

Abstract
Sulfated glycosaminoglycans (GAGs) are linear polysaccharides of repeating disaccharide sequences on which are superimposed highly complex and variable patterns of sulfation, especially in heparan sulfate (HS). HS and the structurally related heparin exert important biological functions, primarily by interacting with proteins and regulating their activities. Evidence is accumulating that these interactions depend on specific saccharide sequences, but the lack of simple, direct techniques for sequencing GAG saccharides has been a major obstacle to progress. We describe how HS and heparin saccharides can be sequenced rapidly by using an integrated strategy with chemical and enzymic steps. Attachment of a reducing-end fluorescent tag establishes a reading frame. Partial selective chemical cleavage at internal N-sulfoglucosamine residues with nitrous acid then creates a set of fragments of defined sizes. Subsequent digestion of these fragments with combinations of exosulfatases and exoglycosidases permits the selective removal of specific sulfates and monosaccharides from their nonreducing ends. PAGE of the products yields a pattern of fluorescent bands from which the saccharide sequence can be read directly. Data are presented on sequencing of heparin tetrasaccharides and hexasaccharides of known structure; these data show the accuracy and versatility of this sequencing strategy. Data also are presented on the application of the strategy to the sequencing of an HS decasaccharide of unknown structure. Application and further development of this sequencing strategy, called integral glycan sequencing, will accelerate progress in defining the structure–activity relationships of these complex GAGs and lead to important insights into their biological functions.
Glycosaminoglycans (GAGs) have a variety of important biological functions, some of which are known to depend on the presence of specific sulfated sequences that confer the ability to bind to protein ligands and influence their activities (1). Heparan sulfate (HS) and heparin have been the focus of much interest, because a huge variety of proteins are known to bind these polysaccharides. They are known to modulate the function of the proteins with which they interact, such as antithrombin III (2) and fibroblast growth factors (3), and some of the specific sequences that confer these functions have been elucidated (4–6). These observations suggest that the biological activity of proteins possessing HS-binding motifs can be modulated by the controlled expression of specific protein-binding HS saccharide sequences. HS and heparin consist of a disaccharide repeat of glucosamine and hexuronic acid {linkage sequence [(1 → 4)α-d-glucosaminyl-(1 → 4)β-d-hexuronosyl]n, where n = 50 to 150}, in which the glucosamine is either N-acetylglucosamine (GlcNAc) or N-sulfoglucosamine (GlcNS) and the hexuronate is present as either glucuronate (GlcA) or its C-5 epimer iduronate (IdoA). Ester (O)-sulfation, which occurs predominantly at C-2 of IdoA and C-6 of the glucosamine residues, but also rarely at C-2 of GlcA and C-3 of glucosamine, adds structural complexity to the polymer chain (7). Critically, at each step, only a fraction of potential substrates are modified, resulting in extensive scope for sequence diversity. Patterns of sulfation and epimerization are controlled tightly but differentially during biosynthesis, and polymer modification is directed to distinct regions of the chains, resulting in an ordered arrangement of structural domains. N- and O-sulfate groups are in close proximity and arranged in relatively discrete clusters called S domains, separated by regions of low sulfation in which the disaccharides are mainly N-acetylated (8). Specific sulfated sequences, predominantly in the modified S domains, form recognition motifs for protein ligands that are essential to biological function.
Determination of GAG structure is a formidable analytical problem that has limited structure–activity studies, and the development of improved methods is necessary for further progress. Current approaches involve separation techniques (such as size-exclusion chromatography, strong anion exchange (SAX)-HPLC, PAGE, and capillary electrophoresis) that permit analysis of disaccharide composition and susceptibility to heparitinases or low pH nitrous acid. These provide important data on composition and domain structure (9–12) but generally yield indirect and incomplete sequence information. Approaches involving the use of NMR and mass spectroscopy provide accurate sequence information for small saccharides (13–16) but require specialist experience and equipment not readily available to many researchers and, in the case of NMR, large amounts of material (nanomolar to micromolar quantities). Because it is now possible to purify HS saccharides to sufficient homogeneity by using SAX-HPLC and PAGE techniques (17, 18) and to identify functional saccharides by affinity chromatography or screening in bioassays (5, 6, 18), there is an urgent need for direct sequencing strategies that can be applied routinely to small amounts of saccharides in any laboratory.
Exoglycosidases have proved invaluable reagents for sequencing carbohydrate chains (19), and their use in sequencing methods with reducing-end fluorescent tags and PAGE separations has been described (20, 21). The recent availability of recombinant exolytic lysosomal enzymes that degrade GAGs (22, 23) allows their exploitation for sequence analysis, because they remove specific sulfate groups or monosaccharide residues selectively from the nonreducing end of saccharides (22, 23). These exoenzymes can be used to degrade saccharides sequentially and, in concert with high resolution PAGE oligosaccharide mapping, provide a direct approach for determining the structure of the nonreducing terminal sequences of purified GAG saccharides (24). Here, we report a strategy that integrates the exoenzymes with a chemical cleavage step and permits rapid sequencing of HS and heparin saccharides. Data are presented that show the principles of this new technique, referred to as integral glycan sequencing (IGS), because it is capable of identifying complete sequence information in a single experiment. IGS requires only small amounts (picomoles) of material and produces accurate sequence information from test saccharides of known structure. We also show the application of IGS to the sequencing of an HS decasaccharide of unknown structure, purified by SAX-HPLC. Some of these data have been described in abstract form (25).
