

Summary
Previous studies have indicated that replication stress can trigger apoptosis-like cell death, accompanied (where tested) by production of reactive oxygen species (ROS), in mammalian cells and budding yeast (Saccharomyces cerevisiae). In mammalian cells, inappropriate entry into mitosis also leads to cell death. Here we report similar responses in fission yeast (Schizosaccharomyces pombe). We used ROS- and death-specific fluorescent stains to measure the effects of mutations in replication initiation and checkpoint genes in fission yeast on the frequencies of ROS production and cell death. We found that certain mutant alleles of each of the four tested replication initiation genes caused elevated ROS and cell death. Where tested, these effects were not enhanced by checkpoint gene mutations. Instead, when cells that were competent for replication but defective in both the replication and damage checkpoints were treated with hydroxyurea, which slows replication fork movement, the frequencies of ROS production and cell death were greatly increased. This was a consequence of elevated CDK activity, which permitted inappropriate entry into mitosis. Thus studies in fission yeast are likely to prove helpful in understanding the pathways that lead both from replication stress and from inappropriate mitosis to cell death in mammalian cells.
Keywords: reactive oxygen species, cell death, checkpoint, replication, mitosis, apoptosis
Introduction
The term “reactive oxygen species (ROS)” applies to any mixture of molecules, ions and free radicals containing derivatives of molecular oxygen that are more reactive than oxygen itself. The ROS formed in living cells commonly include hydrogen peroxide, hydroxyl radical and superoxide anion. The normal process of respiration in mitochondria is a major source of ROS, and production of ROS is enhanced when mitochondrial function is disturbed during apoptosis. In apoptosis, and also in some types of necrotic cell death, unusually large amounts of ROS can be released and can contribute, by extensive oxidation of macromolecules, to the killing of cells. In some types of apoptosis, ROS also serve essential signaling functions (reviewed in (Fleury et al., 2002)).
Apoptosis is not confined to multicellular eukaryotes. Unicellular organisms, including budding yeast and fission yeast, can undergo programmed cell death with many of the features of apoptosis in multicellular organisms (reviewed in (Burhans et al., 2003; Madeo et al., 2004; Rodriquez-Menocal and D’Urso, 2004)). Where tested, ROS production has proved to accompany apoptosis in yeasts and fungi (Balzan et al., 2004; Cheng et al., 2003; Madeo et al., 1999; Zhang et al., 2003), although in some cases it is not required (Balzan et al., 2004; Cheng et al., 2003).
A wide variety of factors can stimulate apoptosis in yeasts (reviewed in (Burhans et al., 2003; Madeo et al., 2004)). In budding yeast these apoptotic triggers include extensive DNA damage (Blanchard et al., 2002; Qi et al., 2003; Weinberger et al., 2005) and defects in the replication initiation proteins Cdc6p (Blanchard et al., 2002) and Orc2p (Weinberger et al., 2005). The same triggers, DNA damage (Norbury and Zhivotovsky, 2004) and defects in replication initiation proteins (Blanchard et al., 2002; Dodson et al., 2004; Feng et al., 2003; Kim et al., 2003; Pelizon et al., 2002; Schories et al., 2004; Shreeram et al., 2002; Yim et al., 2003), can also induce apoptosis in mammalian cells.
In mammalian cells, defects in DNA replication or DNA structure activate replication and damage checkpoints, and, among other consequences, these checkpoints inhibit CDK activity—thus preventing cells from entering mitosis with incompletely replicated or damaged DNA (Sancar et al., 2004). Entry into mitosis with damaged or incompletely replicated DNA usually leads to cell death by an apoptotic mechanism (Castedo et al., 2004). For these reasons it seemed likely to us that mutations in genes important for the DNA damage or replication checkpoints might enhance susceptibility to inappropriate mitosis when combined with mutations in genes affecting replication initiation.
Study of the interaction between checkpoint genes and replication initiation genes in pathways leading to apoptosis would be facilitated by the availability of a model organism capable of undergoing apoptosis and with checkpoint pathways similar to those of mammalian cells but more amenable to genetic analysis than mammalian cells. Fission yeast is such an organism. In contrast to budding yeast, in which checkpoints prevent mitosis primarily by inhibiting spindle elongation or anaphase chromosome separation, checkpoints in fission yeast—as in mammalian cells—prevent entry into mitosis by inhibiting CDK activity (Caspari and Carr, 1999). Furthermore, there is substantial evidence for apoptosis in fission yeast (Brezniceanu et al., 2003; James et al., 1997; Jürgensmeier et al., 1997; Zhang et al., 2003).
Here we report the use of ROS production and cell death assays in fission yeast to test our prediction that checkpoint gene mutations would enhance the effects of replication initiation gene mutations on the frequency of apoptosis-like cell death. We found that mutations in genes encoding replication initiation proteins were likely to stimulate ROS production. Where tested, this ROS production was not further stimulated by loss of checkpoint functions—in contrast to our prediction. However, we found that when replication forks were slowed by treatment with HU, checkpoint mutations dramatically stimulated ROS production and cell death. It is known that checkpoint failure leads to uncontrolled CDK activity and entry into mitosis with unreplicated DNA (Boddy et al., 1998). Cells with constitutively activated CDK also enter mitosis prematurely (Gould and Nurse, 1989), and we found that they produced ROS. These results suggest that several different pathways, with varying degrees of dependence on checkpoints, can lead from replication defects to ROS production and cell death in fission yeast.
Materials and Methods
Cell culture
The strains used in this study are listed in Table S1. Cells were propagated at the indicated temperatures in YES medium (Moreno et al., 1991), with the exception of the dfp1 mutants, which were maintained in Edinburgh Minimal Medium (EMM) (Moreno et al., 1991) containing supplements with the exception of leucine.
ROS and PI-staining assays
Logarithmically growing non-temperature-sensitive strains were incubated at 25°C. Temperature-sensitive strains in log phase were shifted from the permissive temperature (25°C) to 25°, 30°, 35° or 37° for 2, 4 or 6 hours. In indicated cases, HU (USB Corporation; 12 mM) or MG132 (Calbiochem; 250 μM) was added at the beginning of the incubation. At the end of the incubation the ROS indicator dye (DCDHFDA; Molecular Probes) was added (10 μg/ml final), and incubation was continued for 80 minutes. Cells were then harvested in a table-top centrifuge and washed twice with citrate buffer (50 mM sodium citrate, pH 7.0). The pellets were resuspended in an appropriate volume of citrate buffer containing PI (Sigma; 10 μg/ml final) and then analyzed by fluorescence microscopy or flow cytometry. The flow data were further analyzed using FlowJo software (TreeStar, Inc.) as described in Fig. S1.
Generation of double-mutant strains
Double-mutant strains were generated by appropriate crosses followed by selection for indicator phenotypes.
