

Abstract
Klippel-Trenaunay syndrome (KTS) is a disorder primarily characterized by capillary-venous vascular malformations associated with altered limb bulk and/or length. We report the identification of a balanced translocation involving chromosomes 8q22.3 and 14q13 in a patient with a vascular and tissue overgrowth syndrome consistent with KTS. We demonstrated that translocation t(8;14)(q22.3;q13) arose de novo. These data suggest that a pathogenic gene for a vascular and tissue overgrowth syndrome (KTS) may be located at chromosome 8q22.3 or 14q13. Fluorescence in situ hybridization (FISH) analysis was used to define the breakpoint on chromosome 8q22.3 to a <5-cM interval flanked by markers AFMA082TG9 and GATA25E10, and the 14q13 breakpoint within a 1-cM region between STSs WI-6583 and D14S989. This study provides a framework for the fine-mapping and ultimate cloning of a novel vascular gene at 8q22.3 or 14q13.
Klippel-Trenaunay syndrome (KTS) is a disorder comprised of capillary and venous malformations associated with overgrowth of the affected extremity (Klippel and Trenaunay, 1900; Berry et al., 1998; Jacob et al., 1998; Cohen, 2000). The first group of KTS cases was described by the French physicians Klippel and Trenaunay in 1900 (Klippel and Trenaunay, 1900). They noted the combination of three features: cutaneous capillary hemangioma, early onset of varicose veins or venous malformations, and bone and soft tissue hypertrophy (Klippel and Trenaunay, 1900). These three features constitute the primary diagnostic criteria of KTS, although KTS can be diagnosed on the basis of any two of these three findings (Jacob et al., 1998).
The manifestations of KTS are variable. A majority of KTS patients manifest capillary malformations (port-wine stains) (95–100%) (Berry et al., 1998; Jacob et al., 1998). There is a broad spectrum of cutaneous manifestations of KTS. Most commonly there is a port wine stain which can be very light in color to deep maroon. Cutaneous or subcutaneous vascular malformations occur in 40% of patients. Other cutaneous manifestations of KTS include phlebectasias, hyperhidrosis, hyperthermia and hypertrichosis. The venous involvement in KTS can range from subtle abnormalities to massive varicosities and absence of important deep venous structures (Berry et al., 1998; Jacob et al., 1998). Venous malformations and varicosities exist in 75–80% of KTS patients. Lymphatic malformations (LMs) are defects of cutaneous and subcutaneous lymphatic vessels. About 20–35% of patients have cutaneous vesicles which leak lymph.
Limb asymmetry was reported in 65–90% of KTS patients (Gloviczki et al., 1991; Berry et al., 1998; Jacob et al., 1998). In addition to bony hypertrophy, many patients have soft tissue hypertrophy. The soft tissue hypertrophy is usually fatty and contains variable amounts of venous structures.
The pathogenic mechanism underlying vascular overgrowth syndromes such as KTS is not clear. Although three cases of familial occurrence of KTS in very small kindreds and a high frequency of capillary malformations among KTS relatives were reported (Aelvoet et al., 1992; Ceballos-Quintal et al., 1996; Lorda-Sanchez et al., 1998), it remains uncertain whether genetic factors contribute to pathogenesis of KTS. In this study, we identified a de novo translocation, t(8;14)(q22.3; q13), which is associated with a vascular and tissue overgrowth syndrome with the manifestations of KTS clinical features. Identification of de novo translocation t(8;14)(q22.3;q13) may indicate that a pathogenic gene for KTS is located either at chromosome 8q22.3 or at 14q13.
Materials and methods
Genomic DNA samples and linkage analysis
Informed consent was obtained from patient KTS002 and members of his family in accordance with standards established by local institutional review boards.
Genomic DNA was prepared from peripheral blood leukocytes using DNA isolation kits for mammalian blood according to the manufacturer’s instructions (Roche Biochemical, Inc.). For the paternal test, genotyping analysis with ten short tandem repeat (STR) polymorphisms on ten different chromosomes was performed. Amplification of each STR was carried out as previously described (Wang et al., 1995, 1996, 2001).
