved for case-control association studies for CAD/MI. Interestingly, an independent case-control association study was carried out with 1891 MI patients and 1798 controls, also from a Japanese population, but failed to identify any association between SNP 252A/G in intron 1 or p.Thr26Asn in exon 3 of the LTA gene and MI [41]. SNP 252A/G in in-tron 1 was also analyzed in a German population, and no association was detected with CAD or MI or with the risk of restenosis, death, or MI after coronary artery stenting [42,43]. On the positive side, studies of more than 400 parental-proband trios families showed positive association of SNP p.Thr26Asn in exon 3 with CAD in white Europeans [44]. Clearly, more research, both genetic and functional studies, is needed to further test the association between LTA SNPs and CAD/MI.
Lymphtoxin-α-regulatory gene LGALS2 and myocardial infarction
Ozaki et al. [37••] performed a study to identify proteins that interact with LTA and found that galectin-2 (encoded by the LGALS2 gene), a member of the galactose-binding lectin family, binds to LTA and regulates the extracellular secretion of LTA. One SNP, 3279C/T in intron 1, of LGALS2 was found to be significantly associated with MI (odds ratio = 1.57, P = 2.6 × 10−6), and the minor allele has a protective role against MI. Functional studies showed that the minor allele T reduced expression of LGALS2 by 50%. Reduced expression of galectin-2 is expected to decrease the extracellular level of LTA, leading to less inflammation and reduced risk for MI. It will be interesting to test whether the association between LGALS2 and MI can be replicated in another independent Japanese population or other populations.
ALOX5AP (encoding 5-lipoxygenase-activating protein) and myocardial infarction and stroke
A genome-wide linkage scan with 296 Icelandic families identified a suggestive linkage to MI on chromosome 13q12-13 in women — LOD score (log base 10 of the likelihood ratio under the hypotheses of linkage and nonlinkage) = 2.86 [34••] – but not in men (LOD score = 1.51). ALOX5AP encoding 5-lipoxygenase-activating protein (FLAP) is located in the region and became a good candidate gene. In a case-control association study with 779 MI patients and 624 controls, a haplotype (HapA) involving four SNPs in ALOX5AP was found to be significantly associated with MI (relative risk of 1.80, P = 2.3 × 10−5, which remains significant, 0.005, after adjusted for the number of haplotypes tested) and also associated with stroke to a lesser degree [34••]. A different haplotype, HapB, involving four other SNPs of ALOX5AP was significantly associated with MI in a British population (relative risk of 1.95, P = 3.7 × 10−4), which provides supportive evidence for the association between ALOX5AP and MI. Before the ALOX5AP report, the gene for 5-lipoxygenase (5-LO) was already shown to be involved in susceptibility to atherosclerosis in mice [45] and was associated with carotid intimal-medial thickness in a cohort of 470 healthy, middle-aged women and men from the Los Angeles Atherosclerosis Study [46]. 5-Lipoxygenase is an enzyme that mediates the production of leukotrienes, which are inflammatory mediators generated from arachidonic acid. All these studies suggest that the 5-lipoxygenase pathway plays an important role in increasing susceptibility to CAD and MI. It is important to note, however, that when the data from the ALOX5AP study were analyzed separately for women where the original linkage was identified, the significance level of the association with MI was reduced to 9.8 × 10−3, which may become nonsignificant after adjusting for the number of haplotypes tested [34••]. This raises a possibility that there may be another gene that increases susceptibility to MI in women under the 13q12–13 linkage peak.
