

Abstract
Macrophages activate autophagy as an immediate response to Legionella pneumophila infection, but what marks the pathogen phagosome as a target for the autophagy machinery is not known. Because a variety of bacteria, parasites, viruses, and toxins that associate with the endoplasmic reticulum enter host cells by a cholesterol-dependent route, we tested the hypothesis that autophagy is triggered when microbes engage components of lipid raft domains. As the intracellular respiratory pathogen L. pneumophila or the extracellular uropathogen FimH+ Escherichia coli entered macrophages by a cholesterol-sensitive mechanism, they immediatezly resided in vacuoles rich in glycosylphosphatidylinositol moieties and the autophagy enzyme Atg7. As expected for autophagosomes, the vacuoles sequentially acquired the endoplasmic reticulum protein BiP, the autophagy markers Atg8 and monodansyl-cadaverine, and the lysosomal protein LAMP-1. A robust macrophage response to the pathogens was cholesterol-dependent, since fewer Atg7-rich vacuoles were observed when macrophages were pretreated with methyl-β-cyclodextrin or filipin. A model in which macrophages exploit autophagy to capture pathogens within the lipid raft pathway for antigen presentation prior to disposal in lysosomes is discussed.
Keywords: Legionella pneumophila, uropathogenic E. coli, cholesterol, endoplasmic reticulum, lysosomes
INTRODUCTION
As sentinels of the immune system, macrophages efficiently deliver microbes to the toxic phagolysosome pathway and also present antigens to stimulate a specific antibody-or cell-mediated defense. Yet many microbes can avoid immediate digestion in lysosomes by entering host cells through a cholesterol-sensitive pathway.1–6 As a consequence, in macrophage cultures, some pathogens survive in vacuoles that are rich in cholesterol, GM1 gangliosides, and glycosylphosphatidylinositol (GPI) anchored proteins, three characteristics of lipid rafts.7
A variety of particles that engage lipid raft components of host cells subsequently associate with the ER, including Brucella suis, the virus SV40, the eukaryotic parasite Toxoplasma gondii, cholera toxin, and latex beads.4,8–12 Another member of this group is Legionella pneumophila,13–15 a pathogen of macrophages that releases soluble factors that stimulate immediate autophagy.16 In this conserved catabolic process, autophagosomes sequester cytoplasmic material and extraneous organelles within membranes derived from the secretory pathway for delivery to lysosomes.17,18 As such, autophagy could account for the assembly of ER-derived phagosomes that are competent to present vacuolar antigens to the MHCI pathway.19–21 Indeed, numerous microbes interact with the autophagy pathway of host cells.16,22–34 Therefore, we postulated that autophagy of lipid raft-derived vacuoles is a general mechanism for macrophages to capture microbes within ER before their disposal in toxic lysosomes.
To examine whether autophagy is triggered by microbes that engage the lipid raft pathway, we exploited two Gram-negative pathogens that both employ surface structures to enter cholesterol-rich vacuoles. Compared here are Dot/Icm Type IV secretion-competent L. pneumophila,35 an intracellular respiratory pathogen that resides within ER-derived vacuoles,13–15 and FimH+ piliated Escherichia coli, an extracellular uropathogen that enters eukaryotic cells rarely.1,36
MATERIALS AND METHODS
Macrophages
Bone marrow-derived macrophages obtained from A/J mice (The Jackson Laboratory) were maintained in RPMI 1640 containing 10% heat-inactivated fetal bovine serum (RPMI/FBS) as described.37 The susceptibility of A/J mice to L. pneumophila infection is conferred by a mutation in naip5.38,39 Compared to cells obtained from C57BL/6J mice, macrophages derived from A/J mice have sluggish autophagosome maturation,16 a trait that facilitates analysis of the dynamic composition of nascent autophagosomes. In both A/J and C57BL/6J bone marrow-derived macrophages, autophagosomes that contain uropathogenic E. coli mature at rates comparable to the vacuoles induced by either starvation or rapamycin, as judged by the kinetics of Atg8 acquisition (ref. 16 and unpublished data).
Bacteria
L. pneumophila strain Lp02, a virulent thymine auxotrophic derivative of Philadelphia 1, and its dotA mutant derivative were cultured either to the infectious post-exponential phase (PE) or the noninfectious exponential phase (E) as described.37 Phase-locked FimH+ and FimH− E. coli strains CFT073 ON and CFT073 OFF40 and E. coli DH5α were cultured to saturation in Luria broth.