Isolation of PAC and YAC clones
PAC clones were isolated by screening a human genomic PAC library using a PCR-based screening assay of pooled libraries (Genome Systems) with STS primers. YACs were identified by screening the YAC database at the Whitehead Institute/MIT Genome Center and a database at Genethon. The individual PAC and YAC clones were purchased from commercial vendors.
Karyotype and FISH analysis
Chromosomes were prepared from peripheral blood lymphocyte cultures (Bangs and Donlon, 1996). The karyotype analysis was performed using GTG banding (Schreck and Disteche, 1994).
PAC DNA was labeled by nick-translation with biotin d-ATP and used as probes for FISH. FISH analysis was performed as described (Knoll and Lichter, 1994). YAC probes were prepared by Alu-PCR followed by nick-translation with digoxigenin-11-dUTP and the Alu-specific ALE1 primer (5′-GCCTCCCAAAGTGCTGGGATTACAG-3′) and ALE3 primer (5′-CCA [C/T]TGCACTCCAGCCTGGG-3′). Chromosome in situ suppression hybridization was performed on metaphase chromosomes from the patient by using each labeled DNA (500 ng/chromosome slide) as a probe, together with 25-fold excess of human Cot-1 DNA as a competitor. Chromosomes were then counterstained with vectashield, DAPI (Sigma), and hybridization signals were detected with anti-digoxigenin-fluorescein Fab fragments (Roche Biochemical, Inc.). Chromosomes were then photographed under a fluorescence microscope using a CCD camera (Photometrics) and Smartcapture software (Vysis). For two-color FISH experiments, PAC probes were differentially labeled using digoxigenin-dUTP (detected with a rhodamine-conjugated anti-digoxigenin antibody) and biotin-dATP (detected with FITC-conjugated avidin).
Results
Clinical characterization of the patient
Patient KTS002 is a 15-year old boy with manifestations of the three primary clinical features of Klippel-Trenaunay syndrome (KTS): (i) The child has port wine stains on his back and left arm and a pigmented linear nevus-type lesion on his arm. (ii) One varicose vein was observed over the medial aspect of the right knee. Also, he had varicosities removed from the right ankle and leg. He has nodular lesions on his right leg consistent with old superficial venous thromboses. (iii) The patient’s right leg is 1.2 cm longer than the left leg. In 1999 he had a right proximal tibial epiphysiodesis for leg length discrepancy. His left kidney is larger than the right kidney as shown by ultrasonography.
It has been reported that a certain number of KTS patients are associated with other secondary clinical findings including developmental delay, macrocephaly, seizures, glaucoma, intra-vascular coagulopathy, and other features (Berry et al., 1998; Jacob et al., 1998). Similarly, patient KTS002 has gigantism of his hands and feet, gingival hypertrophy, and macrocephaly. An MRI revealed macrocephaly, a Chiari 1 malformation, and a possible slow flow vascular malformation in the right Sylvian fissure. He is mentally retarded with an IQ in the mid 50’s. He has a seizure disorder controlled with medication. He has had documented episodes of cellulitis. Some of these features may be complications resulting from the primary KTS features, and some of the non-typical KTS features may be explained by a gross cytogenetic abnormality (a translocation that may affect the function of multiple genes) identified in the patient (see below).
Identification of translocation t(8;14)(q22.3;q13)
A routine cytogenetic evaluation revealed that patient KTS002 carried a balanced translocation involving chromosomes 8q22.3 and 14q13, t(8;14)(q22.3;q13). The presence of the translocation was further confirmed by our analysis on high resolution GTG-banded chromosomes of cultured peripheral blood lymphocytes (Fig. 1). All 20 cells examined have the translocation.
Fig. 1.