EXPERIMENTAL PROCEDURES
Materials.
Heparin tetrasaccharides F3, ΔHexA(2S)-GlcNS(6S)-GlcA-GlcNS(6S), and F4, ΔHexA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S), [note that ΔHexA(2S) is Δ4,5-unsaturated uronate residue 2-sulfate; GlcNS(6S) is GlcNS 6-sulfate; and IdoA(2S) is IdoA 2-sulfate] were a gift from Bob Linhardt (University of Iowa, Iowa City, IA; ref. 26), and heparin hexasaccharide 6-27 was a gift from Kazuyuki Sugahara (University of Kobe, Kobe, Japan; ref. 16). Recombinant exoenzymes were prepared as described below and are available from Oxford Glycosciences (Abingdon, U.K.), which also supplies kits for 2-aminobenzoic acid (2AA)-labeling and PAGE gel solutions. Porcine mucosal HS was a gift from Organon (Oss, The Netherlands). Formamide and 2AA (99% purity grade) were supplied by Fluka, and sodium cyanoborohydride was supplied by Aldrich. Heparitinase and HS disaccharide standards were gifts from Keiichi Yoshida (Seikagaku Kogyo, Tokyo).
Fluorescent Labeling of Saccharides.
Saccharide samples, typically 1–2 nmol, were dried by centrifugal evaporation in microtubes and then subjected to reductive amination by dissolving the sample in formamide containing 1 M sodium cyanoborohydride and 0.4 M 2AA and incubating for 16–24 h at 37°C. A 1,000-fold molar excess of 2AA over saccharide (e.g., 2.5 μl of reaction mix per 1 nmol of saccharide) was used. Labeled saccharide was separated from free label by gel filtration on Sephadex G-25 Superfine (Amersham Pharmacia). Samples (diluted to 500 μl with H2O) were run on two 5-ml HiTrap desalting columns (Amersham Pharmacia), connected in series, eluted with H2O, and monitored by fluorescence (310-nm excitation/420-nm emission) or UV absorbance (310 nm). Void-volume fractions were pooled and concentrated by centrifugal evaporation, and the amount of fluorescent product was estimated by using a fluorometer with reference to a 2AA standard.
Partial Nitrous Acid Cleavage.
Controlled partial nitrous acid (HNO2) cleavage was carried out as described (27) with modifications. Samples in H2O were made to 20 mM HCl and 2 mM sodium nitrite by addition of ×10 strength solutions and incubated on ice for 1–4 h. At a series of time points (typically, 1, 2, 3, and 4 h), an aliquot of the digest was removed, and the pH was raised to ≈5.0 by the addition of 1/5 volume of 200 mM sodium acetate buffer (pH 6.0). On completion of the reaction, the aliquots were pooled and either used directly for enzyme digests or desalted with a G-25 column.
Mercuric Acetate Cleavage.
Samples were dissolved in 25 μl of H2O, mixed with 25 μl of 30 mM mercuric acetate in 150 mM sodium acetate (pH 5.0; ref. 28), and incubated at 37°C for 60 min. The incubation was followed by the addition of 200 μl of 4 M sodium chloride and desalted with a G-25 column.
Exoenzyme Digests.
Recombinant human iduronate-2-sulfate sulfatase (I2Sase; EC 3.1.6.13; stock activity = 6 units/ml), human α-l-iduronidase (Idase; EC 3.2.1.76; stock activity = 3.6 units/ml), caprine glucosamine-6-sulfate sulfatase (G6Sase; EC 3.1.6.14; stock activity = 810 milliunits/ml), and human α-N-acetylglucosaminidase (NAG; EC 3.2.1.50; stock activity = 175 milliunits/ml) were prepared as described (refs. 29–32; 1 unit of activity is 1 μmol of substrate cleaved per min). Recombinant human sulfamidase (NSase; EC 3.10.1.1; stock activity = 100 milliunits/ml) also was prepared (B. Weber and J. Hopwood, unpublished observations). Bovine-liver β-d-glucuronidase (Gase; EC 3.2.1.31; stock activity = 40 Fishman units/μl) was obtained from Sigma (G-0501). Saccharides (typically, 20–200 pmol) were treated with the exoenzymes at 37°C for 4–16 h in a final volume of 25 μl of 50 mM sodium acetate buffer (pH 5.0) containing 100 μg/ml BSA. Final enzyme concentrations for effective complete digestion were in the range 10–50 milliunits/ml. Multiple digests were carried out by simultaneous addition of all the enzymes, except NSase, which was added after 6Sase digestion was complete, because glucosamine 6-sulfate is a very poor substrate for the G6Sase (33). Enzyme digests were terminated by heating the digest at 100°C for 5 min.
PAGE and Gel Imaging.
