

Summary
After ingestion by macrophages, Legionella pneumophila enter spacious vacuoles that are quickly enveloped by endoplasmic reticulum (ER), then slowly transferred to lysosomes. Here we demonstrate that the macrophage autophagy machinery recognizes the pathogen phagosome as cargo for lysosome delivery. The autophagy conjugation enzyme Atg7 immediately translocated to phagosomes harbouring virulent Legionella. Subsequently, Atg8, a second autophagy enzyme, and monodansyl-cadaverine (MDC), a dye that accumulates in acidic autophagosomes, decorated the pathogen vacuoles. The autophagy machinery responded to 10–30 kDa species released into culture supernatants by Type IV secretion-competent Legionella, as judged by the macrophages’ processing of Atg8 and formation of vacuoles that sequentially acquired Atg7, Atg8 and MDC. When compared with autophagosomes stimulated by rapamycin, Legionella vacuoles acquired Atg7, Atg8 and MDC more slowly, and Atg8 processing was also delayed. Moreover, compared with autophagosomes of Legionella-permissive naip5 mutant A/J macrophages, those of resistant C57BL/6 J macrophages matured quickly, preventing efficient Legionella replication. Accordingly, we discuss a model in which macrophages elevate autophagy as a barrier to infection, a decision influenced by regulatory interactions between Naip proteins and caspases.
Introduction
Autophagy is a conserved membrane traffic pathway by which eukaryotic cells capture cytoplasmic components for delivery to lysosomes. In response to nutrient starvation, autophagy breaks down macromolecules to generate substrates for essential biosynthetic pathways. In addition to cellular homeostasis, this degradative pathway contributes to numerous other physiological processes, including morphogenesis, cellular differentiation, tissue remodelling and antigen presentation (Nimmerjahn et al., 2003; Ogier-Denis and Codogno, 2003). Several pathological processes also involve autophagy, including carcinogenesis, myopathies and neurodegenerative diseases (Ogier-Denis and Codogno, 2003).
A morphological hallmark that distinguishes autophagy from other degradative pathways is its sequestration of cytoplasmic material within a double-membrane sac that fuses to form the nascent autophagosome (Dunn, 1990a; Seglen and Bohley, 1992). Genetic studies in yeast indicate the early secretory pathway contributes membrane to form autophagosomes (Hamasaki et al., 2003). In mammalian cells, proposed origins of the isolating membrane of nascent autophagosomes include the ER, a post-Golgi compartment, or a novel organelle called the phagophore (Hirsimaki, 1983; Dunn, 1990a; Seglen et al., 1990; Ueno et al., 1991; Stromhaug et al., 1998; Rich et al., 2003). As autophagosomes mature, they acquire lysosomal features, including the vacuolar ATPase, an acidic pH, lysosome-associated membrane protein-1 (LAMP1), and lysosomal hydrolases (Hirsimaki, 1983; Dunn, 1990b; Kopitz et al., 1990). Within autophagolysosomes, the sequestered cytosolic components are degraded.
Autophagosome biogenesis is controlled by a unique protein conjugation system composed of several proteins now designated Atg (Klionsky et al., 2003). Atg7 (Apg7/Gsa7) is a broadly conserved ubiquitin E1-like activating enzyme that mediates the homotypic fusion of the isolating membranes to assemble the autophagosome (Mizushima et al., 1998; Tanida et al., 1999; Ohsumi, 2001; Tanida et al., 2001). When autophagy is stimulated, cytosolic Atg7 protein redistributes to the isolation membrane where it first catalyses the conjugation of Atg12 to Atg5, then of Atg8 (Apg8/MAP-LC3) to lipids, thereby promoting the formation and expansion of the nascent autophagosome (Tanida et al., 1999; Kabeya et al., 2000; Ohsumi, 2001). In mammalian cells, autophagosome formation requires class III phosphatidylinositol 3-phosphate kinases (PI3-kinases), because the pathway is blocked by the inhibitor 3-methyladenine (3MA; Seglen and Gordon, 1982). A negative regulator of autophagy is Tor kinase, which can be inhibited pharmacologically by rapamycin (Noda and Ohsumi, 1998). Autophagy is stimulated when pro-apoptotic caspases are inhibited (Martin and Baehrecke, 2004; Yu et al., 2004), suggesting that autophagy and apoptosis are co-ordinated cellular responses to stress.
The intracellular bacterial pathogen Legionella pneumophila (Legionella) initially avoids but subsequently tolerates its delivery to lysosomes by a process that resembles autophagy (Swanson and Fernandez-Moreira, 2002). In the post-exponential (PE) phase, Legionella employ Type IV secretion to enter a lipid-raft-rich spacious vacuole that does not fuse with lysosomes (Horwitz, 1983a; Vogel et al., 1998; Joshi et al., 2001; Watarai et al., 2001). Instead, smooth vesicles from the ER attach to the cytosolic face of vacuoles that harbour virulent bacteria (Horwitz, 1983b; Tilney et al., 2001; Kagan and Roy, 2002). Next, the phagosomal membrane changes thickness, the secretory vesicles fuse, and, by 4 h, most Legionella vacuoles are surrounded by a double membrane of rough ER (Horwitz, 1983b; Swanson and Isberg, 1995; Abu Kwaik, 1996; Tilney et al., 2001; Kagan and Roy, 2002). Within this ER-derived compartment, Legionella differentiate to a replicative form that represses traits that promote transmission, including factors that block phagosome maturation (Molofsky and Swanson, 2003). After a lag period of several hours, the pathogen replicates for more than 15 h within an acidic, endocytic, lysosomal vacuole in macrophages of permissive A/J mice (Sturgill-Koszycki and Swanson, 2000).
Although macrophage delivery of the Legionella phagosome to the lysosomes via an ER intermediate is grossly similar to autophagy, a number of aspects are different (Swanson and Isberg, 1995, Tilney et al., 2001; Ogier-Denis and Codogno, 2003). Autophagosomes decorated with ribosomes have not been observed; indeed, whether the ER contributes to autophagosome formation in mammalian cells remains a matter of debate (Hirsimaki, 1983; Dunn, 1990a; Ueno et al., 1991; Rich et al., 2003). Furthermore, the Legionella vacuole merges with the lysosomes after a delay of several hours (Sturgill-Koszycki and Swanson, 2000), a maturation period considerably longer than that of autophagosomes (Kopitz et al., 1990; Noda et al., 2002; Ogier-Denis and Codogno, 2003). Within the soil amoeba Dictyostelium discoidium, Legionella can replicate in several different autophagy mutant strains (Otto et al., 2004). Accordingly, autophagy itself does not appear to be a prerequisite for intracellular replication of Legionella in all cell types. Therefore, we tested the hypothesis that macrophages activate the autophagy pathway as a defence against Legionella infection by applying a combination of biochemical markers, pharmacological inhibitors, and bacterial and host mutants. The data support a model in which macrophages deliver Legionella vacuoles to lysosomes by autophagy, a cellular defence mechanism that pathogens must modulate to establish infection.
Results
Autophagy enzymes redistribute to Legionella phagosomes
To analyse whether the macrophage autophagy machinery could account for both the sequestration of the Legionella phagosome within ER and its subsequent delivery to the lysosomes, we examined the distribution of three molecular markers of autophagosomes in cells that had phagocytosed bacteria. Immediately after internalization, ~50% of wild-type (WT) bacteria resided in spacious compartments surrounded by Atg7, a conjugating enzyme that initiates formation of the isolation vacuole (Fig. 1A and B; Mizushima et al., 1998; Tanida et al., 1999; Dorn et al., 2001; Ohsumi, 2001; Tanida et al., 2001; Klionsky et al., 2003). Accumulation of the autophagy-initiating enzyme was specific to phagosomes harbouring pathogenic bacteria, because Atg7 was rarely associated with vacuoles containing either PE dotA mutant (Fig. 1A and B) or E WT Legionella or avirulent Escherichia coli DH5α (Fig. 1B), microbes that macrophages efficiently deliver to the endosomal pathway (Berger et al., 1994; Roy et al., 1998; Joshi et al., 2001; Kagan and Roy, 2002). The appearance of Atg7 around vacuoles reflected a recruitment from cytosolic pools, because the amount of Atg7 protein detected by Western analysis did not change during the 30 min infection (A. Amer, unpubl.). As expected for autophagy, redistribution of Atg7 to the pathogen phagosomes required class III PI3-kinases, because Atg7-positive vacuoles were rare when macrophages were infected with Legionella in the presence of 10 mM 3MA (Seglen and Gordon, 1982). By 15 min after infection, the spacious vacuoles harbouring Legionella appeared more condensed (A. Amer, unpubl.), a pattern in good agreement with the previously described short-lived spacious phagosomes that contain Type IV secretion-competent Legionella and are rich in GM1 gangliosides and glycosylphosphatidylinositol-linked proteins (Watarai et al., 2001). The phagosomes progressively shed Atg7; by 3 h, the enzyme was rarely detected near vacuoles (Fig. 1B).