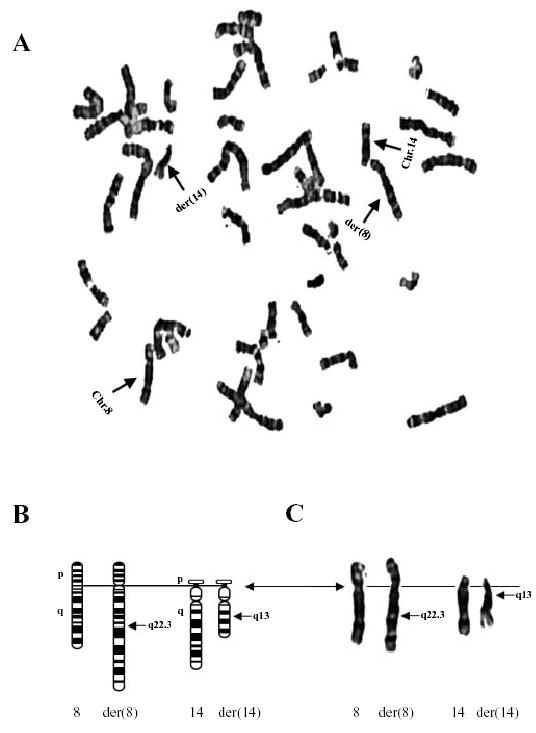
(A) A metaphase spread prepared from blood samples of a patient with Klippel-Trenaunay syndrome (KTS002) and stained by Giemsa banding. A translocation involving chromosomes 8 and 14 (indicated by arrows) was identified. (B, C) Translocation t(8;14)(q22.3;q13) with centromeres as the solid line and breakpoints marked. Ideograms of normal chromosomes 8 and 14 and derivative chromosomes 8 and 14 are shown at the left (B), and G-banded chromosomes are shown at the right (C).
Translocation t(8;14)(q13;q22.3) arose de novo
To further characterize translocation t(8;14)(q22.3;q13), we used karyotype analysis to determine whether other family members of patient KTS002 also carry the translocation. Clinical examinations of the KTS patient’s sister and both parents indicate that they are not affected with KTS. Blood samples were obtained from each of them and high-resolution GTG-banded chromosomes of cultured peripheral blood lymphocytes were examined. Neither the parents nor the sister carry translocation t(8;14) (Fig. 2).
Fig. 2.
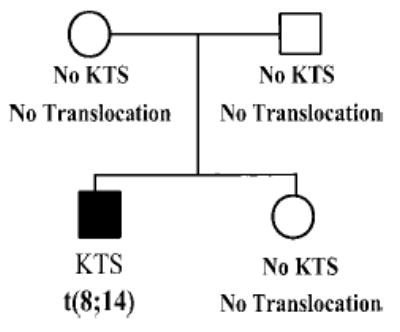
Translocation t(8;14)(q22.3;q13) arose de novo. Pedigree structure of the KTS family with translocation t(8;14)(q22.3;q13). Results from clinical diagnosis of KTS and cytogenetic studies are summarized below each individual. Normal individuals are indicated as empty squares (males) or circles (females), and the affected individual is shown as a filled symbol. KTS, Klippel-Trenaunay syndrome; t(8;14), balanced translocation involving chromosomes 8q22.3; and 14q13.
We performed the paternal test using linkage analysis with DNA samples from all four members. We randomly selected ten PCR-based short tandem repeat (STR) markers that are highly polymorphic and informative (heterozygosity >0.70) (Weber/CHLH Marker Set 8). Genotyping data were scored and analyzed against the pedigree information. Our results confirmed the relationship of all four members in family KTS002 (data not shown).
Because patient KTS002 is affected with KTS and has translocation t(8;14)(q22.3;q13), while both parents and the sister are clinically normal and do not carry the translocation, we conclude that translocation t(8;14) has arisen de novo.
Characterization of angiopoietin-1 (ANGPT1) as a candidate gene for KTS
Possible candidate genes for KTS were identified through the NCBI GeneMap’99 database (http://www.ncbi.nlm.nih.gov/genemap) for genes located on chromosome 8q22.3 and 14q13. The human angipoietin-1 gene (ANGPT1) was identified on chromosome 8q22.3. Angiopoietin-1 is an endothelium-specific ligand for receptor tyrosine kinase Tie2 and it is required for embryonic vascular stabilization, branching morphogenesis, and post-natal angiogenesis (Hanahan, 1997). Based on its location and physiology, ANGPT1 became a candidate gene for KTS.