Cloning of the orp5+ genomic fragment and cDNA and disruption of orp5+
The orp5+ gene was localized to cosmid 855 (Mizukami et al., 1993), which maps just proximal to the moc1+/sds23+ gene on chromosome II. The orp5+ genomic clone was obtained by PCR and confirmed by sequencing. The full length orp5+ cDNA was isolated from a ZapII (Stratagene) S. pombe cDNA library by plaque hybridization with the orp5+ genomic fragment. Sequencing was carried out on both strands.
Isolation of orp5 temperature-sensitive mutants
A 4.1-kb fragment containing the sds23+ and orp5+ genes was amplified by PCR and cloned into pBluescript, and a 2.2-kb fragment containing ura4+ was inserted between the sds23+ and orp5+ open reading frames. The resulting plasmid was used as a template for mutagenic PCR with excess dNTPs to amplify the sds23+, ura4+, orp5+ region. The resulting PCR products were transformed into the fission yeast strain, YM71 (h-, ura4D-18, leu1-32, ade6-704). The ura4+ transformants obtained at 25°C, and in which the chromosomal orp5+-sds23+ region was replaced with a mutated PCR fragment by homologous recombination, were screened for temperature-sensitivity (Ts) at 36.5°C. The resulting Ts mutants were transformed with plasmids expressing either orp5+ or sds23+ to identify orp5 Ts mutation(s). Nine Ts strains were complemented by the orp5+ plasmid but not by the sds23+ plasmid. The orp5-H19, orp5-H30, and orp5-H37 mutants were chosen for further analysis.
Results
Measuring the production of ROS in fission yeast
We used the dye, 2',7'-dichlorodihydrofluorescein diacetate (DCDHFDA), which produces green fluorescence in the presence of ROS (LeBel et al., 1992; Tsuchiya et al., 1994), to measure ROS production in fission yeast cells. This dye is able to penetrate the membranes of living cells (LeBel et al., 1992). Consequently, it can detect ROS in both living cells and in dead and dying cells that lack intact membranes. To distinguish living cells from dead ones, we used a second indicator dye, propidium iodide (PI) (Tsuchiya et al., 1994). PI cannot pass through the intact membranes of living cells. When PI enters a dead cell, it binds to the remaining DNA within the dead cell and fluoresces red. The fluorescence micrographs in Fig. 1 demonstrate that most wild-type fission yeast cells displayed insufficient spontaneous green or red fluorescence to be detected by a cooled charge-coupled device (CCD) camera. In contrast, cells bearing the dfp1Δ13–240 mutation (affecting the gene which encodes the fission yeast homolog of Dbf4p, the regulatory subunit of the Cdc7p kinase, which is essential for initiation of DNA replication; Table 1) displayed variable, but easily detectable, levels of both green and red fluorescence. We interpret the pure green cells as living cells (because they did not stain with PI) that were generating high ROS levels, and we interpret the pure red cells as dead or dying cells that were still sufficiently intact to retain DNA but were either not producing ROS or were unable to retain ROS due to membrane permeability. Some cells displayed both green and red fluorescence and appeared in various shades of yellow. These yellow-tinged cells constituted a significant fraction of the fluorescent cells. The presence of both green and red fluorescence in single cells is consistent with the possibility that ROS production preceded cell death and that the sequence of fluorescence was green to yellow to red. For the experiments reported here, we did not further investigate the possibility that ROS might be a cause of cell death. We simply noted a correlation between these two phenomena, as quantified in the experiments described below.
Fig. 1.

Detection of ROS production and death in fission yeast cells. Wild type and dfp1Δ13–240 mutant cells were incubated for 80 minutes at 25°C in the presence of DCDHFDA to detect ROS. The cells were then harvested, washed, and resuspended in buffer with PI to detect dead cells.
Table 1.
Names of relevant functionally similar genes in fission yeast, budding yeast, and humans
| Fission yeast | Humans | Budding yeast | Protein function |
|---|---|---|---|
| dfp1 | DBF4/ASK1 | DBF4 | Initiation of replication |
| hsk1 | CDC7 | CDC7 | Initiation of replication |
| cdc18 | CDC6 | CDC6 | Formation of pre-replication complex |
| orp2 | ORC2 | ORC2 | Formation of pre-replication complex |
| orp5 | ORC5 | ORC5 | Formation of pre-replication complex |
| pku70 | KU70 | YKU70 | Non-homologous end joining |
| rad32 | MRE11 | MRE11 | Homologous recombination |
| rqh1 | BLM | SGS1 | Replication fork stability |
| rhp9/crb2 | BRCA1 or 53BP1 | RAD9 | DNA damage checkpoint |
| rad3 | ATR | MEC1 | DNA damage and replication checkpoints |
| cds1 | CHK2/CDS1 | RAD53 | DNA replication checkpoint (fission yeast) |
| cdc2 | CDK1/CDC2 | CDC28 | Cyclin-dependent kinase |
This table contains the genes discussed in the text for which the fission yeast name differs from the human name.
We used flow cytometry to quantitate the extents of red and green fluorescence (Fig. S1). In the following figures we report the number of cells per 10,000 examined whose green or red fluorescence exceeded a threshold that was set so as to exclude autofluorescence from unstained cells.
Mutants in the genes encoding the initiation proteins Orp2p, Orp5p and Cdc18p displayed increased ROS production and increased cell death
The first step in the initiation of eukaryotic DNA replication is the association of the six proteins of the Origin Recognition Complex (ORC) with replication origins. This is followed by the additional association of Cdc6p (Cdc18p in fission yeast) and Cdt1p (Bell and Dutta, 2002). We tested the effects of mutations in the fission yeast homologs of some of these proteins on ROS production and PI staining. Fig. 2 displays a time-course and temperature-dependence study of ROS production in fission yeast strains bearing mutations in orp2 (encoding the second largest subunit of ORC; (Kiely et al., 2000; Leatherwood et al., 1996)) or cdc18 (encoding the fission yeast homolog of Cdc6p; (Kelly et al., 1993; Muzi-Falconi et al., 1996)). Several interesting conclusions emerge from these results. First, as in Fig. 1, there was a rough correlation between ROS production and PI staining, consistent with the possibility that ROS production is an important contributor to the cell death detected by PI staining. Second, the two mutant alleles of the orp2 gene had strikingly different effects on ROS production and PI staining. While enhancement of ROS production in the orp2-2 mutant strain was barely detectable even at high temperatures, enhancement in the orp2-7 strain was striking at all temperatures and for all incubation times. Interestingly, at a restrictive temperature (36.5°C), the orp2-2 mutation inhibits initiation of DNA replication more strongly than does the orp2-7 mutation (Kiely et al., 2000). Furthermore, all of these mutations are temperature-sensitive for replication. Cells bearing these mutations can replicate their DNA at 25°C but not at 37°C. Therefore at lower temperatures, at least, the strong effect of the orp2-7 mutation on ROS production and PI staining could not be a direct consequence of its inhibition of initiation of DNA replication.
Fig. 2.