PAGE with Tris-glycine gels and electrotransfer to nylon membranes for purification and recovery of saccharides were performed as described (8, 17). PAGE was also performed with Tris-acetate gels with Tris-Mes running buffer prepared according to the supplier’s instructions (Oxford Glycosciences). Resolving gel formats were either minigel (6-cm) or standard (16-cm) lengths, and the acrylamide concentrations were in the range 25–35%. Running conditions were 200 V for sample loading (30–60 min), followed by 400 V (minigels) or 400–800 V (larger gels) for 2–8 h. Gels were imaged (after removal from the glass plates) on a UV transilluminator (312-nm maximum emission wavelength) fitted with a glass UV band-pass filter (type UG-1 or MUG-2; HV Skan, Solihull, Midlands, U.K.). Images were captured by using grab-it software and a gel-imaging camera (Ultraviolet Products), fitted with a blue filter (≈450-nm wavelength). Note that negative images of the original gels are shown below for ease of viewing.
Saccharide Preparation.
Size-fractionated HS saccharides were prepared by heparitinase treatment of porcine mucosal HS and Bio-Gel P-10 chromatography as described (34). Heparin tetrasaccharides and hexasaccharides were isolated by partial nitrous acid treatment of intact bovine-lung heparin (Sigma) as follows. Nitrous acid reagent was made (9), and a 100-μl portion was added to a heparin sample (400 μl at 12.5 mg/ml) dissolved in distilled H2O. After incubation for 30–60 s the reaction was stopped rapidly by addition of 30 μl of 1 M Na2CO3. The products were separated on a Bio-Gel P10 column, and the tetrasaccharide and hexasaccharide pools were isolated as described (34). After 2AA-labeling, the major tetrasaccharides and hexasaccharides were isolated by preparative PAGE/electrotransfer (24).
SAX-HPLC.
SAX-HPLC of HS saccharides was performed on a Propac PA1 column (4 × 250 mm; Dionex; refs. 18 and 35). Samples were eluted with a linear gradient of 0–1.0 M sodium chloride over 90 min, and selected peaks were desalted with a G-25 column. Analysis of disaccharide composition was achieved by lyase depolymerization and SAX-HPLC as described (36) with modifications. Disaccharides were desalted with a G-25 column, labeled with 2AA, and separated by SAX-HPLC as above by using a gradient of 0–2 M sodium chloride over 120 min, and fluorescence (310/420 nm) was monitored with a Shimadzu RF-551 fluorometer. Identification of peaks was made by comparison with authentic unsaturated disaccharide standards labeled with 2AA.
RESULTS
Fluorescence Labeling and Exosequencing.
Fluorescence labeling is a sensitive method for detection of saccharides and has the advantage of introducing a reading frame for sequencing. Saccharides can be labeled specifically at their reducing ends by reductive amination with the simple fluorophore 2AA (37). Because the coupling reaction for standard sugars was found to be unsuitable for HS saccharides, we developed modified reaction conditions by using formamide as the solvent, which allows near-quantitative coupling of 2AA to their reducing ends (efficiency typically is 70–80%). A gel filtration step allowed rapid purification of tagged saccharide from free tagging reagent. This method resulted in good recovery of the product (typically 70–80%) free of salts that might interfere with subsequent enzymic conditions. When a 2AA-tagged heparin tetrasaccharide was purified and resolved by PAGE (Fig. 1A), fluorescence imaging could detect down to ≈1 pmol per band. Fig. 1B shows the results of treatment of the 2AA-tagged heparin tetrasaccharide with different combinations of exoenzymes and separation of the products by PAGE. Shifts were observed with each of the exoenzymes used, allowing the sequence of the nonreducing terminal disaccharide structure, IdoA(2S)-GlcNS(6S), to be read directly from the banding pattern. Note that, in most cases, the band shifts are downward (increased mobility mainly because of lower molecular mass), whereas loss of the N-sulfate group with NSase caused an upward shift (presumably because of an altered charge:mass ratio). The results show that the exoenzymes produce band shifts that can be observed readily by PAGE mapping.
Figure 1.

Principles of IGS and a simple example. (A) Fluorescence detection of different amounts of a 2AA-tagged heparin tetrasaccharide run on a 33% Tris-acetate minigel. (B) Exosequencing of a 2AA-tagged heparin tetrasaccharide with lysosomal enzymes and separation of the products on a 33% Tris-acetate minigel (15 pmol per lane). After the exoenzyme treatments, the band shifts shown indicate the structure of the nonreducing-end disaccharide unit (lane 1, untreated). (C) Schematic representation of IGS of a hexasaccharide. (D) Actual example of IGS performed on a purified heparin hexasaccharide, corresponding to the scheme in C, with the combinations of partial HNO2 and exoenzyme treatments indicated (lane 1, untreated, 25 pmol; other lanes correspond to ≈200 pmol per lane of starting sample for partial HNO2 digest). Hexasaccharide (purified from bovine-lung heparin) has the putative structure IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S)-IdoA(2S)-anhydromannose 6-sulfate. Electrophoresis was performed on a 16-cm 35% Tris-glycine gel.
IGS Strategy.