STSs were generated based on sequences at 5′-UTR and 3′-UTR of ANGPT1, and used for screening a human genomic PAC library. We identified one PAC for the 5′-end of ANGPT1 and two PACs for the 3′-end. The PACs were used for FISH analysis to determine whether ANGPT1 is disrupted by the translocation. As shown in Fig. 3, FISH signals from the 5′-UTR PAC (212d10) as well as the two 3′-UTR PACs (224e4 and 111i04) were detected on the normal chromosome 8 as well as the derivative chromosome 8. These studies suggest that ANGPT1 is located centromeric to the chromosome 8q22.3 breakpoint and it is not disrupted by translocation t(8;14) (q22.3;q13).
Fig. 3.
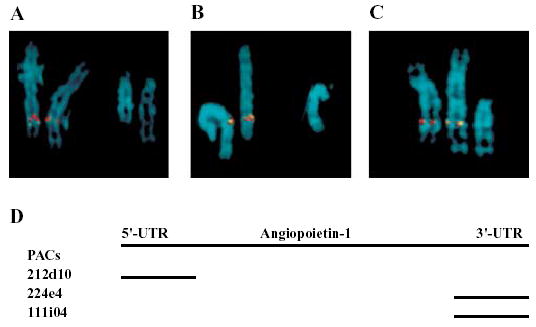
FISH mapping of the angiopoietin-1 gene (ANGPT1) to the vicinity of the 8q22.3 translocation breakpoint. (A–C). Examples of metaphase chromosomes 8, 14, and their derivatives (d8; d14) hybridized with chromosome 8-specific PAC clones 212d10 (A), 224e4 (B), and 111i04 (C). All three PAC probes (biotin-dUTP labeled ) showed hybridization signals (green color; detected with FITC-conjugated avidin) on normal chromosome 8 as well as derivative chromosome 8, but not on derivative chromosome 14. Therefore, ANGPT1 is located proximal to the 8q22.3 breakpoint. PAC 212d10 was then labeled with digoxigenin-dUTP and used in two-color FISH analysis (red signals detected with a rhodamine-conjugated anti-digoxigenin antibody). Both the red and green signals were detected on normal chromosome 8 as well as derivative chromosome 8, but not on derivative chromosome 14. Therefore, ANGPT1 is not disrupted by the 8q22.3 breakpoint. (D). PAC clones specific to ANGPT1. PAC clone 212d10 is specific to the 5′UTR of ANGPT1, and PAC clones 224e4 and 111i04 are specific to 3′UTR.
Definition of translocation breakpoints using fluorescence in situ hybridization (FISH)
In order to characterize translocation t(8;14)(q22.3;q13), we defined the translocation breakpoints within small regions. FISH experiments were performed using nine YACs on chromosome 8q22.3→q23 and 12 YACs on 14q13 (Figs. 4, 5). Positive FISH signals were detected for YACs 771e3, 721a12, 917e5, 916e10, 850a5, and 899a3 on normal chromosome 8 and derivative chromosome 8 (Figs. 4, 5), localizing these YACs on the centromeric side of the 8q22.3 translocation breakpoint. Positive FISH signals for YACs 956e8, 934h8, and 940d10 were detected on normal chromosome 8 and derivative chromosome 14, suggesting that these three YACs are located on the telometric side of the translocation breakpoint. Together, these results defined the 8q22.3 breakpoint in a <5-cM region flanked by marker AFMA082TG9 (YACs 850a5 and 899a3) and marker GATA25E10 (YAC 956e8).
Fig. 4.