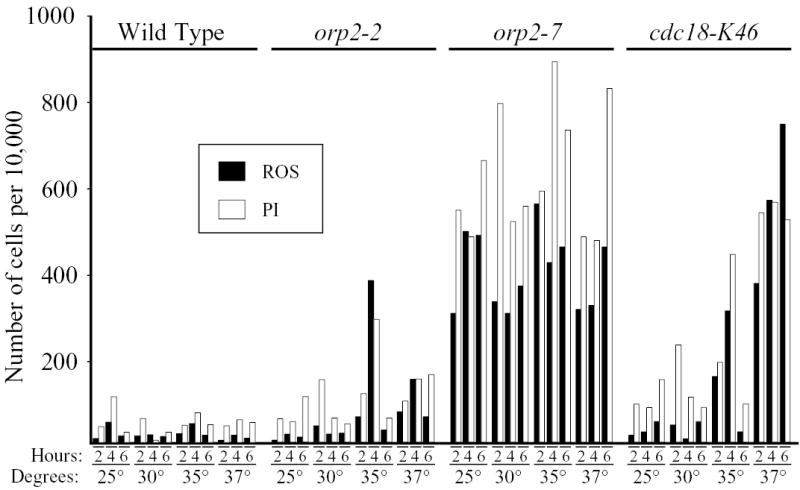
A time- and temperature-dependence study of ROS production and PI staining in cells bearing temperature-sensitive mutations in orp2 or cdc18. The indicated strains, in log phase, were shifted to the indicated temperatures for the indicated numbers of hours. Then DCDHFDA was added, and incubation was continued at the same temperatures for an additional 80 minutes. Other conditions were as in Figs. 1 and S1.
In contrast to the temperature-independent effect of orp2-7 on ROS production and PI staining, the cdc18-K46 mutation had a temperature-dependent effect. It is possible, therefore, that ROS production and PI staining in the cdc18-K46 strain are direct consequences of loss of the ability of Cdc18p to contribute to formation of pre-RCs.
Similar allele-specific effects were detected when we compared mutant alleles of the orp5 gene (which encodes the fifth largest subunit of ORC). ROS production in the orp5-H19 strain was strong only at high temperatures, while ROS production in the orp5-H30 strain was relatively weak at all tested temperatures (Fig. 3). Another allele, orp5-H37, behaved like orp5-H30 (results not shown). As above, there was an imperfect correlation between ROS production and PI staining. However, the tested alleles of orp5 behaved differently from those of orp2 with regard to correlation between ROS production and replication competence. The orp5-H19 allele showed the strongest replication-initiation defect: at 36°C, orp5-H19 cells arrested at the G1-S interface with 1C DNA content. In contrast, orp5-H30 cells arrested in S phase, and orp5-H37 cells showed no obvious replication defect but arrested in G2/M with a possible mitotic defect (results not shown; F. Matsunaga, H. Kato, D. Gong, G. D’Urso, K. Tanaka and Y. Murakami, manuscript in preparation). Thus the orp5 allele with the strongest replication defect generated the most ROS, while the orp2 allele with the strongest replication defect generated the least ROS. This variability is consistent with other results presented in this report, which suggest that there are multiple independent pathways by which defects in replication initiation proteins can stimulate ROS production.
Fig. 3.
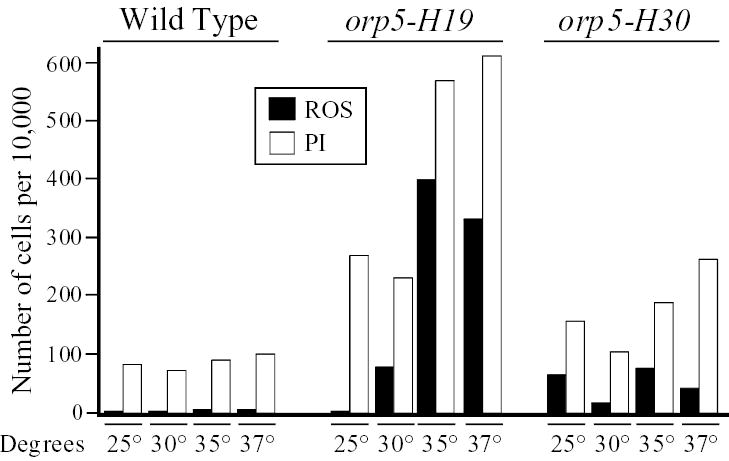
A temperature-dependence study of ROS production and PI staining in cells bearing temperature-sensitive mutations in orp5. This experiment was carried out as in Fig. 2, except that the time of incubation was 4 hours in all cases.
N- and C-terminal deletions in the dfp1 gene led to increased ROS production and PI staining
Dfp1p is the fission yeast homolog of budding yeast Dbf4p. Just as Dbf4p activates the Cdc7p kinase, Dfp1p activates the Hsk1p kinase, which is the fission yeast homolog of Cdc7p. In all tested eukaryotic organisms, the homologues of Cdc7p and Dbf4p are essential for initiation of DNA replication at individual replication origins (Bell and Dutta, 2002). In addition, where tested, these proteins have proved to be important for viability and checkpoint activation in response to DNA damage (reviewed in (Duncker and Brown, 2003; Kim et al., 2003)). Proteins of the Dbf4p family are not well conserved between species, except for three small motifs (N, M and C) in the N-terminal, middle and C-terminal portions of the protein, respectively (Takeda et al., 1999). Of these, motif M is essential for initiation of replication, motif N is important for protection against a wide range of DNA-damaging agents, and motif C is important specifically for protection against alkylation damage (Fung et al., 2002; Ogino et al., 2001; Takeda et al., 1999).
We wondered whether mutations in the non-essential N- and C-terminal portions of Dfp1p might lead to ROS production and cell death even in the absence of exogenous DNA-damaging agents. To test this possibility, we measured the extent of ROS production and PI staining in a series of N- and C-terminal deletion mutants that had been prepared in the laboratory of Grant Brown (Fung et al., 2002). These included three N-terminal deletions, all of which removed motif N (dfp1Δ183–191, dfp1Δ13–193, and dfp1Δ13–240) and two C-terminal deletions, both of which removed motif C (dfp11–376 and dfp11–459). None of the deletions affected the essential motif M. Interestingly, we found that both N- and C-terminal deletion mutants produced ROS and stained with PI at frequencies significantly elevated compared to wild-type cells under the same conditions (Figs. 1, 4).
Fig. 4.
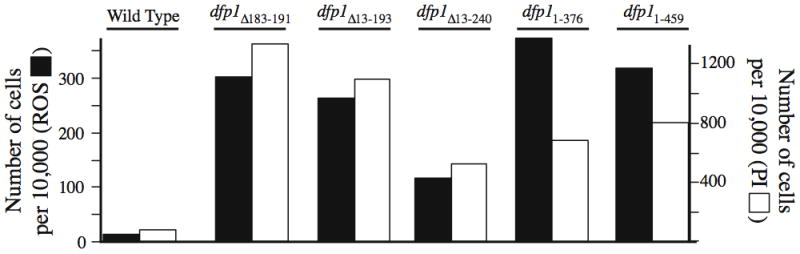
N- and C-terminal deletions in dfp1 lead to ROS production and PI staining. The indicated fission yeast strains were incubated for 80 minutes at 25°C in the presence of DCDHFDA and also stained with PI as in Fig. 1.