Although the exosequencing approach is very effective, it would be laborious to sequence iteratively through longer saccharide sequences, and, in addition, the presence of GlcNS residues would create an obstacle. Although NSase can remove the N-sulfate moiety, a chemical N-acetylation step would be required to create a substrate for NAG, because there is no enzyme available for removal of glucosamine. We, therefore, devised a strategy in which controlled partial HNO2 cleavage of the tagged saccharide is carried out before the exoenzyme digests. This step creates a set of fragments of defined sizes with newly exposed nonreducing ends that correspond to internal sequences of the original saccharide. The sizes of these fragments defines the positions of GlcNS and GlcNAc residues (respectively susceptible and resistant to partial HNO2 cleavage) within the saccharide. Digestion of the fragment set with combinations of exosulfatases and exoglycosidases then permits the selective removal of specific sulfates and monosaccharides from their nonreducing ends. PAGE of the products yields a pattern of fluorescent bands from which the saccharide sequence can be read directly. This strategy, which we have called IGS, has the advantage of being capable of providing complete sequence information in a single run and avoids the need to sequence past GlcNS residues. A simple schematic example of IGS of a hexasaccharide is shown in Fig. 1C, and Fig. 1D shows an actual IGS run of a highly sulfated hexasaccharide purified from bovine-lung heparin, whose sequence corresponds to the schematic example. The putative sequence of the original hexasaccharide is IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S)-IdoA(2S)-anhydromannose 6-sulfate, a typical highly sulfated sequence obtained as a major product from the depolymerization of heparin (38). Treatment of the tagged saccharide with partial HNO2 produced both tetrasaccharide and disaccharide products in addition to some undigested intact saccharide (Fig. 1D, lane 2). This result shows that the saccharide is N-sulfated at each position. Each of the bands was resistant to Gase (Fig. 1D, lane 3) but shifted on digestion with both I2Sase (Fig. 1D, lane 4) and Idase (Fig. 1D, lane 5), thus confirming the expected core structure of repeating IdoA(2S)-GlcNS units. The presence of 6-O-sulfates was not determined in this simple example but can be achieved with an additional digest step with G6Sase (see below). Note that some minor “ghost” bands that migrate more slowly than the major bands are sometimes observed after nitrous acid cleavage (Fig. 1D, lane 2). We have also noted some loss of fluorescence on partial HNO2 treatment (typically 50%), possibly because of instability of the 2AA-saccharide linkage under the acidic conditions. The latter limits the sensitivity of detection, but we have found that neither of these artifacts affect the interpretation of the banding pattern (see Discussion).
IGS of Heparin Saccharides of Known Structure.
To test the accuracy of IGS, we used it to sequence two heparin tetrasaccharides of known structure with internal GlcNS residues but different reducing terminal disaccharide units (26). Partial HNO2 treatment produced the expected disaccharide cleavage product in each case, and subsequent digestion with different combinations of exoenzymes identified sequence information on the disaccharide unit created (data not shown). In the case of tetrasaccharide F3, ΔHexA(2S)-GlcNS(6S)-GlcA-GlcNS(6S), shifts with Gase and then G6Sase were observed for the disaccharide, but none were observed with I2Sase or Idase. In contrast, with the tetrasaccharide F4, ΔHexA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S), a series of shifts with I2Sase, Idase, and G6Sase were observed for the disaccharide, but none were observed for Gase. These data are in complete agreement with the known sequences obtained by NMR (26), and show the efficacy of the combination of partial HNO2 and exoenzyme treatments that form the basis of IGS.
One of the potential difficulties with IGS is the presence of one or more GlcNAc residues (which are resistant to HNO2 cleavage) interrupting contiguous sequences of GlcNS residues. These would result in gaps in the sequence, because partial HNO2 will not create a band corresponding to the adjacent disaccharide unit on the reducing side. However, IGS can sequence structures with internal GlcNAc residues by the additional use of the enzyme NAG. We used IGS to sequence heparin hexasaccharide 6-27 (16) with the structure
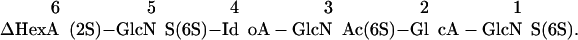 |
Partial HNO2 treatment of heparin hexasaccharide 6-27 created only the tetrasaccharide product because of the cleavage at the GlcNS at position 5, numbering from the reducing end (Fig. 2A, lane 2), but no disaccharide product was created because of the GlcNAc at position 3. Standard IGS digests provided sequence information only on the IdoA-GlcNAc(6S) structure at positions 3 and 4 (Fig. 2A, lanes 1–4). I2Sase did not produce a shift in this tetrasaccharide (data not shown). However, the digestion of the trisaccharide product in Fig. 2A, lane 4, corresponding to the structure GlcNAc-GlcA-GlcNS(6S), with NAG produces the disaccharide (Fig. 2A, lane 5) not released by partial HNO2, and the remaining sequence information then can be obtained by secondary exoenzyme combinations [in this case shifts with Gase (Fig. 2A, lane 6) and G6Sase (Fig. 2A, lane 7)]. Note that the exoenzyme digests must be terminated (by heating to 100°C) before the NAG treatment to prevent their action on the nonreducing disaccharide unit created by the action of NAG.
Figure 2.