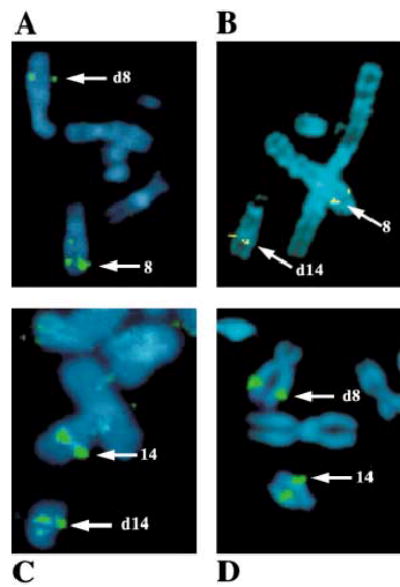
Refined FISH mapping of the t(8;14) breakpoints. (A, B). FISH analysis using YAC probes 899a3 (A) and 956e8 (B) from the chromosome 8q22.3 breakpoint region. Hybridization signals for YAC 899a3 were observed on the normal chromosome 8 (8) and derivative chromosome 8 (d8). For YAC clone 956e8, FISH signals were observed on the normal chromosome 8 and derivative chromosome 14 (d14), indicating that the chromosome 8q22.3 breakpoint is located between YAC 899a3 and YAC 956e8. (C, D). FISH analysis using YAC probes 772d1 (C) and 964b11 (D) from the chromosome 14q13 breakpoint region, respectively. FISH signals were detected on the normal chromosome 14 (14) and derivative chromosome 14 (d14) with YAC 772d1 and on the normal chromosome 14 and derivative chromosome 8 (d8) with YAC 964b11, localizing the chromosome 14q13 breakpoint between YAC 772d1 and YAC964b11. (Metaphase chromosomes appeared to be blue color after DAPI counterstaining.)
Fig. 5.
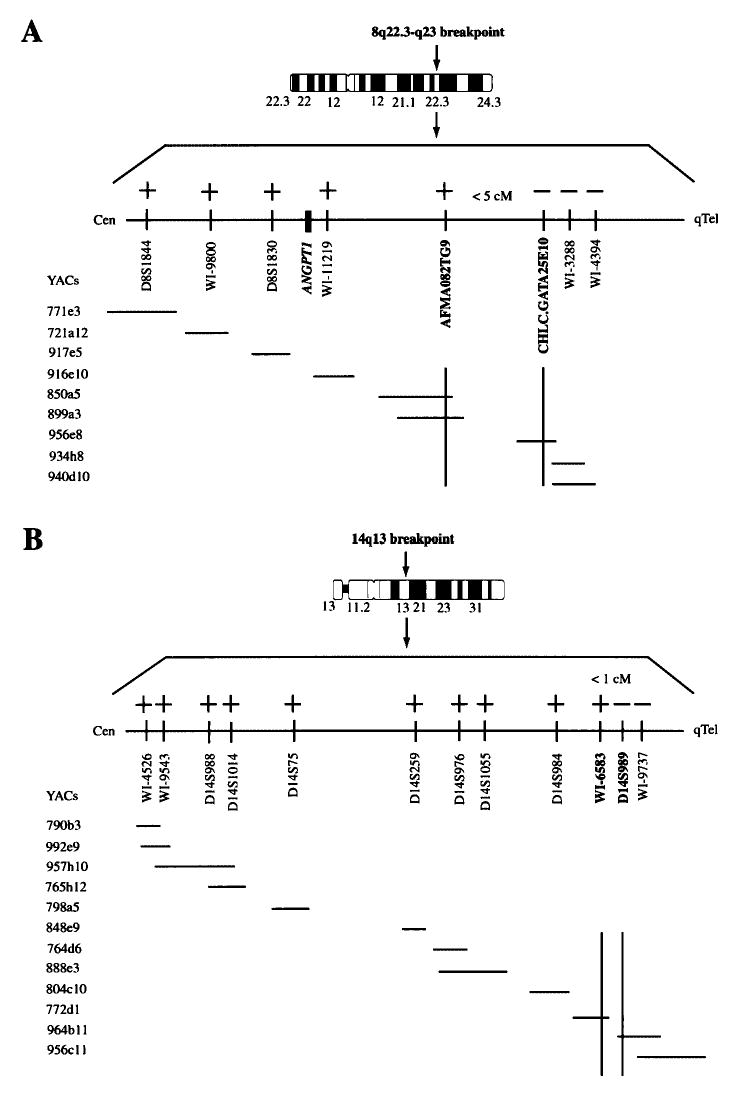
Localization of the 8q22.3 breakpoint (A) and the 14q13 translocation breakpoint (B) by FISH analysis. YACs spanning chromosome regions 8q22.3 (A) and 14q13 (B) were identified by searching the Whitehead Institute/MIT Center for Genome Research YAC database, and purchased from Research Genetics. The “+” symbol indicates that positive FISH signals were obtained on the corresponding derivative chromosome for a specific YAC, and “–” indicates absence of FISH signals on the corresponding derivative chromosome. The 8q22.3 breakpoint is defined within a <5-cM region flanked by markers AFMA082TG9 and GATA25E10, and the 14q13 breakpoint is between STSs WI-6583 and D14S989, <1 cM. ANGPT1, the angiopoietin-1 gene.