Apparent replication-mutant specificity of ROS production
All of the mutations discussed above are in genes essential for initiation of DNA replication. We wanted to know whether DNA metabolism mutants that do not directly affect replication display similarly enhanced ROS production (ROS production in greater than 100 cells per 10,000). We tested the effects of deleting genes encoding proteins required for non-homologous end joining (pku70 and lig4 (Manolis et al., 2001)), for the DNA damage checkpoint (rhp9 (Willson et al., 1997)), and for maintenance of replication fork stability under stress conditions (srs2 and rqh1 (Maftahi et al., 2002; Marchetti et al., 2002)). We also tested genes encoding subunits of the MRN complex (rad32 and rad50), which is involved in homologous recombination and checkpoint activation (Chahwan et al., 2003; Hartsuiker et al., 2001; Tavassoli et al., 1995). As shown in Fig. 5, none of the tested non-replication deletion mutants generated ROS to the same extent as certain mutant alleles of the tested genes encoding replication initiation proteins (Figs. 1–4). These results show that, in the absence of exogenous stressors, replication defects are more likely to lead to ROS production than are recombination or checkpoint defects. The differences between complete deletions of recombination and checkpoint genes (Fig. 5) and point mutations or partial deletions in replication initiation genes (Figs. 1–4) may be a simple consequence of the fact that the replication initiation proteins carry out essential functions in every cell cycle, but the functions of the tested recombination and checkpoint genes may be important only under conditions of DNA damage or replication fork block.
Fig. 5.
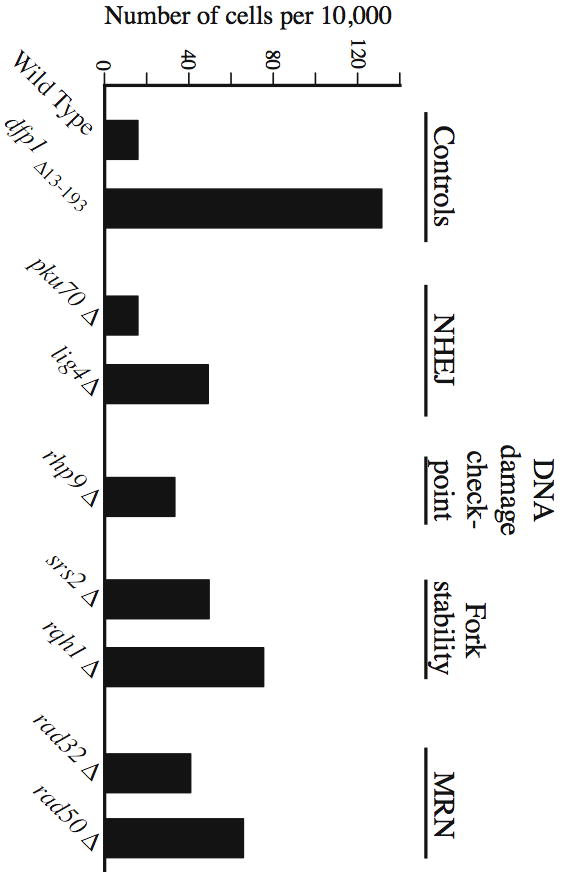
Mutants in pathways affecting non-homologous end joining, the DNA damage checkpoint, replication fork stability, and the MRN complex appear to produce less ROS than mutants in replication proteins. The indicated strains were incubated at 25°C for 80 minutes in the presence of DCDHFDA. Other conditions were as in Figs. 1 and 4.
Checkpoint dependence (or lack thereof) of elevated ROS production and PI staining by replication mutants
We wondered whether the increased ROS production and PI staining evident in certain replication mutant strains would be affected by inactivation of checkpoint pathways. To test this, we deleted the rad3 gene (which encodes Rad3p, the fission yeast ATR homolog, and is essential for the damage and replication checkpoints (Bentley et al., 1996; Furuya and Carr, 2003)) or the cds1 gene (which encodes Cds1p, the fission yeast Chk2p homolog, and is essential for the replication checkpoint (Furuya and Carr, 2003; Lindsay et al., 1998; Murakami and Okayama, 1995)) from cells bearing the orp5-H19, dfp1Δ13–193, or dfp1Δ183–191 mutations. The results in Fig. 6 show that neither the rad3 nor cds1 deletion by itself had a significant effect on ROS production or PI staining. These deletions also had no significant effect on the already elevated ROS production and PI staining of the orp5-H19 strain. In contrast, deleting either rad3 or cds1 inhibited the elevated ROS production and PI staining of the dfp1 mutants. Thus a functional replication checkpoint is required for elevated ROS production by dfp1 N-terminal deletion mutants, but ROS production in the orp5-H19 strain is independent of both the replication and damage checkpoints. We conclude that, although many replication initiation mutants display elevated ROS production and PI staining, the pathways leading from replication defects to ROS production are not all identical. At least two pathways are involved, depending on the replication mutant. One pathway requires replication checkpoint function; the other is checkpoint-independent.
Fig. 6.
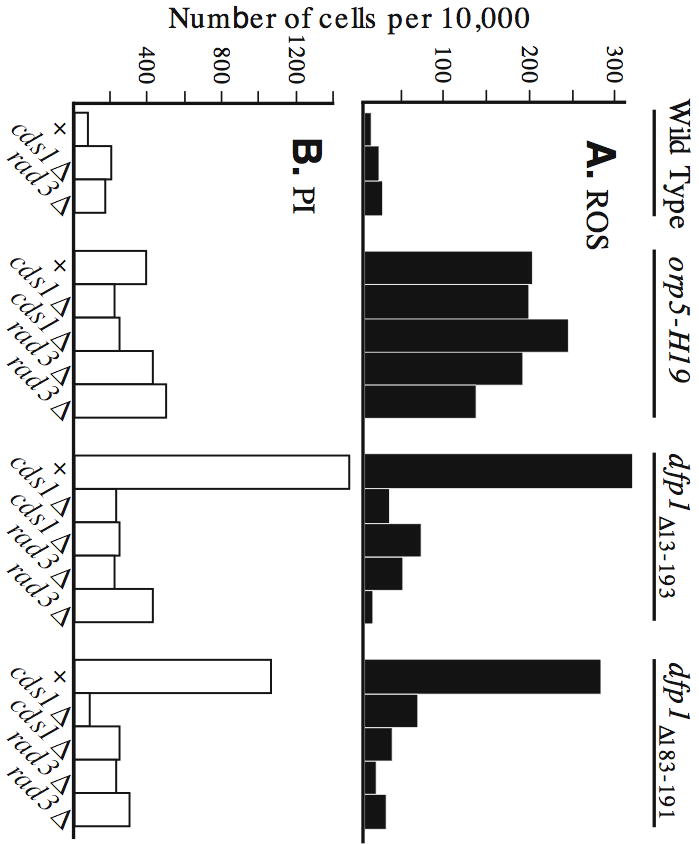
ROS production by dfp1 N-terminal deletion mutants requires a functional Rad3p- and Cds1p-dependent checkpoint pathway, but ROS production by the orp5-H19 strain does not. Double-mutant strains bearing orp5-H19 or dfp1Δ13–193 or dfp1Δ183–191 along with rad3Δ or cds1Δ were generated by crossing. Results are shown for two independently derived isolates of each type. Strains lacking a cds1Δ or rad3Δ mutation are indicated by “+”. Cells in early log phase were incubated at 30°C for six hours. (A) ROS production. (B) PI staining.