IGS of a heparin hexasaccharide of known structure. Heparin hexasaccharide 6-27, with the structure ΔHexA(2S)-GlcNS(6S)-IdoA-GlcNAc(6S)-GlcA-GlcNS(6S), was 2AA-tagged and subjected to sequencing on 16-cm 33% Tris/acetate gels. (A) IGS of heparin hexasaccharide 6-27 with the combinations of partial HNO2 and exoenzyme treatments indicated (lane 1, untreated, 20 pmol; other lanes correspond to ≈90 pmol per lane of starting sample for partial HNO2 digest). (B) Determining the sequence of the nonreducing disaccharide unit of heparin hexasaccharide 6-27 by using the I2Sase, G6Sase, and mercuric acetate (MA) treatments shown (lane 1, untreated; ≈20 pmol per lane).
Because heparin hexasaccharide 6-27 was created by heparinase cleavage, additional treatments are required to obtain complete sequence information on the nonreducing disaccharide unit. We have observed that I2Sase is capable of removing 2-O-sulfate groups from ΔHexA residues (Fig. 2B, lane 2), whereas the resulting unsulfated ΔHexA is resistant to both Gase and Idase (data not shown). However, complete sequencing of the nonreducing terminal unit of lyase-derived saccharides is possible by using mercuric acetate (28) to selectively remove the ΔHexA residue (Fig. 2B, lane 3). Subsequent G6Sase digestion also produced a shift (in this case an upwards shift because of lower mobility; Fig. 2B, lane 4). Together, these data identify the nonreducing disaccharide structure as ΔHexA(2S)-GlcNS(6S). The complete sequence of heparin hexasaccharide 6-27 identified by IGS was in full agreement with the known sequence established by NMR (16).
IGS of a Purified HS Decasaccharide.
To demonstrate the application of IGS to a larger saccharide of unknown structure, an HS decasaccharide was purified by SAX-HPLC (Fig. 3A, arrow) and subjected to IGS (Fig. 3B). Partial HNO2 treatment created bands at each disaccharide unit (Fig. 3B, lane 2), indicating the presence of GlcNS units at each position. Gase digestion did not produce shifts at any position (Fig. 3B, lane 3), whereas I2Sase and Idase digestions resulted in shifts at the internal positions 4, 6, and 8 numbering from the reducing end (Fig. 3B, lanes 4 and 5; see below). The disaccharide product was resistant to I2Sase but shifted with Idase, although the resulting monosaccharide was not clearly evident on the gel. Shifts caused by G6Sase digestion were evident only at position 7 (Fig. 3B, lane 6, arrows). This single experiment provided information on the core sequence of this saccharide, but additional experiments were carried out to complete the sequencing at the reducing and nonreducing ends. Because the presence or absence of a 6-O-sulfate on the reducing terminal was unclear in the IGS run (Fig. 3B, lane 6), we treated some labeled decasaccharide with heparitinase II to digest it to disaccharides and analyzed the products by SAX-HPLC. The only 2AA-labeled disaccharide observed was ΔHexA-GlcNS (data not shown), indicating that there was no 6-O-sulfate on the reducing disaccharide unit. We have found this approach for determining the reducing end disaccharide structure useful, because the shifts achieved with G6Sase on monosaccharides are sometimes incomplete, probably because the fluorophore-tagged monosaccharide is in an open-ring conformation and thus a poor substrate for G6Sase (33). The nonreducing terminal was determined to be ΔHexA-GlcNS(6S), because it was resistant to I2Sase digestion (Fig. 3B, lane 4) but was susceptible to both mercuric acetate and G6Sase digestion (Fig. 3C, lanes 2 and 3, respectively). In total, these sequencing experiments required ≈3 nmol of starting material (≈7.5 μg), and the data indicated the sequence to be
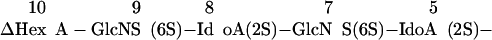 |
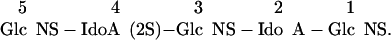 |
Analysis of the disaccharide composition of the decasaccharide by SAX-HPLC (data not shown) indicated the following composition: ΔHexA-GlcNAc, 4%; ΔHexA-GlcNAc(6S), 4%; ΔHexA-GlcNS, 22%; ΔHexA-GlcNS(6S), 20%; ΔHexA(2S)-GlcNS, 34%; and ΔHexA(2S)-GlcNS(6S), 16%. These figures are in close agreement with the composition expected from the IGS sequence data, assuming 100% purity (0%, 0%, 20%, 20%, 40%, and 20%, respectively); the small amounts of other disaccharide units are probably caused by minor contaminants.
Figure 3.

Purification and IGS of an HS decasaccharide. (A) SAX-HPLC of a pool of HS decasaccharides derived by heparitinase treatment of porcine mucosal HS. The arrow indicates the peak selected for sequencing. (B) IGS of the purified HS decasaccharide on a 16-cm 33% Tris-acetate gel with the combinations of partial HNO2 and exoenzyme treatments indicated (lane 1, untreated, 20 pmol; other lanes correspond to ≈400 pmol per lane of starting sample for partial HNO2 digest). (C) Determining the sequence of the nonreducing disaccharide unit of the HS decasaccharide with the mercuric acetate (MA) and G6Sase treatments shown (lane 1, untreated; ≈40 pmol per lane).