Similar analysis defined the chromosome 14q13 translocation breakpoint in a 1-cM region between STS WI-6583 (YAC 772d1) and marker D14S989 (YAC 964b11) (Figs. 4, 5).
Discussion
We report the identification of a balanced translocation involving chromosomes 8q22.3 and 14q13 in a patient affected with a vascular and tissue overgrowth syndrome. There are several overgrowth syndromes affecting humans that have features in common. These syndromes include Klippel-Trenaunay syndrome (KTS), Proteus syndrome, Sturge-Weber syndrome, and Parks-Weber syndrome. Clinically, it may be difficult to determine into which syndrome a particular patient best fits. These syndromes have in common vascular overgrowth (superficial capillary malformations, venous malformations, lymphatic malformations), and a variety of tissue overgrowth. As discussed in the Results section, the overall clinical characteristics of patient KTS002 reported in this study best fit KTS. However, features such as the nevus-type lesion, gigantism of the hands and feet, and megaencephaly are more common with Proteus syndrome (Biesecker et al., 1999). Future molecular genetic studies may be needed to clarify these overlapping overgrowth syndromes.
We demonstrated that the translocation t(8;14)(q22.3;q13) arose de novo. The identification of a de novo translocation associated with KTS implicates a pathogenic gene in the development of this vascular and tissue overgrowth syndrome. This hypothesis is further supported by the report of three cases of familial occurrence of KTS and the high frequency of capillary malformations among relatives (Aelvoet et al., 1992; Ceballos-Quintal et al., 1996; Lorda-Sanchez et al., 1998). Recently, we have started to perform family studies on 34 KTS families. We identified three kindreds with two or more family members affected with KTS (data not shown). In addition, we found that in 16 of 34 families, other family members of the KTS patients have vascular anomalies such as capillary malformations, venous malformations, and/or varicose veins (data not shown). Together, these studies provide evidence for the hypothesis that genetic factors contribute to the pathogenesis of KTS. By contrast, Happle (1986, 1987, 1993) proposed the concept of paradominant inheritance (autosomal lethal genes surviving only in a mosaic state) to explain the genetic basis of KTS and other syndromes that are characterized by sporadic occurrence and a mosaic distribution of lesions. Based on this hypothesis, homozygotes are non-viable. Heterozygotes with a paradominant mutation are phenotypically normal and the mutation can be transmitted unperceived through many generations. KTS develops in the heterozygotes when a somatic mutation occurs during early embryogenesis, leading to the formation of a mosaic population of cells being homozygous or hemizygous for the mutation. The key to distinguishing the above two hypotheses is to identify the genes that cause KTS by characterizing cytogenetic abnormalities such as translocation t(8;14)(q22.3;q13).
A balanced translocation, t(5;11)(q13.3;p15.1), was reported in another patient with KTS (Whelan et al., 1995). The KTS patient associated with translocation t(5;11)(q13.3;p15.1) shares not only the primary KTS features, but also other clinical findings. Both patients had mental retardation. In both translocation patients, the left kidney is larger than the right kidney. Identification of two different chromosome rearrangements associated with KTS presumably implicates different genes in the etiology of this disease, suggesting that KTS is genetically heterogeneous. Chromosome abnormalities of this kind have repeatedly led to the isolation of genes responsible for the phenotype in a wide variety of human genetic diseases. Further characterization of translocations associated with KTS may lead to identification of pathogenic genes responsible for KTS.