Hydroxyurea-dependent ROS production and PI staining in checkpoint mutant strains
Although ROS production and PI staining were not significantly elevated in the cds1 and rad3 deletion strains (Fig. 6), we suspected that they might be elevated when these mutant strains were treated with hydroxyurea (HU), because these strains are highly sensitive to HU (Jimenez et al., 1992; Murakami and Okayama, 1995). HU inhibits ribonucleotide reductase, which leads to depletion of deoxyribonucleoside triphosphates and to the stalling of replication forks. An intact replication checkpoint involving Rad3p and Cds1p is required to maintain the stability of such stalled forks and to prevent eventual cell death (Lindsay et al., 1998; Murakami and Okayama, 1995). In the absence of the replication checkpoint, the DNA damage checkpoint (dependent on Rad3p and Chk1p) is required to inhibit rapid passage through mitosis with unreplicated DNA in the presence of HU (Boddy et al., 1998). In addition to Rad3p, both of these checkpoints require the five additional “checkpoint-Rad” proteins, Rad1p, Rad9p, Rad17p, Rad26p and Hus1p (Al-Khodairy et al., 1994). These proteins are the fission yeast homologs of the mammalian proteins with the same names, except for Rad26p, whose mammalian homolog is ATRIP (reviewed in (Nyberg et al., 2002)).
When we tested deletions of the genes encoding the checkpoint-Rad proteins we found that, in each case, treating the mutant strain with HU led to elevated ROS production and PI staining (Fig. 7). In contrast to the HU dependence of ROS production in checkpoint-Rad-mutant strains, ROS production in dfp1 N-terminal and C-terminal deletion strains, which is high in the absence of HU, could not be significantly further stimulated by addition of HU (Fig. S2). We conclude that one or more checkpoint pathways, which are dependent on the checkpoint-Rad proteins, are required to prevent elevated ROS production and cell death when fission yeast cells are exposed to HU.
Fig. 7.
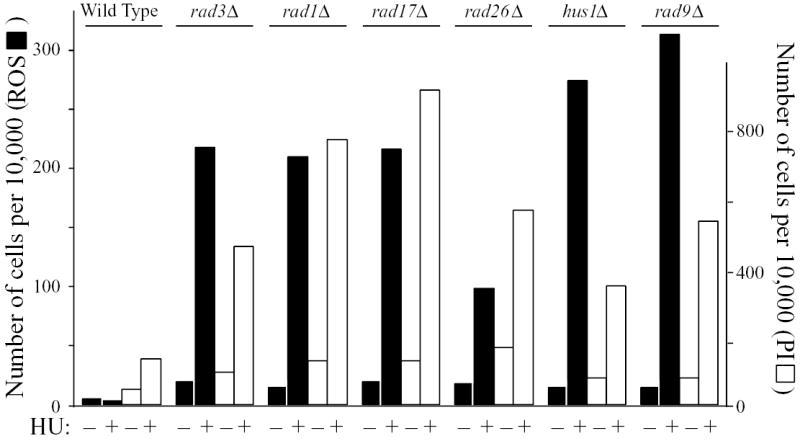
Deletion of any of the checkpoint-Rad genes leads to elevated ROS production and PI staining in the presence of HU. The indicated strains were incubated at 25°C for 4 hours with or without 12 mM HU. Then DCDHFDA was added, and incubation was continued for 80 minutes. The cells were stained with PI and analyzed by flow cytometry as in previous figures.
HU-dependent, checkpoint-mutant-dependent ROS production requires inappropriate entry into mitosis
The two well-characterized checkpoint pathways operating downstream of the checkpoint-Rad proteins are the replication checkpoint, which is dependent on Cds1p (Murakami and Okayama, 1995), and the damage checkpoint, which is dependent on Chk1p (Walworth et al., 1993). To determine which of these checkpoint pathways might be required to prevent HU-induced elevated ROS production, we measured the effects on HU-induced ROS production of deleting the cds1 and chk1 genes. We found that neither of the single deletions facilitated ROS production or cell death during a 4-hour incubation with HU, but the HU-treated double-deletion (cds1Δ chk1Δ) strain produced about as much ROS and PI staining as the HU-treated rad3Δ strain (Fig. 8).
Fig. 8.
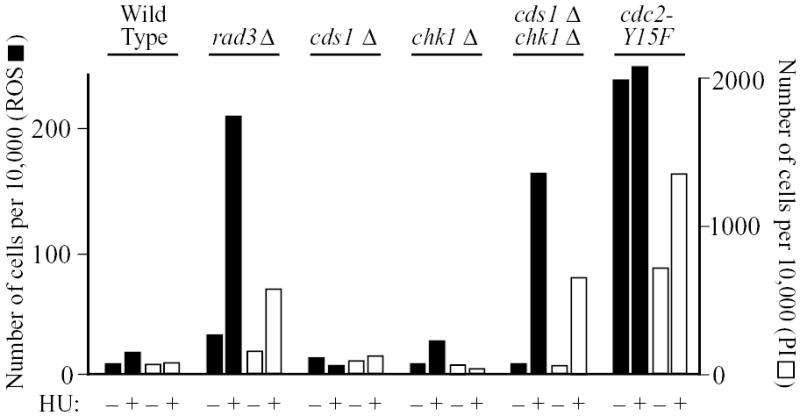
Stimulation of ROS production and PI staining by HU requires abrogation of both the Cds1p- and Chk1p-dependent checkpoint pathways or dysregulation of Cdc2p. The indicated strains were incubated at 25°C for 4 hours with or without 12 mM HU. Then DCDHFDA was added, and incubation was continued for 80 minutes. The cells were stained with PI and analyzed by flow cytometry as in previous figures.