Fig. 1.
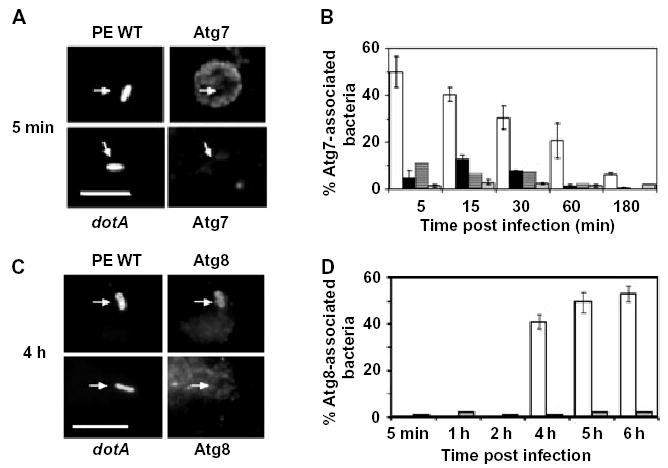
The autophagy enzymes Atg7 and Atg8 redistribute to phagosomes that contain virulent Legionella.
A. Macrophages infected for 5 min with post-exponential wild-type (PE WT) or avirulent dotA Legionella that express GFP were stained with Atg7-specific antibody. Arrows indicate position of bacteria. Scale bar = 10 μm.
B. The percentage Atg7-positive phagosomes was determined after infecting macrophages for the periods indicated with PE WT (white bars), exponential phase WT (black bars), PE dotA mutant (striped bars) or PE WT Legionella after pretreating macrophages with the autophagy inhibitor 3MA (grey bars). Values are the mean percentage colocalization ± SD calculated by scoring 100 bacteria per coverslip in at least three experiments.
C. Macrophages infected for 4 h with PE WT or dotA Legionella that express GFP were stained with Atg8-specific antibody. Arrows indicate position of bacteria. Scale bar = 10 μm.
D. The percentage Atg8-positive phagosomes was determined after infecting macrophages for the periods indicated with PE WT (white bars) or dotA mutant Legionella (grey bars). Values are the mean percentage colocalization ± SD calculated by scoring 100 bacteria per coverslip in two independent experiments.
To examine by another approach whether macrophages handled Legionella phagosomes as cargo for the autophagy pathway, we examined the distribution of Atg8 (LC3), a second conjugating enzyme that is recruited to autophagosomes as they mature (Kabeya et al., 2000). When autophagy was stimulated by incubating macrophages in amino acid-free buffer for 1 h, numerous Atg8-positive vacuoles were detected (Fig. 2D). Beginning ~4 h after infection of macrophages, Atg8 colocalized with >40% of the vacuoles that contained PE WT Legionella, but not those that carried either PE dotA mutants (Fig. 1C and D) or avirulent E WT Legionella or E. coli (A. Amer, unpubl.). The transient association with Atg7 and the subsequent accumulation of Atg8 around vacuoles harbouring virulent Legionella is characteristic of autophagosome maturation (Kopitz et al., 1990; Noda et al., 2002; Ogier-Denis and Codogno, 2003; Yoshimori, 2004).
Fig. 2.
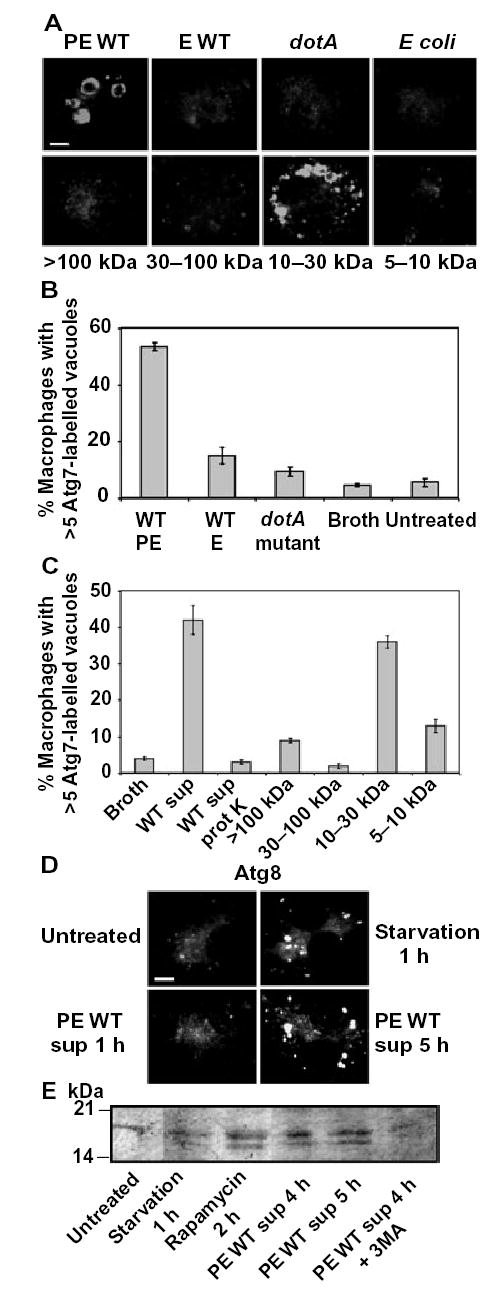
The macrophage autophagy machinery responds to 10–30 kDa molecules shed by virulent Legionella.
A. Formation of Atg7-positive vacuoles was analysed by immunofluorescence microscopy in macrophages incubated for 15 min either with supernatants prepared from cultures of PE WT, E WT or PE dotA mutant Legionella or PE E. coli or with samples obtained after size fractionation of the PE WT Legionella culture supernatant. Scale bar = 10 μm.
B. The mean percentage of macrophages harbouring >5 Atg7-labelled vacuoles 15 min after infection with PE WT, E WT or PE dotA mutant Legionella, incubation with sterile broth, or untreated, was calculated from three experiments, ± SD.
C. The mean percentage of macrophages harbouring >5 Atg7-labelled vacuoles 15 min after treatment either with broth, PE Legionella culture supernatant (WT sup), proteinase K-treated PE Legionella culture supernatant (WT sup protK), or samples obtained after size fractionation of the WT PE supernatant was calculated from two experiments, ± SD.
D. Formation of Atg8-positive vacuoles was analysed by immunofluorescence microscopy in macrophages that were untreated or were incubated with amino acid-free buffer for 1 h or with PE WT Legionella supernatants (PE WT sup) for 1 or 5 h. Scale bar = 10 μm.
E. Lipidation of Atg8 protein was evaluated by Western analysis of macrophages that either were untreated or were incubated with amino acid-free buffer for 1 h (starvation), with rapamycin for 2 h, or with PE WT Legionella supernatant for 4 or 5 h, in the absence or presence of 3MA.
To analyse the interaction of the autophagy pathway with Legionella phagosomes by a third method, we utilized monodansyl-cadaverine (MDC), a fluorescent compound that accumulates in acidic autophagic vacuoles (Biederbick et al., 1995; Niemann et al., 2000; Munafo and Colombo, 2001). The results of several control assays verified that MDC behaved as a valid marker of macrophage autophagosomes. By 6 h after infection, MDC accumulated in ~30% of the vacuoles that harboured virulent PE Legionella, but not avirulent dotA mutants (Fig. 3A and B). MDC persisted within mature replication vacuoles even 24 h after infection (Fig. 3A), a period when these organelles are acidic (Sturgill-Koszycki and Swanson, 2000). As expected for autophagy, formation of the MDC-positive vacuoles by infected cells was inhibited by 3MA (Fig. 3C). However, compared with autophagosomes induced by amino acid-free buffer, the pathogen vacuoles accumulated MDC more slowly (Fig. 3B and C). Therefore, macrophages appeared to handle phagosomes that contained virulent Legionella as cargo for autophagy, as judged by their sequential acquisition by a 3MA-sensitive process of the autophagy-initiating enzyme Atg7, the conjugating enzyme Atg8 and the fluorescent marker of mature acidic autophagosomes MDC. However, compared with typical autophagosomes, the pathogen-induced vacuoles matured more slowly.
Fig. 3.
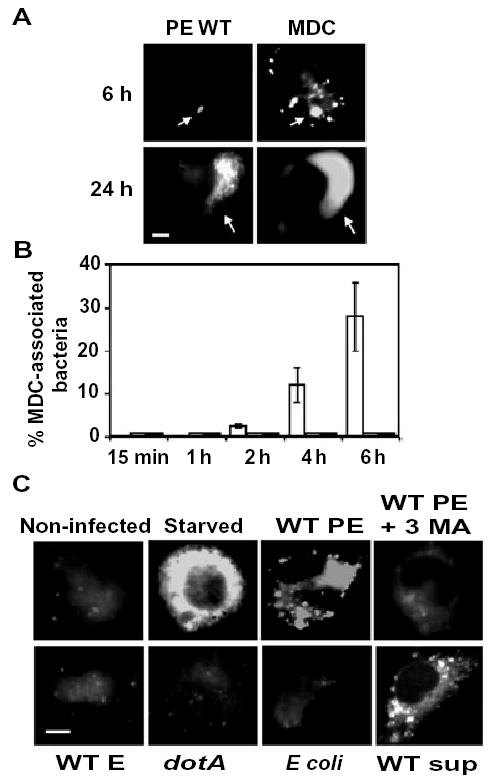
The autophagosome marker monodansyl-cadaverine accumulates in vacuoles that contain virulent Legionella.