DISCUSSION
Sequence analysis of carbohydrates with glycosidases is a well established approach (19), and their use in combination with electrophoretic separation methods is a developing field (20, 21). Although some studies have used exoenzymes to identify particular sulfate groups or monosaccharides in HS saccharides (39) and antithrombin-binding heparin saccharides (40–42), exoenzymes have not been used previously in a concerted manner for sequencing. Here, we have demonstrated a strategy, IGS, which involves a fluorescent detection tag, a combination of chemical and enzymic digestion steps, and separation and imaging of the products by PAGE. The techniques involved are relatively simple, and the equipment required is available in most laboratories. The data described here on IGS of heparin saccharides of known structure show the basic principles involved and indicate that the strategy permits rapid and accurate sequencing of a variety of sulfated sequences encompassing the major structural variations that occur in these molecules. In many cases, the complete sequence can be deduced in a single run, but we have also shown how additional treatments can easily provide sequence information for the structures at the nonreducing and reducing ends of saccharides. Furthermore, we have shown that IGS can be used effectively to sequence HS saccharides of unknown structure, which can be purified to a high degree of homogeneity by SAX-HPLC. Because complimentary methods for identifying bioactive HS saccharides are also well established (5, 6, 18), the availability of IGS will permit extensive and detailed structure–activity studies on HS saccharides. IGS should be particularly useful for sequencing binding saccharides obtained by affinity purification, and it even may be possible to fractionate labeled saccharides (although caution would need to be exercised to ensure that binding properties are not altered by the label).
As with any emerging technique, there are both limitations and room for further development, and we anticipate a number of improvements in IGS. One limitation is the ability of PAGE to produce shifts because of the action of the exoenzymes, particularly the exosulfatases. The examples with isocratic acrylamide gels encompass the sizes of some known protein-binding sequences and show the immediate applicability of the technique. To date, we have found that minigels and standard gels are effective for sequencing up to hexasaccharides and decasaccharides, respectively. However, sequencing of larger saccharides (up to ≈10 disaccharide units) should be readily achievable with the resolving capacity of longer and/or gradient PAGE gels that offer improved resolution (43, 44). We also envisage that IGS will be applied with alternative high-resolution techniques such as HPLC (18, 35), capillary electrophoresis (11, 12), and mass spectrometry (11). A further limitation is the sensitivity of detection; the tag described here, 2AA, is a very simple fluorophore that is compatible with the procedures involved in IGS. However, its fluorescence yield (with a detection limit in the low picomolar range with simple gel-imaging equipment) is not particularly high and has some drawbacks in terms of stability (e.g., some loss of signal with partial HNO2 treatment). A number of alternative tags proved incompatible either because of loss of fluorescence in the partial HNO2 step (probably caused by reactions with amino groups essential for fluorescence) or because of inadequate gel separation (caused by excess hydrophobicity). Fluorophores that display up to 25-fold higher fluorescence yields are in use in other applications; thus, enhanced sensitivity (into the femtomolar range) can be expected, as can the development of improved fluorescent tags that are fully compatible with IGS. Furthermore, HPLC and capillary electrophoresis would allow a more sensitive detection of fluorescence than does gel imaging. In addition, the use of radioactive end labels such as 3H are also feasible (6) and could be used in gel and HPLC separations. The practical limitations of IGS remain to be explored fully in future work.
We have observed some artifacts during IGS that should be noted. One is the generation by partial HNO2 of minor ghost bands that migrate behind the major bands. We have some evidence that this is caused by the loss of the N-sulfate moiety from the reducing end of saccharides (data not shown), and the removal of this moiety tends to cause an upward band shift (Fig. 1B). We also have noted in some cases (particularly when 6-O-sulfate groups are removed) that gel shifts can be either up or down, depending on the size and charge of the saccharide, on the position of the group within the saccharide, and on the gel format used. It is likely that this result is caused by effects on charge:mass ratios and the complex migration process of these saccharides through acrylamide gels. One possible solution would be to use gradient PAGE gels in which separation is predominantly mass-dependent (43, 44). We have found that these artifacts have not caused problems in deducing the correct sequence, but the artifacts should be borne in mind when interpreting IGS data. Nevertheless, confirming agreement with independent data, such as disaccharide composition or the molecular mass of the saccharide (e.g., by matrix-assisted laser desorption ionization-mass spectroscopy; ref. 11) would ensure that potential sequencing errors are avoided.
Another important issue is the purification of saccharides for sequencing. We have shown here (Fig. 3A) that SAX-HPLC can purify decasaccharides to a level of homogeneity sufficient for sequencing. SAX-HPLC can be used to purify larger species (18), and PAGE also can be used in a preparative mode for purifying saccharides (17, 24). Although a high degree of purity of the starting sample is ideal, we anticipate that IGS will be able to identify and deal with heterogeneity. A saccharide preparation that initially seems to be a single species would result in divergences in the IGS banding pattern caused by the structural differences between the subspecies. It may be possible to deduce the individual sequences in such cases, but such deductions may be precluded by very complex patterns involving more than two species. A more likely option would be to repurify the individual tagged saccharides at their first divergence point in the banding pattern (i.e., the highest molecular mass species of each that shows different structures). This option could be achieved by preparative PAGE (24) and by IGS repeated on the individual saccharides to obtain the complete sequences.