Since HU-treated rad3Δ or cds1Δ chk1Δ cells enter mitosis with unreplicated DNA, while HU-treated wild-type or single-mutant (cds1Δ or chk1Δ) cells do not do so after 4 hours of HU treatment (Boddy et al., 1998; Murakami and Okayama, 1995), these results suggested that the critical event triggering ROS production might be inappropriate entry into mitosis. To test this, we took advantage of the fact that inappropriate entry into mitosis can also be induced by the cdc2-Y15F mutation, in which tyrosine 15 of the Cdc2p cyclin-dependent kinase is replaced by phenylalanine, rendering the site non-phosphorylatable. Since phosphorylation of tyrosine 15 is a major means of negatively regulating Cdc2p kinase activity, Cdc2-Y15Fp is constitutively active. It promotes premature entry into mitosis, giving rise to shortened cells (Gould and Nurse, 1989). If inappropriate entry into mitosis is a trigger for ROS production and cell death, then cdc2-Y15F cells should display constitutively elevated ROS production and cell death even in the absence of HU. Indeed, that is what we found in the experiment of Fig. 8. In other cases, however, we found that—although the cdc2-Y15F mutation led to elevated ROS production— ROS production could be further enhanced by treatment with HU (example in Fig. S3).
Discussion
Defects in replication initiation proteins can induce ROS production and cell death
It is striking that at least one mutant allele of each of the tested genes encoding replication initiation proteins (cdc18, dfp1, orp2 and orp5) proved capable of inducing significant ROS production and cell death (Figs. 1–4, 6). In contrast, deletions of genes encoding proteins involved in non-homologous end joining (pku70 and lig4), homologous recombination (rad32 and rad50), replication fork stability (rqh1 and srs2), and the DNA damage checkpoint (rhp9) had little or no effect on ROS production (Fig. 5).
The reason for this difference between replication initiation proteins and proteins involved in other aspects of DNA metabolism is not clear. Defects in both sets of proteins would be expected to lead to increased levels of DNA damage. It is possible that the types of damage generated by replication initiation defects are distinguishable from the types of damage generated by defects in the other pathways that we studied. It is also possible that the high-ROS-generating mutant alleles of replication initiation genes produce more spontaneous DNA damage than do deletions of the tested recombination, fork stability and checkpoint genes.
ROS production stimulated by defects in replication initiation proteins is not further enhanced by checkpoint mutations
Rad3p and Cds1p are both essential for DNA-integrity checkpoints during S phase in fission yeast (Furuya and Carr, 2003; Rhind and Russell, 2000). We suspected that these replication checkpoint proteins would help to protect cells from problems generated by defects in replication initiation proteins. However, when deletions of the rad3 or cds1 gene were combined with mutations affecting replication initiation proteins (Orp5p or Dfp1p), no enhancement of ROS production or cell death was detected (Fig. 6). In fact, in one case (Dfp1p), deletion of rad3 or cds1 reduced both ROS production and cell death to near background levels, implying that ROS production and cell death in these dfp1 mutant cells required a functional replication checkpoint. Since the rad3 and cds1 deletions had no effect on ROS production and cell death when combined with the orp5-H19 mutation (Fig. 6), we conclude that defects in replication initiation proteins lead to ROS production and cell death by at least two different pathways. One pathway requires a functional replication checkpoint and the other does not. The pathway that requires a functional replication checkpoint is reminiscent of the checkpoint-dependent pathways that, in response to replication stress, induce apoptosis in mammalian cells and thus prevent cancer (reviewed in (Venkitaraman, 2005)).
HU-induced replication stress, combined with checkpoint mutations, leads to ROS production and cell death
We did, however, find one mechanism by which checkpoint mutations could stimulate ROS production and cell death. When replication forks were slowed by treating cells with HU, then deletion of any of the checkpoint-Rad genes, or chk1 plus cds1 together, led to a striking increase of ROS production and cell death (Figs. 7, 8). The fact that significant ROS production required simultaneous deletion of cds1 and chk1 suggested that both the Cds1p-dependent replication checkpoint and the Chk1p-dependent damage checkpoint were individually capable of preventing ROS production. Since each checkpoint by itself is capable of down-regulating the Cdc2p cyclin-dependent kinase sufficiently to prevent premature entry into mitosis during a 4-hour incubation with HU (Boddy et al., 1998; Caspari and Carr, 1999), it seemed likely that inappropriate entry into mitosis in cells with unreplicated DNA might be the ultimate cause of the elevated ROS production and cell death induced by HU treatment of checkpoint-defective cells (Figs. 7, 8).
CDK dysregulation induces ROS production
To test this possibility, we measured ROS production in a fission yeast strain carrying an altered version of the gene encoding Cdc2p (the fission yeast CDK), in which tyrosine 15 had been mutated to phenylalanine (cdc2-Y15F). This form of Cdc2p is constitutively active (Gould and Nurse, 1989) and cannot be down-regulated by the Cds1p- or Chk1p-dependent checkpoint pathways. We found that cells bearing this mutation frequently produced elevated ROS and died even in the absence of HU (Figs. 8, S3). These results imply that dysregulation of CDK activity and inappropriate entry into mitosis can be significant causes of ROS production and cell death in fission yeast cells, just as they are in mammalian cells (Castedo et al., 2004; Castedo et al., 2002).
Utility of fission yeast for study of ROS production and cell death
Whether the ROS production and cell death that take place in certain replication initiation mutants and in HU-treated checkpoint-deficient cells are due to apoptosis or necrosis, they seem sufficiently similar to the ROS production and apoptosis that take place in mammalian and budding yeast cells in response to initiation defects and replication stress that it is worth considering the possible utility of fission yeast as a model organism for further studies of these cell death responses. Fission yeast offers several advantages for such studies. Like budding yeast, it is a genetically tractable organism with a completely sequenced genome. In fission yeast the pathways leading to cell death include one pathway that is initiated by inappropriate mitosis and two that are initiated by defects in replication initiation proteins (Fig. 9). The first pathway is strikingly enhanced by simultaneous defects in the replication and damage checkpoints, while the second and third pathways are checkpoint-independent and dependent on a functional replication checkpoint, respectively. This multiplicity of cell death pathways in fission yeast reminds one of the multiplicity of cell death pathways in mammalian cells. Further examination of the mechanisms of these pathways in fission yeast is likely to shed light on the mechanisms by which inappropriate mitosis and defects in replication initiation proteins lead to cell death in mammalian cells.
Fig. 9.
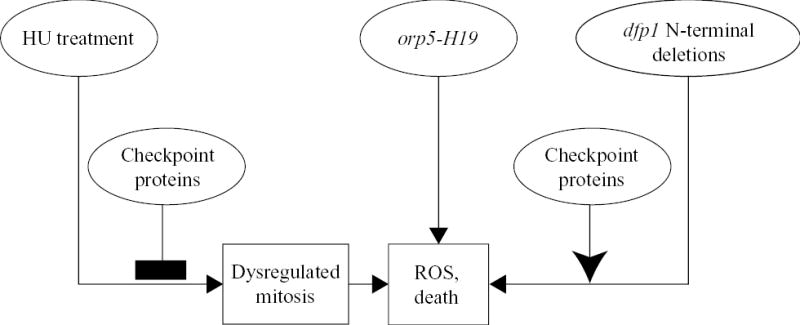
Checkpoint-inhibited, checkpoint-stimulated, and checkpoint-independent pathways leading to production of ROS and cell death in fission yeast. This figure provides a simplified diagrammatic summary of the pathways leading from mutations in genes encoding replication initiation proteins, or from HU treatment, to elevated ROS production and death.