A. Macrophages infected for 6 or 24 h with PE WT Legionella that express GFP were incubated for 10 min with the fluorescent dye MDC and examined immediately. Arrows indicate position of bacteria. Scale bar = 10 μm.
B. The mean percentage of MDC-positive bacterial vacuoles in macrophages infected for the periods indicated with PE WT (white bars), E WT (grey bars), or PE dotA mutant Legionella (black bars) was determined in three independent experiments, ± SD. Bacterial vacuoles were scored as positive when fluorescence was detected within their lumens.
C. Macrophages that were either non-infected, starved for 2 h, or infected for 6 h with WT PE, WT E, dotA PE Legionella, or E. coli were incubated with MDC as in (A). Also shown are macrophages that were pretreated with the autophagy inhibitor 3MA before infection for 6 h with PE WT Legionella (WT PE + 3MA) or that were incubated for 6 h with sterile supernatants obtained from cultures of WT PE Legionella (WT sup). Scale bar = 10 μm.
The macrophage autophagy machinery responds to factors shed from virulent Legionella
Macrophages appeared to activate the autophagy machinery independently of phagocytosis of the pathogen. First, after a 5 min incubation with PE Legionella, only 21 ± 0.5% of the macrophages had ingested a bacterium, yet 54 ± 3% of the cells contained numerous Atg7-labelled donut-shaped compartments. Second, when actin polymerization was disrupted with cytochalasin D, phagocytosis of Legionella was decreased dramatically as expected (Elliott and Winn, 1986), yet 42 ± 1.7% of the macrophages still contained multiple Atg7-decorated vacuoles within 5 min of infection, a frequency significantly greater than the 5.5 ± 1.5% of the uninfected control cells that did so (Fig. 2B). Therefore, we tested the hypothesis that macrophages activate the autophagy machinery in response to soluble bacterial products.
Cell-free supernatant prepared from cultures of PE Legionella stimulated the autophagy machinery. After a treatment for 15 min, >50% of the macrophages contained multiple Atg7-labelled compartments (Fig. 2A and C). After a 5 h incubation with the sterile supernatant, multiple Atg8-labelled vesicles (Fig. 2D) were detected in 25 ± 2.5% macrophages, and by 6 h MDC-positive vacuoles were apparent (Fig. 3C). The maturation rate of the vacuoles was similar to that observed for phagosomes that contain virulent Legionella (Figs 1–3). In contrast, neither sterile bacteriological broth treated in the same manner nor supernatants obtained from cultures of non-infectious microbes, namely E Legionella, PE dotA mutants, or DH5α E. coli, induced formation of vacuoles labelled by Atg7, Atg8, or MDC (Fig. 2A; A. Amer, unpubl.). Stimulation of the autophagy machinery by Legionella soluble products was not accompanied by cytotoxicity, because macrophages remained viable after a 20 h incubation with supernatants that had been concentrated 20-fold (Experimental procedures; A. Amer, unpubl.).
To investigate by a biochemical approach whether soluble Legionella products stimulate autophagosome formation, we examined by Western assay processing of Atg8, a protein that the Atg7 enzyme conjugates to phosphatidylethanolamine during autophagosome biogenesis (Kabeya et al., 2000; Ichimura et al., 2000). Unlike control cells, macrophages that had been incubated with amino acid-free buffer, rapamycin, or Legionella sterile supernaants processed the Atg8 protein, as judged by the appearance of the membrane-associated lipidated species, whose electrophoretic mobility is greater than the cytosolic form (Fig. 2E). As expected, when autophagosome formation was inhibited by pretreating macrophages with 3MA, Legionella supernatants did not stimulate Atg8 lipidation (Fig. 2E). The conjugated Atg8 species was detected within 1 h of amino acid starvation or rapamycin treatment; in contrast, a 4 h incubation with Legionella culture supernatants was required for macrophages to process Atg8 (Fig. 2E; A. Amer, unpubl.). Thus, macrophages activate the autophagy pathway in response to soluble products released by Type IV secretion-competent PE Legionella by a mechanism that does not require phagocytosis of the bacteria.
To begin to characterize biochemically the Legionella autophagosome stimulating factor(s), which we shall refer to as ASF, three experiments were performed. ASF activity did not appear to reflect bacterial lysis, because the protein profile of concentrated culture supernatants was distinct from that of total bacterial lysates, as judged by SDS-PAGE (A. Amer, unpubl.). Treating PE culture supernatants with proteinase K dramatically reduced ASF activity (Fig. 2C). By fractionating the supernatants with filters that exclude either 5, 10, 30 or 100 kDa species, we determined that bacterial products of 10–30 kDa stimulated robust autophagosome formation, whereas fractions that contained molecules >30 or <10 kDa had minimal effect (Fig. 2A and C). The active fraction of 10–30 kDa molecules was complex, as nine species were apparent after separation by SDS-PAGE and staining with Coo-massie Blue. Thus, additional biochemical and molecular analysis can determine how macrophages activate the autophagy machinery in response to soluble 10–30 kDa proteinaceous molecules that are shed by Type IV secretion-competent PE phase Legionella.
Secretory traffic promotes association of Atg7 with the Legionella vacuole
Vesicles from the early secretory pathway are recruited to Legionella phagosomes (Horwitz, 1983b; Swanson and Isberg, 1995; Tilney et al., 2001; Derre and Isberg, 2004). Secretory vesicles are also thought to be assembled by the Atg7 enzyme into the isolation membrane of nascent autophagosomes (Hirsimaki, 1983; Dunn, 1990a; Ueno et al., 1991; Hamasaki et al., 2003). Therefore, we postulated that the accumulation of the Atg7 protein around Legionella phagosomes requires vesicular traffic from the ER. Because vesicle production from the ER depends on the sequential action of the Sar1/COPII and ARF/COPI systems, it can be disrupted by brefeldin A (BFA) (Klausner et al., 1992; Aridor et al., 1995; Scales et al., 1997; Kagan and Roy, 2002). When macrophages were infected for 15 min with Legionella, both Atg7 and the ER lumenal protein Bip were observed in close proximity to 94 ± 1% of the pathogen vacuoles (Fig. 4A), as expected (Fig. 1; Tilney et al., 2001; Kagan and Roy, 2002). In contrast, when vesicular traffic from the ER was inhibited by treating macrophages for 1 h before infection with BFA, accumulation of Atg7 on Legionella vacuoles was reduced by ~50% (Fig. 4B). The inhibition by BFA was reversible, because removal of the drug at the time of infection restored to control levels colocalization of Atg7 with Legionella vacuoles aged 15 min (A. Amer, unpubl.). These and previous observations are consistent with a model in which secretory vesicles dock on the cytoplasmic face of the Legionella phagosome, then undergo Atg7-mediated homotypic fusion to form and expand the double-membrane ER envelope (Swanson and Isberg, 1995; Abu Kwaik, 1996; Tilney et al., 2001; Kagan and Roy, 2002). Accordingly, biogenesis of the Legionella replication vacuole is analogous to the nucleation-assembly elongation process that has been proposed for autophagosome formation (Noda et al., 2002).
Fig. 4.
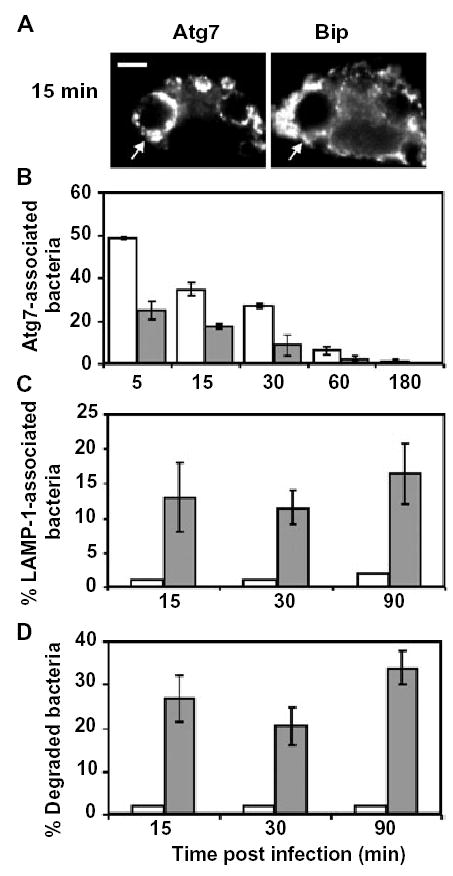
Atg7 accumulation correlates with engulfment of the Legionella phagosome by ER and evasion of lysosomes.