Phosphodiesterase 4D gene and stroke
A genome-wide linkage scan in an Icelandic population mapped a susceptibility locus for stroke on chromosome 5q12 (LOD score = 4.40) [31]. The susceptibility gene at this locus was identified as PDE4D, a gene that encodes the phosphodiesterase 4D [32•]. The strongest association was identified between different haplotypes of PDE4D and combined carotid and cardiogenic strokes, the forms of stroke related to atherosclerosis. The role of PDE4D in ischemic stroke is not clear, but the PDE4D protein degrades the second messenger cAMP, a key signaling molecule involved in inflammatory responses of vascular cells to oxidized lipids [32•]. The link between PDE4D and stroke is also supported by the finding that PDE4D is involved in susceptibility to ischemic brain damage in animal models [47]. As with other case-control association studies, replication in an independent population will strengthen the association between PDE4D and stroke.
Two other genetic loci have been identified for MI on chromosomes 1p and 14q, and four significant linkages were reported for CAD on chromosomes 2q, 3q, 16p, and Xq (Table 1), but the specific genes at these loci remain to be identified.
Disease-linked genes for coronary artery disease and myocardial infarction
New genomic and proteomic approaches have begun to identify genes whose expression is linked to CAD and MI. Microarray analysis allows simultaneous analysis of expression of thousands of genes in CAD tissues vs non-CAD tissues. As an example, expression of 49 genes was newly linked to CAD, and these genes include intercellular adhesion molecule-2, PIM2, ECGF1, fusin, B cell activator (BL34, GOS8), Rho GTPase activating protein-4, retinoic acid receptor responder, β2-arrestin, and many others [48]. Many other genes have been linked to CAD by microarray analysis and have been extensively reviewed [49].
The first proteomic study of CAD was reported recently by the author’s group [50]. Proteins from CAD and non-CAD coronary arteries were separated by two-dimensional gel electrophoresis. Protein spots that showed different expression levels in two tissues were excised from the gels and identified by mass spectrometric analysis. The results from the two-dimensional gel analysis were confirmed by Western blot analysis. Expression of the ferritin light chain was found to be significantly increased in the diseased coronary arteries by about twofold. These results link the ferritin light chain gene to CAD and supports the ‘iron hypothesis’ that proposes an association between excessive iron storage and a high risk of CAD. It remains to be determined whether an elevated ferritin level is a contributor or causative factor for atherosclerotic CAD or is merely associated with the disease process. Nevertheless, increased ferritin expression in coronary arteries may become a significant biomarker for atherosclerotic CAD and may be developed as a diagnostic marker for the disease with more studies in the future.
Conclusion
Significant advances have been achieved in the genetic studies of CAD and MI in the past 2 years. These studies have provided fundamental insights into the pathogenesis of CAD and MI. First, genetic linkage studies with single large families appear to be a very effective approach for identifying the disease-causing genes for CAD and MI. The identification of the first disease-causing gene, MEF2A, for autosomal dominant CAD and MI revealed a new signaling pathway for the pathogenesis of CAD and MI and implicated abnormal function or development of the endothelium as an early trigger for the disease. In the next 5–10 years, we will witness the identification of other new disease-causing genes for CAD and MI using linkage analysis with large families. Furthermore, future studies will also focus on identifying the detailed molecular mechanisms by which disease-causing genes promote development of CAD and MI.
Second, genome-wide association studies and linkage analysis with hundreds of small nuclear families have identified new susceptibility genes for atherosclerosis, CAD, and MI, including LTA and LGALS2 for MI, ALOX5AP for MI and stroke, and PDE4D for ischemic stroke. Future studies will focus on identification of new susceptibility genes and on replication studies of known genes in different populations.
Third, new genomic and proteomic approaches will continue to be employed to identify genes whose expression is linked to CAD and MI. These studies generate novel hypotheses related to the pathogenesis of CAD and MI, and follow-up studies with a variety of studies including molecular biology, cell biology, and animal models will be used to identify new molecular determinants and novel molecular mechanisms for the pathogenesis of CAD and MI.