Supplementary Material
Acknowledgments
Hiroaki Kato and Katsunori Tanaka contributed to the identification, characterization and mutagenesis of orp5. Joseph Goldbeck assisted with the initial ROS experiments. Andrew Phillips and members of the Huberman and Burhans laboratories provided useful comments on the manuscript. Grant Brown, Tony Carr, Kathy Gould, Tom Kelly, Janet Leatherwood, and Hiroto Okayama provided strains. Research in the Murakami laboratory was supported by a Grant-in-Aid for Scientific Research (B) and a Grant-in-Aid for Scientific Research on Priority Areas (A). Research in the Burhans and Huberman laboratories was supported by NIH grants CA084086 (WCB), CA095908 (JAH) and GM070566 (JAH), as well as by the Cancer Center Support Grant (P30 CA016056) to Roswell Park Cancer Institute, which partially supports the Flow Cytometry Facility employed in this study.
References
- Al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJ, Lehmann AR, Carr AM. Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol Biol Cell. 1994;5:147–160. doi: 10.1091/mbc.5.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzan R, Sapienza K, Galea DR, Vassallo N, Frey H, Bannister WH. Aspirin commits yeast cells to apoptosis depending on carbon source. Microbiology. 2004;150:109–115. doi: 10.1099/mic.0.26578-0. [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Bentley NJ, Holtzman DA, Flaggs G, Keegan KS, DeMaggio A, Ford JC, Hoekstra M, Carr AM. The Schizosaccharomyces pombe rad3 checkpoint gene. The EMBO Journal. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Blanchard F, Rusiniak ME, Sharma K, Sun X, Todorov I, Castellano MM, Gutierrez C, Baumann H, Burhans WC. Targeted destruction of DNA replication protein Cdc6 by cell death pathways in mammals and yeast. Mol Biol Cell. 2002;13:1536–1549. doi: 10.1091/mbc.02-02-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Furnari B, Mondesert O, Russell P. Replication checkpoint enforced by kinases Cds1 and Chk1. Science. 1998;280:909–912. doi: 10.1126/science.280.5365.909. [DOI] [PubMed] [Google Scholar]
- Brezniceanu ML, Völp K, Bösser S, Solbach C, Lichter P, Joos S, Zörnig M. HMGB1 inhibitis cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J. 2003;17:1295–1297. doi: 10.1096/fj.02-0621fje. [DOI] [PubMed] [Google Scholar]
- Burhans WC, Weinberger M, Marchetti MA, Ramachandran L, D’Urso G, Huberman JA. Apoptosis-like yeast cell death in response to DNA damage and replication defects. Mutation Research. 2003;532:227–243. doi: 10.1016/j.mrfmmm.2003.08.019. [DOI] [PubMed] [Google Scholar]
- Caspari T, Carr AM. DNA structure checkpoint pathways in Schizosaccharomyces pombe. Biochimie. 1999;81:173–181. doi: 10.1016/s0300-9084(99)80050-9. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9:1287–1293. doi: 10.1038/sj.cdd.4401130. [DOI] [PubMed] [Google Scholar]
- Chahwan C, Nakamura TM, Sivakumar S, Russell P, Rhind N. The fission yeast Rad32 (Mre11)-Rad50-Nbs1 complex is required for the S-phase DNA damage checkpoint. Mol Cell Biol. 2003;23:6564–6573. doi: 10.1128/MCB.23.18.6564-6573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Park TS, Chio LC, Fischl AS, Yi XS. Induction of apoptosis by sphingoid long-chain bases in Aspergillus nidulans. Mol Cell Biol. 2003;23:163–177. doi: 10.1128/MCB.23.1.163-177.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson GE, Shi Y, Tibbetts RS. DNA replication defects, spontaneous DNA damage, and ATM-dependent checkpoint activation in replication protein A-deficient cells. J Biol Chem. 2004;279:34010–34014. doi: 10.1074/jbc.C400242200. [DOI] [PubMed] [Google Scholar]
- Duncker BP, Brown GW. Cdc7 kinases (DDKs) and checkpoint responses: lessons from two yeasts. Mutation Research. 2003;532:21–27. doi: 10.1016/j.mrfmmm.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Feng D, Tu Z, Wu W, Liang C. Inhibiting the expression of DNA replication-initiation proteins induces apoptosis in human cancer cells. Cancer Res. 2003;63:7356–7364. [PubMed] [Google Scholar]
- Fleury C, Mignotte B, Vayssière JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141. doi: 10.1016/s0300-9084(02)01369-x. [DOI] [PubMed] [Google Scholar]
- Fung AD, Ou J, Bueler S, Brown GW. A conserved domain of Schizosaccharomyces pombe dfp1+ is uniquely required for chromosome stability following alkylation damage during S phase. Mol Cell Biol. 2002;22:4477–4490. doi: 10.1128/MCB.22.13.4477-4490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya K, Carr AM. DNA checkpoints in fission yeast. J Cell Sci. 2003;116:3847–3848. doi: 10.1242/jcs.00790. [DOI] [PubMed] [Google Scholar]
- Gould KL, Nurse P. Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature. 1989;342:39. doi: 10.1038/342039a0. [DOI] [PubMed] [Google Scholar]
- Hartsuiker E, Vaessen E, Carr AM, Kohli J. Fission yeast Rad50 stimulates sister chromatid recombination and links cohesion with repair. The EMBO Journal. 2001;20:6660–6671. doi: 10.1093/emboj/20.23.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James C, Gschmeissner S, Fraser A, Evan GI. CED-4 induces chromatin condensation in Schizosaccharomyces pombe and is inhibited by direct physical association with CED-9. Curr Biol. 1997;7:246–252. doi: 10.1016/s0960-9822(06)00120-5. [DOI] [PubMed] [Google Scholar]
- Jimenez G, Yucel J, Rowley R, Subramani S. The rad3+ gene of Schizosaccharomyces pombe is involved in multiple checkpoint functions and in DNA repair. Proceedings of the National Academy of Sciences USA. 1992;89:4952–4956. doi: 10.1073/pnas.89.11.4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgensmeier JM, Krajewski S, Armstrong RC, Wilson GM, Oltersdorf T, Fritz LC, Reed JC, Ottilie S. Bax- and Bak-induced cell death in the fission yeast Schizosaccharomyces pombe. Mol Biol Cell. 1997;8:325–339. doi: 10.1091/mbc.8.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TJ, Martin GS, Forsburg SL, Stephen RJ, Russo A, Nurse P. The fission yeast cdc18+ gene product couples S phase to START and mitosis. Cell. 1993;74:371–382. doi: 10.1016/0092-8674(93)90427-r. [DOI] [PubMed] [Google Scholar]