A. Macrophages infected for 15 min with PE WT Legionella were stained with Atg7- and Bip-specific antibodies. Arrows point to a phagosome.
B. Control macrophages (white bars) or macrophages pretreated for 1 h with BFA (grey bars) were infected with PE WT Legionella for the periods shown, then colocalization of phagosomes with the autophagy enzyme Atg7 was scored. The mean percentage Atg7- associated bacteria that were stained with both DAPI and Legionella-specific antibody was calculated from three experiments ± SD.
C. Control macrophages (white bars) or macrophages pretreated for 1 h with BFA (grey bars) were infected with PE WT Legionella for the periods shown, then colocalization of phagosomes with the late endosomal and lysosomal protein LAMP-1 was scored. The mean percentage of LAMP-1-associated bacteria that were stained with both DAPI and Legionella-specific antibody was calculated, ± SD, from three experiments.
D. Control macrophages (white bars) or macrophages pretreated for 1 h with BFA (grey bars) were infected with PE WT Legionella for the periods shown, then stained with Legionella-specific antibody. A degraded bacterium was defined by the absence of a rod shape and presence of smaller particles stained by Legionella-specific antibody. The mean percent of degraded bacteria, ± SD, was calculated by scoring 100 bacteria per coverslip in three experiments.
Colocalization of pathogenic Legionella with autophagosomal vacuoles correlates with immediate evasion of the endocytic pathway
Legionella residence in ER-derived vacuoles is correlated with its immediate evasion of the endosomal pathway (Berger et al., 1994; Swanson and Isberg, 1995; Kagan and Roy, 2002; Molofsky and Swanson, 2004). Therefore, if the autophagy machinery assembles the ER-derived vacuole, drugs that inhibit autophagy are predicted to increase lysosomal degradation of Legionella. To test whether there is a reciprocal relationship between the autophagosome and phagolysosome pathways, we utilized two pharmacological inhibitors of autophagosome assembly. When accumulation of secretory vesicles and Atg7 by pathogen vacuoles was inhibited by BFA (Kagan and Roy, 2002), LAMP-1 was detected on ~15% of the vacuoles harbouring Legionella (Fig. 4C), and ~30% of the intracellular Legionella were degraded as early as 15 min after infection (Fig. 4D). Likewise, when autophagy was blocked by 3MA (Figs 1B and 3C), ~30% of intracellular bacteria were visibly degraded and ~40% were killed by 2 h after infection (Fig. 5A and B). The decrease in pathogen survival was not a consequence of 3MA toxicity, because the drug had no effect on either the viability of macrophages or Legionella (Experimental procedures; A. Amer, unpubl.), or the ability of macrophages to kill E Legionella, or the intracellular survival of dotA Legionella (Fig. 5B). Thus, both genetic and pharmacological studies of Legionella trafficking in macrophages suggest a reciprocal relationship between delivery of phagosomes to the autophagy pathway or directly to the lysosomes.
Fig. 5.
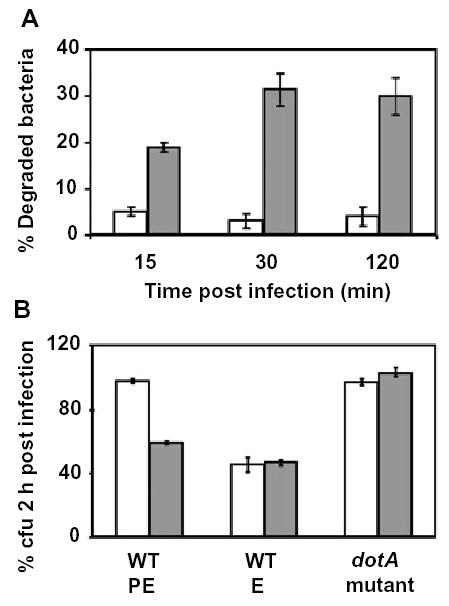
Legionella survival in the autophagosomal pathway.
A. Control macrophages (white bars) or macrophages pretreated and maintained with the autophagy inhibitor 3MA (grey bars) were infected with PE WT Legionella for the periods indicated, then the mean percentage degraded bacteria, ± SD, was determined as for Fig. 4D.
B. Control macrophages (white bars) or macrophages pretreated and maintained with the autophagy inhibitor 3MA (grey bars) were infected with PE WT, E WT, or PE dotA mutant Legionella, then survival of intracellular bacteria 2 h after infection was calculated as the percentage of cell-associated colony-forming units (cfu) = (cfu associated with macrophages @2 h)/(cfu associated with macrophages @15 min) × 100.
Slow autophagosome maturation correlates with survival of Legionella
Autophagy is a major catabolic pathway by which eukaryotic cells sequester organelles for lysosomal degradation, yet residence in autophagosomal vacuoles promoted Legionella survival in macrophage cultures (Figs 4 and 5). To account for this paradox, we postulated that to establish infection Legionella perturbs maturation of its autophagosome. Indeed, when autophagy was stimulated by treating macrophages with either rapamycin or amino acid-free buffer, the organelles matured considerably faster than vacuoles that contained the pathogen, as judged by comparing the kinetics of Atg7 and Atg8 colocalization with vacuoles and phagosomes (Fig. 6B and D; A. Amer, unpubl.). Thus, Legionella appears to encode factors that inhibit autophagosome maturation.
Fig. 6.
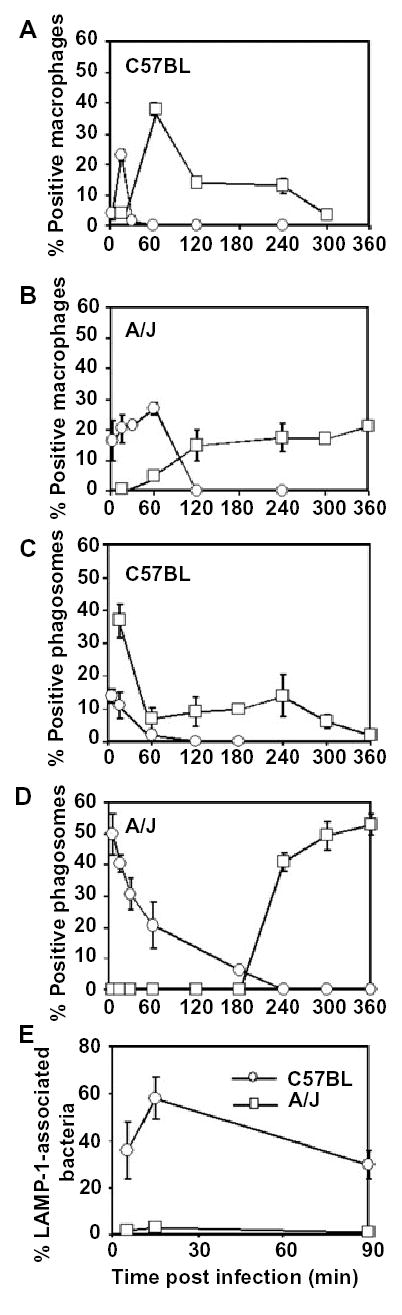
The mouse naip5 mutation, which confers permissiveness to Legionella infection, correlates with retarded autophagosome maturation.
A and B. Macrophages derived from the bone marrow of resistant C57BL/6 J mice (A) or permissive A/J mice (B) were incubated with the autophagy-inducer rapamycin for the periods indicated, then stained with Atg7- or Atg8-specific antibodies. The mean percentage of macrophages that contained >5 Atg7 labelled vacuoles (circles) or multiple Atg8-labelled vesicles (squares) was calculated, ± SD, by scoring 100 macrophages in each of two experiments. In untreated C57BL/6 J mice-derived macrophages, 2 ± 0.7% and 1.5 ± 0.35% of the cells contained >5 Atg7 labelled vacuoles and multiple Atg8-labelled vesicles, respectively; in untreated A/J mice-derived macrophages, 7 ± 2.8% and 3.5 ± 1.8% of cells contained >5 Atg7 labelled vacuoles and multiple Atg8-labelled vesicles respectively.
C and D. Macrophages obtained from resistant C57BL/6 J mice (C) or permissive A/J mice (D) were infected for 5 min with PE WT Legionella, then colocalization of Atg7 (circles) and Atg8 (squares) with the phagosomes was determined at the times indicated. The mean percentage of positive phagosomes, ± SD, was calculated by scoring 100 bacteria per coverslip in two experiments.
E. Macrophages obtained from resistant C57BL/6 J (circles) or permissive A/J mice (squares) were infected with PE WT Legionella for periods indicated, then acquisition of the late endosomal and lysosomal protein LAMP-1 by Legionella phagosomes was determined. The mean percentage LAMP-1-associated bacteria, ± SD, was calculated by scoring 100 bacteria per coverslip in two experiments.