A number of other improvements and adaptations of IGS are envisaged. The detection of rare sulfate groups with the exoenzymes glucosamine-3-O- and GlcA-2-O-sulfatases (22, 40–42, 45) will enhance the scope of IGS. Also, glucosamine residues are known to occur in HS (46); they would be resistant to HNO2 and NAG and would block complete sequencing. High pH HNO2 could be used to cleave at these residues, or, alternatively, chemical N-acetylation would make them susceptible to NAG. In addition, the initial partial scission step need not be chemical; for example, IGS also will be applicable to other GAGs, such as chondroitin and dermatan sulfate, through the use of bacterial lyase enzymes. Modified versions of IGS also may have applications in the sequencing of other non-GAG saccharides. We anticipate that application of IGS and its further development will dramatically accelerate the pace of structural analysis of HS and heparin saccharides, compared with current methods, and lead to significant advances in our understanding of the structure–activity relationships of these complex polysaccharides.
Acknowledgments
We thank Kaye Beckman, Julie Bielicki, Peter Clements, and Birget Weber for preparation of the exoenzymes; Craig Freeman for helpful discussions; Bob Linhardt, Kazuyuki Sugahara, and Keiichi Yoshida for providing saccharide standards; James Niland for technical assistance; and Oxford Glycosciences for providing gel solutions. Funding was provided by the Cancer Research Campaign (to J.T.G. and J.E.T.), the Medical Research Council (Senior Fellowship to J.E.T.), the National Health and Medical Research Council of Australia (to J.J.H.), and The Royal Society (to J.E.T.).
ABBREVIATIONS
- 2AA
2-aminobenzoic acid
- G6Sase
glucosamine-6-sulfate sulfatase
- GAG
glycosaminoglycan
- Gase
β-d-glucuronidase
- GlcA
glucuronate
- GlcNAc
N-acetylglucosamine
- GlcNS
N-sulfoglucosamine
- GlcNS(6S)
GlcNS 6-sulfate
- ΔHexA
Δ4,5-unsaturated uronate residue
- ΔHexA(2S)
ΔHexA 2-sulfate
- I2Sase
iduronate-2-sulfate sulfatase
- Idase
α-l-iduronidase
- IdoA
iduronate
- IdoA(2S)
IdoA 2-sulfate
- HS
heparan sulfate
- IGS
integral glycan sequencing
- NAG
α-N-acetylglucosaminidase
- NSase
sulfamidase
- SAX
strong anion exchange
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Spillman D, Lindahl U. Curr Opin Struct Biol. 1994;4:677–682. [Google Scholar]
- 2.Bourin M, Lindahl U. Biochem J. 1993;289:313–330. doi: 10.1042/bj2890313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turnbull J E, Gallagher J T. Biochem Soc Trans. 1993;21:477–482. doi: 10.1042/bst0210477. [DOI] [PubMed] [Google Scholar]
- 4.Lindahl U, Thunberg L, Bäckström G, Riesenfeld J, Nordling K, Björk I. J Biol Chem. 1984;259:12368–12376. [PubMed] [Google Scholar]
- 5.Turnbull J E, Fernig D, Ke Y, Wilkinson M C, Gallagher J T. J Biol Chem. 1992;267:10337–10341. [PubMed] [Google Scholar]
- 6.Maccarana M, Casu B, Lindahl U. J Biol Chem. 1993;268:23898–23905. [PubMed] [Google Scholar]
- 7.Gallagher J T, Turnbull J E, Lyon M. Int J Biochem. 1992;24:553–556. doi: 10.1016/0020-711x(92)90326-v. [DOI] [PubMed] [Google Scholar]
- 8.Turnbull J E, Gallagher J T. Biochem J. 1991;273:553–559. doi: 10.1042/bj2730553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turnbull J E, Gallagher J T. Biochem J. 1990;265:715–724. doi: 10.1042/bj2650715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnbull J E, Lyon M, Gallagher J T. In: Extracellular Matrix: A Practical Approach. Haralson M, Hassell J, editors. Oxford: IRL; 1995. pp. 199–220. [Google Scholar]
- 11.Rhomberg A J, Ernst S, Sasisekharan R, Biemann K. Proc Natl Acad Sci USA. 1998;95:4176–4181. doi: 10.1073/pnas.95.8.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai U, Wang H, Ampofo S, Linhardt R J. Anal Biochem. 1993;213:120–127. doi: 10.1006/abio.1993.1394. [DOI] [PubMed] [Google Scholar]
- 13.Mallis L, Wang H, Loganathan D, Linhardt R. Anal Chem. 1989;61:1453–1458. doi: 10.1021/ac00188a030. [DOI] [PubMed] [Google Scholar]
- 14.Yamada S, Yoshida K, Sugiura M, Sugahara K, Khoo K, Morris H, Dell A. J Biol Chem. 1993;268:4780–4787. [PubMed] [Google Scholar]
- 15.Pervin A, Gallo C, Jandik K, Han X, Linhardt R. Glycobiology. 1995;5:83–95. doi: 10.1093/glycob/5.1.83. [DOI] [PubMed] [Google Scholar]