Future genetic studies will promise to revolutionize the early diagnosis, treatment, and prevention of CAD and MI. A unique advantage for the management of cardiovascular disease is that a significant number of cases are potentially preventable. The early diagnosis by genetic testing will force lifestyle modifications in individuals with risk genetic factors, which alone or in combination with other therapeutic options may delay the onset of the disease or prevent MI. A genetic testing kit for CAD and MI can now be easily developed based on MEF2A and other new disease-causing genes, which may identify many patients with a very high risk of developing CAD and MI. New drugs can also be developed using CAD and MI genes as targets, which may drive the paradigm shift in modern medicine to personalized medicine, i.e., the right medicine/therapy for the right patient.
Footnotes
This study was supported by NIH grants R01 HL73817, R01 HL65630, R01 HL66251, the Chinese Ministry of Science and Technology National High Technology 863 Project No 2002BA711A07, and an American Heart Association Established Investigator award (all to Q W).
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Pyeritz RE, Seidman CE, Basson CT. Genetic studies of myocardial and vascular disease. In: Topol EJ, editor. Textbook of cardiovascular medicine. edn 2. Philadelphia: Lippincott Williams & Wilkins; 2002. pp. 1967–1989. [Google Scholar]
- 3.Yong S, Tian X, Wang Q. LQT4 gene: the missing ankyrin. Mol Interv. 2003;3:131–136. doi: 10.1124/mi.3.3.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vincent GM. The molecular genetics of the long QT syndrome: genes causing fainting and sudden death. Annu Rev Med. 1998;49:263–274. doi: 10.1146/annurev.med.49.1.263. [DOI] [PubMed] [Google Scholar]
- 5.Colditz GA, Stampfer MJ, Willett WC, et al. A prospective study of parental history of myocardial infarction and coronary heart disease in women. Am J Epidemiol. 1986;123:48–58. doi: 10.1093/oxfordjournals.aje.a114223. [DOI] [PubMed] [Google Scholar]
- 6.Lewis D, Wang Q, Topol EJ. Ischaemic heart disease. Nature Encyclopedia of Life Sciences. 2002;10:508–515. [Google Scholar]
- 7.Shen G, Archacki SR, Wang Q. The molecular genetics of coronary artery disease and myocardial infarction. Acute Coronary Syndrome. 2004;6:129–141. [Google Scholar]
- 8.Slack J, Evans KA. The increased risk of death from ischaemic heart disease in first degree relatives of 121 men and 96 women with ischaemic heart disease. J Med Genet. 1966;2:239–257. doi: 10.1136/jmg.3.4.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q, Pyeritz RE. Molecular genetics of cardiovascular disease. In: Topol EJ, editor. Textbook of cardiovascular medicine. edn 1. New York: Lippincott Williams & Wilkins; 2000. pp. 1–12. [Google Scholar]
- 10.Wang Q, Chen Q. Cardiovascular disease and congenital defects. Nature Encyclopedia of Life Sciences. 2000;3:646–657. [Google Scholar]
- 11.Wang Q, Chen Q. Cardiovascular disease and congenital heart defects. Nature Encyclopedia of Human Genome. 2003;1:396–411. [Google Scholar]
- 12.Schildkraut JM, Myers RH, Cupples LA, et al. Coronary risk associated with age and sex of parental heart disease in the Framingham Study. Am J Cardiol. 1989;64:555–559. doi: 10.1016/0002-9149(89)90477-3. [DOI] [PubMed] [Google Scholar]
- 13.Marenberg ME, Risch N, Berkman LF, et al. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–1046. doi: 10.1056/NEJM199404143301503. [DOI] [PubMed] [Google Scholar]
- 14.Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac ar-rhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 16.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 17.Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24:133–170. doi: 10.1146/annurev.ge.24.120190.001025. [DOI] [PubMed] [Google Scholar]