To investigate by a genetic approach whether the rate of autophagosome maturation affects infection by Legionella, we utilized macrophages obtained from a congenic mouse strain that is resistant to infection. Unlike permissive A/J mice, which harbour a naip5 mutation that reduces the amount of Naip proteins, neither C57BL/6 J mice nor their isolated macrophages support Legionella replication (Dietrich et al., 1995; Diez et al., 2000; Diez et al., 2003; Wright et al., 2003). When autophagy was induced by rapamycin or starvation, vacuoles formed by restrictive C57BL/6 J macrophages more quickly acquired and then shed both Atg7 and Atg8 than did the vacuoles of permissive A/J macrophages (Fig. 6A and B; A. Amer, unpubl.). Likewise, after infection with PE Legionella, autophagosomes formed by resistant C57BL/6 J macrophages matured more rapidly than those of permissive A/J macrophages, as judged by the kinetics of both Atg7 and Atg8 colocalization (Fig. 6C and D). As expected for efficient autophagosome maturation, ~50% of the Legionella phagosomes formed by resistant C57BL/6 J macrophages accumulated the late endosomal and lysosomal protein LAMP-1 within 15 min of infection (Fig. 6E). In comparison, in permissive A/J macrophages, LAMP-1 is not detected on Legionella phagosomes until ~8 h after infection (Sturgill-Koszycki and Swanson, 2000). Thus, macrophages with efficient autophagosome maturation were resistant to infection, whereas those with a sluggish autophagy pathway were susceptible to Legionella infection.
Discussion
Here we report morphological, biochemical and genetic evidence that macrophages activate the autophagy machinery as an immediate response to infection by an intracellular pathogen. The autophagy pathway is activated by soluble factors released by infectious Legionella by a mechanism that does not require phagocytosis of the bacteria. However, vacuoles that harbour pathogens mature more slowly than autophagosomes stimulated by either rapamycin or amino acid-free buffer. Furthermore, the autophagy response of macrophages from mice that are permissive for Legionella infection is more lethargic than that of phagocytes from resistant mice. Accordingly, we postulate that macrophages activate autophagy as a barrier to infection; consequently, certain intracellular pathogens perturb autophagosome maturation to avoid or delay their delivery to toxic lysosomes.
The interpretation that macrophages handle Legionella phagosomes as cargo for the autophagy pathway is supported by a large body of morphological and genetic data presented here and elsewhere. When immature, both autophagosomes and vacuoles that contain Legionella are limited by a double membrane that forms by homotypic fusion of numerous small vesicles derived from the secretory pathway, a process described in the autophagy field as nucleation-assembly elongation (Noda et al., 2002). Dependent on the sequential activities of the Sar1/COPII and ARF/COPI systems, vesicle production from the ER promotes biogenesis of both Legionella replication vacuoles and yeast autophagosomes (Aridor et al., 1995; Scales et al., 1997; Kagan and Roy, 2002; Hamasaki et al., 2003; Derre and Isberg, 2004). As they mature, both organelles colocalize first the autophagy enzyme Atg7, next Atg8, and then the fluorescent dye MDC (Mizushima et al., 1998; Tanida et al., 2001). At any given time, only a subset of the vacuoles colocalized with a particular autophagy marker, most likely because the associations are transient, phagosome formation is not synchronous, and the fluorescence signal is limiting. Finally, in their terminal stage of development, both autophagosomes and Legionella vacuoles acquire lysosomal features, including LAMP-1, the acid hydrolase cathepsin D, and an acidic pH (Hirsimaki, 1983; Dunn, 1990b; Sturgill-Koszycki and Swanson, 2000).
Analysis of Legionella trafficking indicates that membranes derived from the secretory pathway contribute to autophagosome biogenesis by macrophages. The ER resident proteins Bip and calnexin, the recombinant ER marker KDEL-YFP, the small GTPases ARF1 and Rab1, the v-SNARE Sec22, and ribosomes all colocalize with the Legionella vacuole, and its membrane acquires a thickness characteristic of ER (Horwitz, 1983b; Swanson and Isberg, 1995; Tilney et al., 2001; Kagan and Roy, 2002; Nagai et al., 2002; Molofsky and Swanson, 2004). Furthermore, when vesicular traffic from the ER is inhibited by BFA, association of the autophagy enzyme Atg7 with Legionella vacuoles is reduced (Fig. 4B). Therefore, studies of Legionella–macrophage interactions provide another example where ER is the source of autophagosome isolation membranes (Hirsimaki, 1983; Dunn, 1990a; Ueno et al., 1991; Rich et al., 2003). In particular, we predict that Atg7 binds to the secretory vesicles that attach to the cytoplasmic face of the L. pneumophila phagosome to mediate their homotypic fusion, thereby generating an isolation membrane around the pathogen phagosome (Fig. 7), a model compatible with the ultra-structural studies of Tilney and colleagues (Tilney et al., 2001).
Fig. 7.
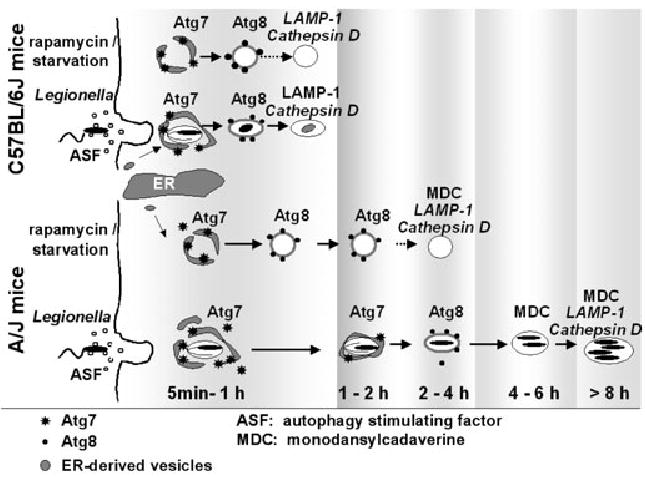
Autophagosomes of A/J macrophages mature slowly. In C57BL/6 J derived macrophages, autophagosomes induced by either rapamycin, starvation or Legionella mature at similar rates. In A/J mouse macrophages, autophagosomes induced by starvation or rapamycin mature at a slower rate than their counterparts in the C57BL/6 J derived cells. Even more sluggish is the maturation of Legionella-containing autophagosomes, which require several hours to mature into an acidic lysosomal compartment. Vacuolar accumulation of markers assayed here are shown; events described in the literature are indicated by dotted arrows and italics.
Microbial traffic along the autophagy pathway is not unique to Legionella (Kirkegaard et al., 2004). Both the picoranviruses poliovirus and herpes simplex virus type 1 encode proteins that induce the formation of double membrane vesicles that resemble autophagosomes (Talloczy et al., 2002; Kirkegaard et al., 2004). In HeLa cells, Brucella abortus is engulfed by autophagosome-like vacuoles that contain the ER components sec61β and calnexin and also accumulate MDC (Pizarro-Cerda et al., 1998a; 1998b). In endothelial cells, Porphyromonas gingivalis reside in vacuoles that colocalize with Atg7 and Bip (Dorn et al., 2001). Leishmania spp. and Coxiella burnetii, pathogens that multiply in lysosomes, may be delivered to their replicative niche by autophagy (Schaible et al., 1999; Beron et al., 2002). When caspase 1-deficient macrophages are infected with Salmonella that express the type III secretion system substrate SipB, numerous autophagosomes accumulate (Hernandez et al., 2003). At least in tissue culture models of infection, the intracellular pathogens Legionella, P. gingivalis, C. burnetii and B. abortus benefit from autophagy, because these bacteria survive poorly in cells treated with 3MA (Pizarro-Cerda et al., 1998b; Dorn et al., 2001; Beron et al., 2002). On the other hand, the autophagy machinery can effectively eliminate cytoplasmic Group A Streptococcus and inhibit the survival of Mycobacterium tuberculosis (Gutierrez et al., 2004; Nakagawa et al., 2004). Likewise, when intracellular Listeria monocytogenes are killed by antibiotic treatment, the cytoplasmic bacteria are engulfed and eliminated within double-membrane vacuoles that contain the ER protein disulphide isomerase (Rich et al., 2003). Moreover, efficient autophagy appears to prevent Legionella replication in C57BL/6 J macrophages (Fig. 6). Therefore, autophagy can be a defence mechanism against intracellular microbes that some pathogens acquired the means to overcome.