As the first step of the positional cloning of a novel vascular gene affected by the t(8:14) translocation, we have defined the 8q22.3 translocation breakpoint within a <5-cM interval flanked by markers AFMA082TG9 and GATA25E10, and defined the 14q13 breakpoint within a 1-cM region between STSs WI-6583 and D14S989 (Figs. 4, 5). These data provide a framework for the cloning of the translocation breakpoints and identification of a novel vascular gene at chromosome 8q22.3 or 14q13. The 8q22.3 breakpoint region contains the angiopoietin-1 gene encoding an ~75-kDa secreted angiogenic factor (ANGPT1) that binds to and signals through the endothelial cell-specific Tie2 receptor typrosine kinase (Hanahan, 1997). Mice deficient in ANGPT1 developed severe vascular abnormalities in the developing embryo (Suri et al., 1996). ANGPT1 has also been over-expressed in the skin of mice, and this resulted in a dramatic increase in the number, size, and branching pattern of blood vessels (Suri et al., 1998). Thus, ANGPT1 represents a candidate gene for KTS. Although chromosome-banding studies showed that ANGPT1 was located in the same cytogenetic band, 8q22.3, as the translocation breakpoint (data not shown), the ANGPT1 gene is not disrupted by the 8q22.3 translocation breakpoint (Fig. 3). Based on the NCBI (http://www.ncbi.nlm.nih.gov) and ENSEMBL (http://www.ensembl.org) databases, ANGPT1 is located approximately 3–5 cM from AFMA082TG9, the centromeric flanking marker of the 8q22.3 breakpoint (Fig. 5). These data suggest that ANGPT1 is no longer a promising candidate gene for KTS, but it is uncertain whether the long-range positional effect associated with the translocation may affect the function of ANGPT1. Several other candidate genes for KTS in the breakpoint regions include COL14A1 encoding a large extracellular matrix (ECM) mosaic glycoprotein related to collagen type XIV (undulin), MATN2 (matrilin-2) thought to be involved in the formation of filamentous networks in the extracellular matrices of various tissues, and SDC2 (syndecan-2, transmembrane heparan sulfate proteoglycan) involved in the formation of bone. Further characterization of the 8q22.3 and 14q13 breakpoint regions and the candidate genes in these regions will lead to the identification of a novel vascular gene and characterization of the novel vascular gene may provide insight into the molecular mechanism underlying pathogenesis of vascular and tissue overgrowth syndromes such as KTS.
Acknowledgments
We are grateful to the patient and family members for their cooperation in this study. We thank M. McCane and G. Hoeltge at the Department of Pathology and Laboratory Medicine of the Cleveland Clinic Foundation for help with karyotyping analysis of the KTS patient, and Baylor Kleberg Cytogenetics Laboratory directed by L.G. Shaffer for karyotyping analysis for the family members of the KTS patient in this study.
Footnotes
Supported in part by NIH grant R01 HL65630 (Q.W.).
Present address of J.C.: Department of Cancer Genetics, Roswell Park Cancer Institute, Buffalo, New York (USA).