- Kiely J, Haase SB, Russell P, Leatherwood J. Functions of fission yeast Orp2 in DNA replication and checkpoint control. Genetics. 2000;154:599–607. doi: 10.1093/genetics/154.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Yamada M, Masai H. Functions of mammalian Cdc7 kinase in initiation/monitoring of DNA replication and development. Mutation Research. 2003;532:29–40. doi: 10.1016/j.mrfmmm.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Leatherwood J, Lopez-Girona A, Russell P. Interaction of Cdc2 and Cdc18 with a fission yeast ORC2-like protein. Nature. 1996;379:360–363. doi: 10.1038/379360a0. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2',7'-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Lindsay HD, Griffiths DJF, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998;12:382–395. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F, Fröhlich E, Ligr M, Grey M, Sigrist SJ, Wolf DH, Fröhlich KU. Oxygen stress: a regulator of apoptosis in yeast. J Cell Biol. 1999;145:757–767. doi: 10.1083/jcb.145.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F, Herker E, Wissing S, Jungwirth H, Eisenberg T, Fröhlich KU. Apoptosis in yeast. Curr Opinion Microbiol. 2004;7:655–660. doi: 10.1016/j.mib.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Maftahi M, Hope JC, Delgado-Cruzata L, Han CS, Freyer GA. The severe slow growth of Δsrs2 Δrqh1 in Schizosaccharomyces pombe is suppressed by loss of recombination and checkpoint genes. Nucleic Acids Research. 2002;30:4781–4792. doi: 10.1093/nar/gkf581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolis KG, Nimmo ER, Hartsuiker E, Carr AM, Jeggo PA, Allshire RC. Novel functional requirements for non-homologous DNA and joining in Schizosaccharomyces pombe. The EMBO Journal. 2001;20:210–221. doi: 10.1093/emboj/20.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti MA, Kumar S, Hartsuiker E, Maftahi M, Carr AM, Freyer GA, Burhans WC, Huberman JA. A single unbranched S-phase DNA damage and replication forkk blockage checkpoint pathway. Proceedings of the National Academy of Sciences USA. 2002;99:7472–7477. doi: 10.1073/pnas.112702399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukami T, Chang WI, Garkavtsev I, Kaplan N, Lombardi D, Matsumoto T, Niwa O, Kounosu A, Yanagida M, Marr TG, et al. A 13 kb resolution cosmid map of the 14 Mb fission yeast genome by nonrandom sequence-tagged site mapping. Cell. 1993;73:121–132. doi: 10.1016/0092-8674(93)90165-m. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast, Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S phase. Nature. 1995;374:817–819. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- Muzi-Falconi M, Brown GW, Kelly TJ. cdc18+ regulates initiation of DNA replication in Schizosaccharomyces pombe. Proceedings of the National Academy of Sciences USA. 1996;93:1566–1570. doi: 10.1073/pnas.93.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CJ, Zhivotovsky B. DNA damage-induced apoptosis. Oncogene. 2004;23:2797–2808. doi: 10.1038/sj.onc.1207532. [DOI] [PubMed] [Google Scholar]
- Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. TOWARD MAINTAINING THE GENOME: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- Ogino K, Takeda T, Matsui E, Iiyama H, Taniyama C, Arai K-i, Masai H. Bipartite binding of a kinase activator activates Cdc7-related kinase essential for S phase. J Biol Chem. 2001;276:31376–31387. doi: 10.1074/jbc.M102197200. [DOI] [PubMed] [Google Scholar]
- Pelizon C, d’Adda di Fagagna F, Farrace L, Laskey RA. Human replication protein Cdc6 is selectively cleaved by caspase during apoptosis. EMBO Rep. 2002;3:780–784. doi: 10.1093/embo-reports/kvf161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Li TK, Kuo D, Nur-E-Kamal A, Liu LF. Inactivation of Cdc13p triggers MEC1-dependent apoptotic signals in yeast. J Biol Chem. 2003;278:15136–15141. doi: 10.1074/jbc.M212808200. [DOI] [PubMed] [Google Scholar]
- Rhind N, Russell P. Checkpoints: It takes more than time to heal some wounds. Curr Biol. 2000;10:R908–R911. doi: 10.1016/s0960-9822(00)00849-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriquez-Menocal L, D’Urso G. Programmed cell death in fission yeast. FEMS Yeast Research. 2004;5:111–117. doi: 10.1016/j.femsyr.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Schories B, Engel K, Dorken B, Gossen M, Bommert K. Characterization of apoptosis-induced Mcm3 and Cdc6 cleavage reveals a proapoptotic effect for one Mcm3 fragment. Cell Death Differ. 2004;11:940–942. doi: 10.1038/sj.cdd.4401411. [DOI] [PubMed] [Google Scholar]
- Shreeram S, Sparks A, Lane DP, Blow JJ. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene. 2002;21:6624–6632. doi: 10.1038/sj.onc.1205910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda T, Ogino K, Matsui E, Cho MK, Kumagai H, Miyake T, Arai K-i, Masai H. A fission yeast gene, him1+/dfp1+, encoding a regulatory subunit for Hsk1 kinase, plays essential roles in S-phase initiation as well as in S-phase checkpoint control and recovery from DNA damage. Mol Cell Biol. 1999;19:5535–5547. doi: 10.1128/mcb.19.8.5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli M, Shayeghi M, Nasim A, Watts FZ. Cloning and characterisation of the Schizosaccharomyces pombe rad32 gene: a gene required for repair of double strand breaks and recombination. Nucleic Acids Research. 1995;23:383–388. doi: 10.1093/nar/23.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya M, Suematsu M, Suzuki H. In vivo visualization of oxygen radical-dependent photoemission. Methods Enzymol. 1994;233:128–140. doi: 10.1016/s0076-6879(94)33015-8. [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR. Aborting the birth of cancer. Nature. 2005;434:829–830. doi: 10.1038/434829a. [DOI] [PubMed] [Google Scholar]
- Walworth N, Davey S, Beach D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature. 1993;363:368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- Weinberger, M., Ramachandran, L., Feng, L., Sharma, K., Sun, X., Marchetti, M. A., Huberman, J. A. and Burhans, W. C. (2005). Apoptosis in budding yeast caused by defects in initiation of DNA replication. J. Cell Sci. In press. [DOI] [PubMed]
- Willson J, Wilson S, Warr N, Watts FZ. Isolation and characterization of the Schizosaccharomyces pombe rhp9 gene: a gene required for the DNA damage checkpoint but not the replication checkpoint. Nucleic Acids Research. 1997;25:2138–2145. doi: 10.1093/nar/25.11.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim H, Jin YH, Park BD, Choi HJ, Lee SK. Caspase-3-mediated cleavage of Cdc6 induces nuclear localization of p49-truncated Cdc6 and apoptosis. Mol Biol Cell. 2003;14:4250–4259. doi: 10.1091/mbc.E03-01-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Chieu HK, Low CP, Zhang S, Heng CK, Yang H. Schizosaccharomyces pombe cells deficient in triacylglycerols synthesis undergo apoptosis upon entry into the stationary phase. J Biol Chem. 2003;278:47145–47155. doi: 10.1074/jbc.M306998200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.