A clue to how Legionella tolerates the catabolic autophagy pathway is the slow maturation of its vacuole (summarized in Fig. 7; Noda et al., 2002; Ogier-Denis and Codogno, 2003). Whereas Atg7-labelled vacuoles are difficult to detect in cells treated with rapamycin or amino acid free buffer, the enzyme remained associated with some Legionella vacuoles as long as 60 min (Figs 1B and 6A; A. Amer, unpubl.). Second, macrophages accumulated Atg8-labelled vesicles within 1 h of rapamycin treatment or amino acid deprivation (Fig. 6B; A. Amer, unpubl.), but the pathogen compartments did not colocalize with Atg8 for 4 h (Figs 1D and 6D). Third, vesicular MDC was detected 2 h after macrophage autophagy was stimulated by starvation or rapamycin (Fig. 3C; A. Amer, unpubl.), but the dye was not visible within Legionella vacuoles for 4–6 h (Fig. 3B). By similar criteria, the autophagic vacuoles stimulated by soluble factors released by Legionella also matured slowly (Figs 2D and 3C). Finally, A/J-derived macrophages, which are permissive for Legionella infection, have a slower rate of autophagosome maturation than cells from C57BL/6 J mice (Fig. 6). Their lethargic maturation likely explains why Legionella autophagosomal vacuoles accumulate ribosomes; normally, the ER-derived isolation vacuole rapidly converts into an autophagolysosome, masking its origin as a biosynthetic membrane. Presumably, when confronted by the autophagy response of its host phagocyte, Legionella must retard maturation of its vacuole to secure time to differentiate to an acid-tolerant form (Sturgill-Koszycki and Swanson, 2000; Molofsky and Swanson, 2004). Herpes Simplex Virus applies a different strategy to evade degradation in autophagolysosomes: the neurovirulence viral protein ICP34.5 antagonizes eukaryotic initiation factor-2-α (eIF2α) kinase-dependent autophagy (Talloczy et al., 2002; Kirkegaard et al., 2004).
Analysis of Legionella infection of macrophages strengthens the emerging concept of a regulatory link between autophagy and programmed cell death, two cellular responses to stress (Gozuacik and Kimchi, 2004; Martin and Baehrecke, 2004; Yu et al., 2004; Xue et al., 1999). In mice, susceptibility to Legionella infection is conferred by a mutation in naip5, a gene originally identified as neuronal apoptosis inhibitor protein (Diez et al., 2003; Wright et al., 2003; Dietrich et al., 1995). The Naip5 protein is a member of a large family of endogenous caspase inhibitors, based both on its BIR, NOD and LRR domains (Liston et al., 2003) and the ability of BIR domains from human Naip to bind to and inhibit caspase 3 and caspase 7 (Maier et al., 2002). The data presented here are consistent with a number of previous observations that together suggest that Naip proteins promote efficient autophagosome maturation, perhaps by inhibiting caspase-3. First, macrophages from both A/J and C57Bl mice increase expression of Naip proteins after ingesting Legionella, Salmonella typhimurium, or latex beads (Diez et al., 2000). However, compared with permissive naip5 mutant macrophages from A/J mice, C57BL/6 J-derived macrophages express more Naip protein, and their autophagosomes more rapidly merge with lysosomes (Fig. 6C and D). Second, when human monocytes are infected by Legionella, the cells activate caspase-3 and engulf the pathogen within ER, but the vacuoles do not fuse with lysosomes for at least 2 h, perhaps because Rabaptin-5 is degraded (Molmeret et al., 2004). In contrast, when caspase-3 activity is inhibited pharmacologically, the phagocytes instead rapidly deliver Legionella to lysosomes, preventing their replication (Molmeret et al., 2004). Third, the pan-caspase inhibitor zVAD triggers toxic levels of autophagy in human monocytic cells or mouse peritoneal macrophages; a similar cell response is observed when caspase 8 protein is reduced by RNA interference (Yu et al., 2004). The existing data support the idea that autophagy functions as an adaptive response to infection that is efficient when caspase activity is low, whereas programmed cell death is an emergency response triggered when caspase activity is high. How Naip proteins regulate both autophagy and apoptosis can be examined using Legionella infection of macrophages as an experimental system.
Analysis of Legionella trafficking in macrophages has also revealed a reciprocal relationship between delivery of phagosomes directly to the endosomal pathway or their capture by autophagy. When autophagosome formation is inhibited by 3MA or by BFA, or production of ER-derived vesicles is decreased by dominant-interfering SNARE proteins, macrophages efficiently deliver Legionella phagosomes to the endosomal pathway, preventing bacterial replication (Figs 4–6; Watarai et al., 2001; Kagan and Roy, 2002; Kagan et al., 2004). Accordingly, as one means to evade immediate destruction in lysosomes, PE Legionella may release its autophagosome-stimulating factor, ASF (Fig. 3). Although more analysis is required to understand the impact of ASF on Legionella pathogenesis, it is the first activity shown to be released by a Dot/Icm Type IV secretion system-dependent mechanism and also to alter macrophage membrane traffic.
Rather than function as a virulence factor, Legionella ASF can be considered a candidate Pathogen-Associated Molecular Pattern molecule because ASF triggers autophagy, an alternate route to toxic lysosomes. Consistent with the view that autophagy is a barrier to infection, when compared with C57BL/6 J mice, A/J mice are more susceptible to several other intracellular pathogens including Trypanosoma cruzi, Plasmodium chabaudi and Listeria monocytogenes (Stevenson and Tam, 1993; Gonçalves da Costa et al., 2002; Czuprynski et al., 2003). Future studies can examine directly the role of autophagy in other intracellular infections. Furthermore, genetic studies of Legionella growth in D. discoidium indicate that autophagy per se is not required for bacterial replication (Otto et al., 2004); instead, the pathogen may replicate within acidic lysosomal vacuoles not only in A/J macrophages (Sturgill-Koszycki and Swanson, 2000) but also in soil amoebae. Compared with mammalian macrophages, D. discoidium appear to have a more robust phagolysosome pathway, based on the smaller fraction of virulent Legionella that survive phagocytosis, the death of dot/icm mutants, and the prolonged lag period before Legionella replication can be detected (Otto et al., 2004). It is also noteworthy that, unlike mammalian macrophages, D. discoidium lacks caspases, and its single paracaspase gene is not required for programmed cell death (Roisin-Bouffay et al., 2004). Therefore, D. discoidium is a valuable experimental model to analyse some aspects of Legionella biology, but it may not mimic the macrophage response to pathogens.
A clue to how the macrophage autophagy machinery may detect microbes is their predilection for lipid rafts. Numerous pathogens and toxins that associate with either the ER or the autophagy pathway are known to enter host cells via cholesterol-rich domains, including Legionella, B. abortus, S. typhimurium, Polyoma virus, SV40 and cholera toxin (Anderson et al., 1996; Richterova et al., 2001; Watarai et al., 2001; Naroeni and Porte, 2002; Fujinaga et al., 2003). The recent identification of several components of the mammalian autophagy machinery (Klionsky and Emr, 2000; Ohsumi, 2001; Klionsky et al., 2003) will facilitate direct tests of the hypothesis that macrophages exploit autophagy for surveillance against intracellular microbes.
Experimental procedures
Bacteria
Legionella pneumophila strain Lp02, a thymine auxotrophic derivative of Philadelphia 1 and the dotA mutant strain Lp03 (Berger et al., 1994), engineered to express green fluorescent protein (Berger et al., 1994; Sturgill-Koszycki and Swanson, 2000), were cultured as described previously (Byrne and Swanson, 1998) to either the PE phase or the exponential phase. Escherichia coli strain DH5α was cultured to saturation in Luria–Bertani broth.
Macrophages
Bone marrow-derived macrophages from either A/J or C57BL/6 J mice (The Jackson Laboratory) were cultured as described previously (Swanson and Isberg, 1995). Unlike cells from humans and A/J mice, C57BL/6 J macrophages are resistant to Legionella infection. The susceptibility of A/J mice to Legionella infection is conferred by a mutation in naip5 that reduces the amount of Naip proteins (Dietrich et al., 1995; Diez et al., 2003; Wright et al., 2003).
Autophagy stimulation
Autophagy was stimulated by incubating macrophages for the periods indicated either with amino acid-free buffer as described (Swanson and Isberg, 1995) or with 0.2 μg rapamycin (LC Laboratories) per ml RPMI/FBS (Noda and Ohsumi, 1998). To infect macrophages synchronously, PE Legionella were added at a multiplicity of infection (MOI) <5 to ice-cold macrophages, then the preparations were centrifuged 10 min at 400 g at 4°C, transferred to a 37°C water bath for 5 min, then washed three times with warm RPMI/FBS to remove extracellular bacteria.