- 18.Lund-Katz S, Laplaud PM, Phillips MC, Chapman MJ. Apolipoprotein B- 100 conformation and particle surface charge in human LDL subspecies: implication for LDL receptor interaction. Biochemistry. 1998;37:12867–12874. doi: 10.1021/bi980828m. [DOI] [PubMed] [Google Scholar]
- 19.Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 20.Ouguerram K, Chetiveaux M, Zair Y, et al. Apolipoprotein B100 metabolism in autosomal-dominant hypercholesterolemia related to mutations in PCSK9. Arterioscler Thromb Vasc Biol. 2004;24:1448–1453. doi: 10.1161/01.ATV.0000133684.77013.88. [DOI] [PubMed] [Google Scholar]
- 21.Pullinger CR, Eng C, Salen G, et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109–117. doi: 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia CK, Wilund K, Arca M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292:1394–1398. doi: 10.1126/science.1060458. [DOI] [PubMed] [Google Scholar]
- 23.Bodzioch M, Orso E, Klucken J, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 24.Brooks-Wilson A, Marcil M, Clee SM, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 25.Wang L, Fan C, Topol SE, et al. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science. 2003;302:1578–1581. doi: 10.1126/science.1088477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26••.Bhagavatula MR, Fan C, Shen GQ, et al. Transcription factor MEF2A mutations in patients with coronary artery disease. Hum Mol Genet. 2004;13:3181–3188. doi: 10.1093/hmg/ddh329. This paper reports that up to 2% of CAD/MI patients may carry a mutation in MEF2A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q, Rao S, Shen GQ, et al. Premature myocardial infarction novel susceptibility locus on chromosome 1P34-36 identified by genomewide linkage analysis. Am J Hum Genet. 2004;74:262–271. doi: 10.1086/381560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pajukanta P, Cargill M, Viitanen L, et al. Two loci on chromosomes 2 and X for premature coronary heart disease identified in early- and late-settlement populations of Finland. Am J Hum Genet. 2000;67:1481–1493. doi: 10.1086/316902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrap SB, Zammit KS, Wong ZY, et al. Genome-wide linkage analysis of the acute coronary syndrome suggests a locus on chromosome 2. Arterioscler Thromb Vasc Biol. 2002;22:874–878. doi: 10.1161/01.atv.0000016258.40568.f1. [DOI] [PubMed] [Google Scholar]
- 30.Hauser ER, Crossman DC, Granger CB, et al. A genomewide scan for early-onset coronary artery disease in 438 families: the GENECARD study. Am J Hum Genet. 2004;75:436–447. doi: 10.1086/423900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gretarsdottir S, Sveinbjornsdottir S, Jonsson HH, et al. Localization of a susceptibility gene for common forms of stroke to 5q12. Am J Hum Genet. 2002;70:593–603. doi: 10.1086/339252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gretarsdottir S, Thorleifsson G, Reynisdottir ST, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003;35:131–138. doi: 10.1038/ng1245. [DOI] [PubMed] [Google Scholar]
- 33.Ozaki K, Ohnishi Y, Iida A, et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet. 2002;32:650–654. doi: 10.1038/ng1047. [DOI] [PubMed] [Google Scholar]
- 34••.Helgadottir A, Manolescu A, Thorleifsson G, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36:233–239. doi: 10.1038/ng1311. This paper reports that ALOX5AP SNPs are associated with MI in both Icelandic and British populations. [DOI] [PubMed] [Google Scholar]
- 35.Broeckel U, Hengstenberg C, Mayer B, et al. A comprehensive linkage analysis for myocardial infarction and its related risk factors. Nat Genet. 2002;30:210–214. doi: 10.1038/ng827. [DOI] [PubMed] [Google Scholar]