References
- Aelvoet GE, Jorens PG, Roelen LM. Genetic aspects of the Klippel-Trenaunay syndrome. Br J Dermatol. 1992;126:603–607. doi: 10.1111/j.1365-2133.1992.tb00107.x. [DOI] [PubMed] [Google Scholar]
- Bangs CD, Donlon TA. Chromosome preparation from cultured peripheral blood cells. In: Dracopoli NC, Haines JL, Korf BR, Moir DT, Morton CC, Seidman CE, Seidman JG, Smith DR, Boyle ALB, editors. Current Protocols in Human Genetics. John Wiley & Sons, Inc.; New York: 1996. pp. 4.1.1–4.1.19. [DOI] [PubMed] [Google Scholar]
- Berry SA, Peterson C, Mize W, Bloom K, Zachary C, Blasco P, Hunter D. Klippel-Trenaunay syndrome. Am J med Genet. 1998;79:319–326. [PubMed] [Google Scholar]
- Biesecker LG, Happle R, Mulliken JB, Weksberg R, Graham JM, Jr, Viljoen DL, Cohen MM., Jr Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J med Genet. 1999;84:389–395. doi: 10.1002/(sici)1096-8628(19990611)84:5<389::aid-ajmg1>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Ceballos-Quintal JM, Pinto-Escalante D, Castillo-Zapata I. A new case of Klippel-Trenaunay-Weber (KTW) syndrome: evidence of autosomal dominant inheritance. Am J med Genet. 1996;63:426–427. doi: 10.1002/(SICI)1096-8628(19960614)63:3<426::AID-AJMG2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr Klippel-Trenaunay syndrome. Am J med Genet. 2000;93:171–175. doi: 10.1002/1096-8628(20000731)93:3<171::aid-ajmg1>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Gloviczki P, Stanson AW, Stickler GB, Johnson CM, Toomey BJ, Meland NB, Rooke TW, Cherry KJ., Jr Klippel-Trenaunay syndrome: the risks and benefits of vascular interventions. Surgery. 1991;110:469–479. [PubMed] [Google Scholar]
- Hanahan D. Signaling vascular morphogenesis and maintenance. Science. 1997;277:48–50. doi: 10.1126/science.277.5322.48. [DOI] [PubMed] [Google Scholar]
- Happle R. Cutaneous manifestation of lethal genes. Hum Genet. 1986;72:280. doi: 10.1007/BF00291899. [DOI] [PubMed] [Google Scholar]
- Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- Happle R. Klippel-Trenaunay syndrome: is it a para-dominant trait? Br J Dermatol. 1993;128:465–466. doi: 10.1111/j.1365-2133.1993.tb00214.x. [DOI] [PubMed] [Google Scholar]
- Jacob AG, Driscoll DJ, Shaughnessy WJ, Stanson AW, Clay RP, Gloviczki P. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc. 1998;73:28–36. doi: 10.1016/S0025-6196(11)63615-X. [DOI] [PubMed] [Google Scholar]
- Klippel M, Trenaunay P. Du naevus variqueux osteo-hypertrophique. Arch Gen Med. 1900;185:641–672. [Google Scholar]
- Knoll JNM, Lichter P. In situ hybridization to meta-phase chromosomes and interphase nuclei. In: Dracopoli NC, Haines JL, Korf BR, Moir DT, Morton CC, Seidman CE, Seidman JG, Smith DR, Boyle ALB, editors. Current Protocols in Human Genetics. John Wiley & Sons, Inc.; New York: 1994. pp. 4.3.1–4.3.28. [Google Scholar]
- Lorda-Sanchez I, Prieto L, Rodriguez-Pinilla E, Martinez-Frias ML. Increased parental age and number of pregnancies in Klippel-Trenaunay-Weber syndrome. Ann hum Genet. 1998;62:235–239. doi: 10.1046/j.1469-1809.1998.6230235.x. [DOI] [PubMed] [Google Scholar]
- Schreck RR, Disteche CM. Chromosome banding techniques. In: Dracopoli NC, Haines JL, Korf BR, Moir DT, Morton CC, Seidman CE, Seidman JG, Smith DR, Boyle ALB, editors. Current Protocols in Human Genetics. John Wiley & Sons, Inc.; New York: 1994. pp. 4.2.1–4.2.36. [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- Suri C, McClain J, Thurston G, McDonald DM, Zhou H, Oldmixon EH, Sato TN, Yancopoulos GD. Increased vascularization in mice overexpressing angiopoietin-1. Science. 1998;282:468–471. doi: 10.1126/science.282.5388.468. [DOI] [PubMed] [Google Scholar]
- Wang Q, Bond M, Elston RC, Tian X. Molecular genetics. In: Topol EJ, editor. Textbook of Cardiovascular Medicine. Lippincott Williams & Wilkins; Philadelphia: in press. [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nature Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- Whelan AJ, Watson MS, Porter FD, Steiner RD. Klippel-Trenaunay-Weber syndrome associated with a 5:11 balanced translocation. Am J med Genet. 1995;59:492–494. doi: 10.1002/ajmg.1320590416. [DOI] [PubMed] [Google Scholar]