Fluorescence microscopy
For localization of markers on Legionella phagosomes, 3.5 × 105 macrophages cultured on 12 mm glass coverslips in 24 well tissue culture plates were infected synchronously as described above. At the times designated, the preparations were fixed and permeabilized as described (Swanson and Isberg, 1995). Antibodies were diluted into PBS with 5% goat serum (PBS/GS) as follows: rabbit anti-Legionella (a gift from Dr Ralph Isberg, Tufts University School of Medicine), 1:100; rat antilysosomal-associated membrane protein 1 (LAMP1; 1D4B; Developmental Hybridoma Bank), 100%; rabbit anti-Atg7 (Dorn et al., 2001), 1:100; rabbit anti-Atg8/LC3 (a gift from Dr T. Yoshimori, National Institute of Genetics, Japan), 1:200; rat anti-Bip (SantaCruz), 1:100; all fluorescent secondary antibodies (Molecular Probes) were diluted 1:3000. Non-specific antibody binding was reduced by preincubating preparations overnight with PBS/GS. Cells were incubated with antibodies for 1 h at 37°C, then washed three times in PBS/GS. Bacteria were stained with either anti-Legionella antibody or 0.1 μM of the nucleic acid dye 4′,6′-diamino-2-phenylindole (DAPI; Molecular Probes) in PBS. After staining with specific antibodies, mounted coverslips were examined as previously described (Berger et al., 1994; Sturgill-Koszycki and Swanson, 2000). Colocalization with Atg7 was confirmed with confocal microscopy. To score autophagosome formation, a macrophage was defined as positive if it contained >5 donut-or C-shaped Atg7-labelled structures. Autophagosomes were labelled with MDC as previously described (Biederbick et al., 1995).
To ascertain whether MDC behaves as a specific marker for autophagosomes in our experimental model, several control assays were performed. First, compared to untreated cells, macrophages in which autophagy was stimulated for 2 h by either amino acid-free buffer or rapamycin stained brightly with MDC while the untreated macrophages showed a fine reticular background staining with the MDC (Fig. 2C; Amer et al., 2005). To distinguish the characteristic vesicular distribution of MDC in induced autophagosomes, each experiment was performed and scored in comparison with positive and negative controls. Formation of vacuoles that accumulate MDC required class III PI3-kinases, since the organelles were rare when macrophages were starved in the presence of 3MA. When uninfected macrophages whose lysosomes had been labeled by endocytosis of Texas Red-ovalbumin were incubated with MDC, the dye was rarely detected in the abundant lysosomal network (Amer et al., 2005). Nor was appreciable MDC retained by macrophages that were fed E. coli or E phase Legionella, microbes that are efficiently digested in lysosomes, or dotA mutant Legionella, bacteria that persist in non-lysosomal LAMP-1-positive compartments. Thus, MDC did not readily accumulate in acidic lysosomes during our treatment regimen but instead behaved as a valid marker of macrophage autophagosomes.
Supernatant preparation
The supernatants of E or PE phase Legionella cultures were collected by centrifugation at 7000 g for 15 min, sterilized with 0.22 μm filters, then concentrated 20× by centrifugation in 5 kDa Ultrafree-4 centrifugal filters (Vivascience). Supernatant preparations were diluted to 2× final concentration with RPMI/FBS. Sterile AYE broth concentrated similarly served as a control. Size fractionation was achieved by centrifugation of culture supernatants in Amicon Ultra Centrifugal Filter Devices (Millipore) with 5, 10, 30, 100 kDa molecular weight limits. Proteins were digested by treating PE phase supernatants with 1 μg proteinase K per 2.5 μg of protein for 12 h at 37°C.
Western analysis
A/J mouse macrophages treated to the conditions indicated were lysed with Mammalian Protein Extraction Reagent (PIERCE). Samples were separated by SDS-PAGE, transferred to nitrocellulose membranes (Bio-Rad), incubated with Atg8-specific antibody, then developed with WesternBreeze Chromogenic Western Blot Immunodetection Kit (Invitrogen).
3-methyladenine, cytochalasin D and BFA treatment
To inhibit autophagy, macrophages were incubated for 60 min before and during the infection with 10 mM 3-methyladenine (Sigma), as 2 and 5 mM 3MA had no detectable effect (Seglen and Gordon, 1982). At 90 min after infection, the cells were washed three times with RPMI/FBS then incubated for the additional period indicated. To inhibit phagocytosis by depolymerizing actin filaments, macrophages were incubated with 1 μg cytochalasin D per ml RPMI/FBS for 45 min before infection. Dimethyl sulphoxide (0.1%), which was used to dissolve the cytochalasin D, did not alter the growth of the bacteria in macrophage cultures nor affect macrophage viability (A. Amer, unpubl.). To inhibit vesicular traffic from the ER, BFA (Klausner et al., 1992) was added to the macrophage cultures 45 min before infection with Legionella and maintained throughout the incubation period. A 1 h incubation of uninfected macrophages with 3MA, BFA, or cytochalasin D at 5× the working concentration had no effect on macrophage viability as judged by Alamar Blue reduction (Byrne and Swanson, 1998).
Infections
The quantification of viable bacteria and enumeration of the colony-forming units (cfu) were performed as described previously (Swanson and Isberg, 1995; Sturgill-Koszycki and Swanson, 2000).
Acknowledgments
Much gratitude is given to Dr William Dunn (College of Medicine, University of Florida) and Dr Tamotsu Yoshimori (National Institute of Genetics, Japan) for generously providing antibodies used in this work. We thank Brenda Byrne for technical assistance, Dr Atsuki Nara (National Institute of Genetics, Japan) for insightful discussions and John-Demian Sauer for helpful comments on the manuscript. Our research is supported by the National Institute of Allergy and Infectious Diseases of the National Institute of Health, Grant 2 R01 AI040694-06AI.
References
- Abu Kwaik Y. The phagosome containing Legionella pneumophila within the protozoan Hartmonella vermiformis is surrounded by the rough endoplasmic reticulum. Appl Environ Microbiol. 1996;62:2022–2028. doi: 10.1128/aem.62.6.2022-2028.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer AO, Byrne BG, Swanson MS. Macrophages rapidly transfer pathogens from lipid raft vacuoles to autophagosomes. Autophagy. 2005;1 doi: 10.4161/auto.1.1.1589. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson HA, Chen Y, Norkin LC. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol Biol Cell. 1996;7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Bannykh SI, Rowe T, Balch WE. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J Cell Biol. 1995;131:875–893. doi: 10.1083/jcb.131.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger KH, Merriam JJ, Isberg RI. Altered intracellular targeting properties associated with mutations in the Legionella pneumophila dotA gene. Mol Microbiol. 1994;14:809–822. doi: 10.1111/j.1365-2958.1994.tb01317.x. [DOI] [PubMed] [Google Scholar]
- Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002;70:5816–5821. doi: 10.1128/IAI.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14. [PubMed] [Google Scholar]
- Byrne B, Swanson MS. Expression of Legionella pneumophila virulence traits in response to growth conditions. Infect Immun. 1998;66:3029–3034. doi: 10.1128/iai.66.7.3029-3034.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuprynski CJ, Faith NG, Steinberg H. A/J mice are susceptible and C57BL/6 mice are resistant to Listeria monocytogenes infection by intragastric inoculation. Infect Immun. 2003;71:682–689. doi: 10.1128/IAI.71.2.682-689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derre I, Isberg RR. Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect Immun. 2004;72:3048–3053. doi: 10.1128/IAI.72.5.3048-3053.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WF, Damron DM, Isberg RR, Lander ES, Swanson MS. Lgn1, a gene that determines susceptibility to Legionella pneumophila, maps to mouse chromosome 13. Genomics. 1995;26:443–450. doi: 10.1016/0888-7543(95)80161-e. [DOI] [PubMed] [Google Scholar]
- Diez E, Yaraghi Z, MacKenzie A, Gros P. The neuronal apoptosis inhibitory protein (Naip) is expressed in macrophages and is modulated after phagocytosis and during intracellular infection with Legionella pneumophila. J Immunol. 2000;164:1470–1477. doi: 10.4049/jimmunol.164.3.1470. [DOI] [PubMed] [Google Scholar]
- Diez E, Lee SH, Gauthier S, Yaraghi Z, Tremblay M, Vidal S, Gros P. Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat Genet. 2003;33:55–60. doi: 10.1038/ng1065. [DOI] [PubMed] [Google Scholar]
- Dorn BR, Dunn WA, Jr, Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun. 2001;69:5698–5708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WA. Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol. 1990a;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WA. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol. 1990b;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JA, Winn WC. Treatment of alveolar macrophages with cytochalasin D inhibits uptake and subsequent growth of Legionella pneumophila. Infect Immun. 1986;51:31–36. doi: 10.1128/iai.51.1.31-36.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga Y, Wolf A, Rodighiero C, Wheeler H, Tsai B, Allen L, et al. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to ER. Mol Biol Cell. 2003;14:4783–4793. doi: 10.1091/mbc.E03-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves da Costa SC, Calabrese KS, Zaverucha do Valle T, Lagrange PH. Trypanosoma cruzi: infection patterns in intact and athymic mice of susceptible and resistant genotypes. Histol Histopathol. 2002;17:837–844. doi: 10.14670/HH-17.837. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Hamasaki M, Noda T, Ohsumi Y. The early secretory pathway contributes to autophagy in yeast. Cell Struct Funct. 2003;28:49–54. doi: 10.1247/csf.28.49. [DOI] [PubMed] [Google Scholar]