- 16.Yamada S, Yamane Y, Tsuda H, Yoshida K, Sugahara K. J Biol Chem. 1998;273:1863–1871. doi: 10.1074/jbc.273.4.1863. [DOI] [PubMed] [Google Scholar]
- 17.Turnbull J E. Methods Mol Biol. 1993;19:253–267. doi: 10.1385/0-89603-236-1:253. [DOI] [PubMed] [Google Scholar]
- 18.Pye D A, Vives R, Turnbull J E, Hyde P A, Gallagher J T. J Biol Chem. 1998;273:22936–22942. doi: 10.1074/jbc.273.36.22936. [DOI] [PubMed] [Google Scholar]
- 19.Kobata A. Anal Biochem. 1979;100:1–14. doi: 10.1016/0003-2697(79)90102-7. [DOI] [PubMed] [Google Scholar]
- 20.Jackson P. Biochem J. 1990;270:705–713. doi: 10.1042/bj2700705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee K, Al-Hakim A, Loganathan D, Linhardt R. Carbohydr Res. 1991;214:155–168. doi: 10.1016/s0008-6215(00)90538-x. [DOI] [PubMed] [Google Scholar]
- 22.Hopwood J J. In: Heparin. Lane D, Lindahl U, editors. London: Edward Arnold; 1989. pp. 191–227. [Google Scholar]
- 23.Freeman C, Hopwood J J. Adv Exp Med Biol. 1992;313:121–134. doi: 10.1007/978-1-4899-2444-5_13. [DOI] [PubMed] [Google Scholar]
- 24.Turnbull J E, Hopwood J J, Gallagher J T. In: A Laboratory Guide to Glycoconjugate Analysis. Jackson P, Gallagher J T, editors. Basel: Birkhauser; 1997. pp. 261–277. [Google Scholar]
- 25.Turnbull J E, Hopwood J J, Gallagher J T. Glycoconj J. 1997;14:S94. (abstr.). [Google Scholar]
- 26.Linhardt R J, Rice K, Kim Y, Lohse D, Wang H, Loganathan D. Biochem J. 1988;254:781–787. doi: 10.1042/bj2540781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radoff S, Danishefsky I. J Biol Chem. 1984;259:166–172. [PubMed] [Google Scholar]
- 28.Ludwigs U, Elgavish A, Esko J, Meezan E, Roden L. Biochem J. 1987;245:795–804. doi: 10.1042/bj2450795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bielicki J, Hopwood J J, Wilson P, Anson D. Biochem J. 1993;289:241–246. doi: 10.1042/bj2890241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unger E, Durrant J, Anson D S, Hopwood J J. Biochem J. 1994;304:43–49. doi: 10.1042/bj3040043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Litjens T, Bielicki J, Anson D S, Friderici K, Jones M Z, Hopwood J J. Biochem J. 1997;311:89–94. doi: 10.1042/bj3270089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bielicki J, Hopwood J J, Melville E L, Anson D S. Biochem J. 1998;329:145–150. doi: 10.1042/bj3290145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freeman C, Hopwood J J. Biochem J. 1987;246:355–365. doi: 10.1042/bj2460355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker A, Turnbull J E, Gallagher J T. J Biol Chem. 1994;269:931–935. [PubMed] [Google Scholar]
- 35.Sanderson R D, Turnbull J E, Gallagher J T, Lander A D. J Biol Chem. 1994;269:13100–13106. [PubMed] [Google Scholar]
- 36.Kato M, Wang H-M, Bernfield M, Gallagher J T, Turnbull J E. J Biol Chem. 1994;269:18881–18890. [PubMed] [Google Scholar]
- 37.Bigge J, Patel T, Bruce J, Goulding P, Charles S, Parekh R. Anal Biochem. 1995;230:229–238. doi: 10.1006/abio.1995.1468. [DOI] [PubMed] [Google Scholar]
- 38.Tyrrell D J, Ishihara M, Rao N, Horne A, Kiefer M C, Stauber G B, Lam L H, Stack R J. J Biol Chem. 1993;268:4684–4689. [PubMed] [Google Scholar]
- 39.Linker A. Biochem J. 1979;183:711–720. doi: 10.1042/bj1830711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindahl U, Bäckström G, Thunberg L, Leder I. Proc Natl Acad Sci USA. 1980;77:6551–6555. doi: 10.1073/pnas.77.11.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thunberg L, Bäckström G, Lindahl U. Carbohydr Res. 1982;100:393–410. doi: 10.1016/s0008-6215(00)81050-2. [DOI] [PubMed] [Google Scholar]
- 42.Oosta G, Favreau L, Beeler D, Rosenberg R. J Biol Chem. 1982;257:11249–11255. [PubMed] [Google Scholar]
- 43.Rice R, Rottink M, Linhardt R J. Biochem J. 1987;244:515–522. doi: 10.1042/bj2440515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turnbull J E, Gallagher J T. Biochem J. 1988;251:597–608. doi: 10.1042/bj2510597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freeman C, Hopwood J J. Biochem J. 1989;259:209–216. doi: 10.1042/bj2590209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van den Born J, Gunnarsson K, Bakker M A, Kjellen L, Kusche-Gullberg M, Maccarana M, Berden J H, Lindahl U. J Biol Chem. 1995;270:31303–31309. doi: 10.1074/jbc.270.52.31303. [DOI] [PubMed] [Google Scholar]