- 36.Francke S, Manraj M, Lacquemant C, et al. A genome-wide scan for coronary heart disease suggests in Indo-Mauritians a susceptibility locus on chromosome 16p13 and replicates linkage with the metabolic syndrome on 3q27. Hum Mol Genet. 2001;10:2751–2765. doi: 10.1093/hmg/10.24.2751. [DOI] [PubMed] [Google Scholar]
- 37••.Ozaki K, Inoue K, Sato H, et al. Functional variation in LGALS2 confers risk of myocardial infarction and regulates lymphotoxin-alpha secretion in vitro. Nature. 2004;429:72–75. doi: 10.1038/nature02502. This paper identifies LGALS2 as a gene that interacts with LTA and regulates secretion of LTA, and an SNP of LGALS2 is associated with a decreased risk of MI. [DOI] [PubMed] [Google Scholar]
- 38.Pajukanta P, Nuotio I, Terwilliger JD, et al. Linkage of familial combined hyperlipidaemia to chromosome 1q21-q23. Nat Genet. 1998;18:369–373. doi: 10.1038/ng0498-369. [DOI] [PubMed] [Google Scholar]
- 39•.Pajukanta P, Lilja HE, Sinsheimer JS, et al. Familial combined hyperlipidemia is associated with upstream transcription factor 1 (USF1) Nat Genet. 2004;36:371–376. doi: 10.1038/ng1320. This paper reports that SNPs in transcriptional factor USF1 gene are significantly associated with ischemic stroke. [DOI] [PubMed] [Google Scholar]
- 40.Freimer N, Sabatti C. The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat Genet. 2004;36:1045–1051. doi: 10.1038/ng1433. [DOI] [PubMed] [Google Scholar]
- 41.Yamada A, Ichihara S, Murase Y, et al. Lack of association of polymorphisms of the lymphotoxin alpha gene with myocardial infarction in Japanese. J Mol Med. 2004;82:477–483. doi: 10.1007/s00109-004-0556-x. [DOI] [PubMed] [Google Scholar]
- 42.Koch W, Kastrati A, Bottiger C, et al. Interleukin-10 and tumor necrosis factor gene polymorphisms and risk of coronary artery disease and myocardial infarction. Atherosclerosis. 2001;159:137–144. doi: 10.1016/s0021-9150(01)00467-1. [DOI] [PubMed] [Google Scholar]
- 43.Koch W, Tiroch K, von BN, et al. Tumor necrosis factor-alpha, lymphotoxin-alpha, and interleukin-10 gene polymorphisms and restenosis after coronary artery stenting. Cytokine. 2003;24:161–171. doi: 10.1016/j.cyto.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 44.PROCARDIS Consortium. A trio family study showing association of the lymphotoxin-alpha N26 (804A) allele with coronary artery disease. Eur J Hum Genet. 2004;12:770–774. doi: 10.1038/sj.ejhg.5201244. [DOI] [PubMed] [Google Scholar]
- 45.Mehrabian M, Allayee H, Wong J, et al. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91:120–126. doi: 10.1161/01.res.0000028008.99774.7f. [DOI] [PubMed] [Google Scholar]
- 46.Dwyer JH, Allayee H, Dwyer KM, et al. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med. 2004;350:29–37. doi: 10.1056/NEJMoa025079. [DOI] [PubMed] [Google Scholar]
- 47.Funalot B, Varenne O, Mas JL. A call for accurate phenotype definition in the study of complex disorders. Nat Genet. 2004;36:3–4. doi: 10.1038/ng0104-3a. [DOI] [PubMed] [Google Scholar]
- 48.Archacki SR, Angheloiu G, Tian XL, et al. Identification of new genes differentially expressed in coronary artery disease by expression profiling. Physiol Genomics. 2003;15:65–74. doi: 10.1152/physiolgenomics.00181.2002. [DOI] [PubMed] [Google Scholar]
- 49.Archacki S, Wang Q. Expression profiling of cardiovascular disease. Hum Genomics. 2004;1:355–370. doi: 10.1186/1479-7364-1-5-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.You SA, Archacki SR, Angheloiu G, et al. Proteomic approach to coronary atherosclerosis shows ferritin light chain as a significant marker: evidence consistent with iron hypothesis in atherosclerosis. Physiol Genomics. 2003;13:25–30. doi: 10.1152/physiolgenomics.00124.2002. [DOI] [PubMed] [Google Scholar]