- Hernandez LD, Pypaert M, Flavell RA, Galan JE. A Salmonella protein causes macrophage cell death by inducing autophagy. J Cell Biol. 2003;163:1123–1131. doi: 10.1083/jcb.200309161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsimaki P. Studies on vinblastine-induced autophagocytosis in mouse liver. II. Origin of membranes and acquisition of acid phosphatase. Histochem. 1983;79:59–67. doi: 10.1007/BF00494342. [DOI] [PubMed] [Google Scholar]
- Horwitz MA. The Legionnaires’ disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J Exp Med. 1983a;158:2108–2126. doi: 10.1084/jem.158.6.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz MA. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med. 1983b;158:1319–1331. doi: 10.1084/jem.158.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- Joshi AD, Sturgill-Koszycki S, Swanson MS. Evidence that Dot-dependent and – independent factors isolate the Legionella pneumophila phagosome from the endocytic network in mouse macrophages. Cell Microbiol. 2001;3:99–114. doi: 10.1046/j.1462-5822.2001.00093.x. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol. 2002;4:945–954. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Stein MP, Pypaert M, Roy CR. Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J Exp Med. 2004;199:1201–1211. doi: 10.1084/jem.20031706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–314. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner R, Donaldson J, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Cregg JM, Dunn AW, Emr SD, Sakai Y, Sandoval IV, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- Kopitz J, Kisen GO, Gordon PB, Bohley P, Seglen PO. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J Cell Biol. 1990;111:941–953. doi: 10.1083/jcb.111.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston P, Fong WG, Korneluk RG. The inhibitors of apoptosis: there is more to life than Bcl2. Oncogene. 2003;22:8568–8580. doi: 10.1038/sj.onc.1207101. [DOI] [PubMed] [Google Scholar]
- Maier JKX, Lahoua Z, Gendron NH, Fetni R, Johnston A, Davoodi J, et al. The neuronal apoptosis inhibitory protein is a direct inhibitor of caspases 3 and 7. J Neurosci. 2002;22:2035–2043. doi: 10.1523/JNEUROSCI.22-06-02035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DN, Baehrecke EH. Caspases function in autophagic programmed cell death in Drosophila. Development. 2004;131:275–284. doi: 10.1242/dev.00933. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Molmeret M, Zink SD, Han L, Abu-Zant A, Asari R, Bitar DM, Abu Kwaik Y. Activation of caspase-3 by the Dot/Icm virulence system is essential for arrested biogenesis of the Legionella containing phagosome. Cell Microbiol. 2004;6:33–48. doi: 10.1046/j.1462-5822.2003.00335.x. [DOI] [PubMed] [Google Scholar]
- Molofsky A, Swanson MS. Legionella pneumphila CsrA is a pivotal repressor of transmission traits and activator of replication. Mol Microbiol. 2003;50:445–461. doi: 10.1046/j.1365-2958.2003.03706.x. [DOI] [PubMed] [Google Scholar]
- Molofsky AB, Swanson MS. Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol Microbiol. 2004;53:29–40. doi: 10.1111/j.1365-2958.2004.04129.x. [DOI] [PubMed] [Google Scholar]
- Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. 2002;295:679–682. doi: 10.1126/science.1067025. [DOI] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamato A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- Naroeni A, Porte F. Role of cholesterol and the ganglioside GM1 in entry and short term survival of Brucella suis in murine macrophages. Infect Immun. 2002;70:1640–1644. doi: 10.1128/IAI.70.3.1640-1644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann A, Takatsuki A, Elsasser HP. The lysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J Histochem Cytochem. 2000;48:251–258. doi: 10.1177/002215540004800210. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Milosevic S, Behrends U, Jaffee EM, Pardoll DM, Bornkamm GW, Mautner J. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur J Immunol. 2003;33:1250–1259. doi: 10.1002/eji.200323730. [DOI] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Noda T, Suzuki K, Ohsumi Y. Yeast autophagosomes: de novo formation of a membrane structure. Trends Cell Biol. 2002;12:231–235. doi: 10.1016/s0962-8924(02)02278-x. [DOI] [PubMed] [Google Scholar]
- Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta. 2003;1603:113–128. doi: 10.1016/s0304-419x(03)00004-0. [DOI] [PubMed] [Google Scholar]
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- Otto GP, Wu MY, Clarke M, Lu H, Anderson OR, Hilbi H, et al. Macroautophagy is dispensable for intracellular replication of Legionella pneumophila in Dictyostelium discoideum. Mol Microbiol. 2004;51:63–72. doi: 10.1046/j.1365-2958.2003.03826.x. [DOI] [PubMed] [Google Scholar]
- Pizarro-Cerda J, Meresse S, Parton RG, van der Goot G, Sola-Landa A, Lopez-Goni I, et al. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect Immun. 1998a;66:5711–5724. doi: 10.1128/iai.66.12.5711-5724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerda J, Moreno E, Sanguedolce V, Mege JL, Gorvel JP. Virulent Brucella abortus prevents lysosome fusion and is distributed within autophagosome-like compartments. Infect Immun. 1998b;66:2387–2392. doi: 10.1128/iai.66.5.2387-2392.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5:455–468. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- Richterova Z, Liebl D, Horak M, Palkova Z, Stokrova J, Hozak P. Caveolae are involved in the trafficking of mouse polyoma virus virions and artificial VP1 pseudocapsids toward cell nuclei. J Virol. 2001;75:10880–10891. doi: 10.1128/JVI.75.22.10880-10891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roisin-Bouffay C, Luciani MF, Klein G, Levraud JP, Adam M, Golstein P. Developmental cell death in Dictyostelium does not require paracaspase. J Biol Chem. 2004;279:11489–11494. doi: 10.1074/jbc.M312741200. [DOI] [PubMed] [Google Scholar]
- Roy CR, Berger KH, Isberg RR. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol Microbiol. 1998;28:663–674. doi: 10.1046/j.1365-2958.1998.00841.x. [DOI] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Schaible UE, Schlesinger PH, Steinberg TH, Mangel WF, Kobayashi T, Russell DG. Parasitophorous vacuoles of Leishmania mexicanao acquire macromolecules from the host cell cytosol via two independent routes. J Cell Sci. 1999;112:681–693. doi: 10.1242/jcs.112.5.681. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen PO, Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia. 1992;48:158–172. doi: 10.1007/BF01923509. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB, Holen I. Nonselective autophagy. Semin Cell Dev Biol. 1990;1:441–448. [PubMed] [Google Scholar]
- Stevenson MM, Tam MF. Differential induction of helper T cell subsets during blood-stage Plasmodium chabaudi AS infection in resistant and susceptible mice. Clin Exp Immunol. 1993;92:77–83. doi: 10.1111/j.1365-2249.1993.tb05951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromhaug PE, Berg TO, Fengsrud M, Seglen PO. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J. 1998;335:217–224. doi: 10.1042/bj3350217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill-Koszycki S, Swanson MS. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J Exp Med. 2000;192:1261–1272. doi: 10.1084/jem.192.9.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63:3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Fernandez-Moreira E. A microbial strategy to multiply in macrophages: the pregnant pause. Traffic. 2002;3:170–177. doi: 10.1034/j.1600-0854.2002.030302.x. [DOI] [PubMed] [Google Scholar]
- Talloczy Z, Jiang W, Virgin HWT, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation-and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y, Kominami E. Apg7p/Cvt2p: a novel protein-activating enzyme essential for autophagy. Mol Biol Cell. 1999;10:1367–1379. doi: 10.1091/mbc.10.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Ohsumi M, Ohsumi Y, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem. 2001;276:9846–9854. doi: 10.1074/jbc.C000752200. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Harb OS, Connelly PS, Robinson CG, Roy CR. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J Cell Sci. 2001;114:4637–4650. doi: 10.1242/jcs.114.24.4637. [DOI] [PubMed] [Google Scholar]
- Ueno T, Muno D, Kominami E. Membrane markers of endoplasmic reticulum preserved in autophagic vacuolar membranes isolated from leupeptin-administered rat liver. J Biol Chem. 1991;266:18995–18999. [PubMed] [Google Scholar]
- Vogel JP, Andrews HL, Wong SK, Isberg RR. Conjugative transfer by the virulence system of Legionella pneumophila. Science. 1998;279:873–876. doi: 10.1126/science.279.5352.873. [DOI] [PubMed] [Google Scholar]
- Watarai M, Derre I, Kirby J, Growney JD, Dietrich WF, Isberg RR. Legionella pneumophila is internalized by a macropinocytotic uptake pathway controlled by the Dot/Icm system and the mouse Lgn1 locus. J Exp Med. 2001;194:1081–1096. doi: 10.1084/jem.194.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright EK, Goodart SA, Growney JD, Hadinoto V, Endrizzi MG, Long EM, et al. Naip5 affects host suceptibility to the intracellular pathogen Legionella pneumophila. Curr Biol. 2003;13:27–36. doi: 10.1016/s0960-9822(02)01359-3. [DOI] [PubMed] [Google Scholar]
- Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004;313:453–458. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]