Fluorescence microscopy
To promote synchrony, E. coli or L. pneumophila were added to macrophages at a multiplicity of infection (MOI) of < 5, then the samples were centrifuged for 10 min, transferred to a 37°C water bath for 5 min, washed 3 times with 37°C RPMI/FBS, treated for 6 min with 10 μg gentamicin per ml RPMI/FBS at 37°C, rinsed 3 times with RPMI/FBS, then incubated at 37°C and 5% CO2. Under these conditions, the majority of macrophages contained one bacterium. After the periods designated, the preparations were fixed, permeabilized, and stained with Atg7-, Atg8-, BiP-, LAMP-1-specific antibodies or monodansyl-cadaverine as previously described.23,40,14,16,36 To detect GPI-anchored proteins, fixed and permeabilized samples were incubated with 2 μg Alexa594-aerolysin (Protox Biotech) per ml PBS for 1 h at 37°C. Bacteria were stained with DAPI or NHS-carboxy-fluorescein as described previously.14,42 Preparations from two or more independent experiments were examined and scored as previously described.16 Because the majority of intracellular FimH− E. coli were degraded within 1 h, the colocalization of 25–50 of these intracellular bacteria with BiP and with LAMP-1 was scored. To quantify the percentage of macrophages that were infected, cells were infected synchronously as described above. At the times shown, extracellular bacteria were removed by washing the monolayers three times with PBS, then the preparations were fixed, and the fraction of 100 macrophages that contained at least one intact bacillus was enumerated in three or more independent experiments.
Autophagy
To stimulate autophagy, macrophages were cultured for 1 or 2 h with rapamycin (0.2 μg per ml RPMI/FBS), which relieves autophagy inhibition by Tor kinase.17 To determine the effect of autophagy on bacterial survival, macrophages cultured for 1 h without or with 10 mM 3-methy-ladenine17 in RPMI/FBS were infected synchronously as described above at an MOI of < 10 in the presence or absence of 3MA, then incubated at 37°C for an additional 5 or 60 min in the presence or absence of 3MA. To remove 3MA after 1 h of infection, all samples were washed with RPMI/FBS, then incubated for an additional 1 h in RPMI/FBS. To quantify viable bacteria after the 5 min and the 2 h infection period, the supernatant and macrophage lysate from each well were collected and pooled, then aliquots were plated onto nutrient agar to enumerate the colony forming units (CFU) as described previously.14,42 The % CFU at 2 h post infection was calculated from duplicate samples in two independent experiments as [(intracellular CFU at 2 h post-infection)/(intracellular CFU at 5 min post-infection)] x 100. Treatment with 3MA did not prevent cholesterol-dependent entry of bacteria, as judged by comparing the cell-associated CFU at 15 min post-infection in the presence and absence of drug, and control experiments demonstrated that 3MA was not toxic to either the bacteria or macrophages (unpublished data).
Cholesterol depletion or sequestration
Macrophages treated for 1 h at 37°C with the concentrations shown of methyl-β-cyclodextrin or filipin (Sigma) in RPMI/FBS were infected at an MOI < 5, centrifuged for 10 min, then transferred to a 37°C water bath for 5 or 15 min. Preparations were washed 3 times with RPMI/FBS at 37°C, then fixed, permeabilized, and stained with anti-Legionella antibody. The number of bacteria associated with 100 macrophages was enumerated microscopically in three independent duplicate experiments. The number of bacteria observed in 200 untreated macrophages ranged from 40–80. Control experiments determined that macrophages remained viable after a 1 hr incubation with 50 mM methyl-β-cyclodextrin or 25 μg/ml filipin.
RESULTS
Like L. pneumophila, uropathogenic E. coli stimulate autophagosome formation
When autophagy was stimulated by incubating macrophages with either rapamycin or infectious PE L. pneumophila, the cells rapidly formed numerous vacuoles decorated with Atg7 (Fig. 1A and B), a ubiquitin E1-like activating enzyme that initiates autophagosome formation.18,43 Smaller numbers of similar autophagosomal structures were detected when macrophages were incubated with FimH+ E. coli, an extracellular pathogen of the urinary tract (Fig. 1A and B). Macrophages accumulated Atg7-positive vacuoles as a response to pathogenic microbes, since the organelles were rarely observed in cells incubated with either sterile medium or the corresponding avirulent bacteria: exponential phase (E) wild-type L. pneumophila37 or FimH− E. coli.1 Because ~90% of the Atg7-positive vacuoles stimulated by PE L. pneumophila were also rich in GPI-linked molecules (unpublished data), we next examined whether the accumulation of autophagy enzymes was more pronounced on lipid raft-derived phagosomes that harbored pathogenic bacteria.
Figure 1.
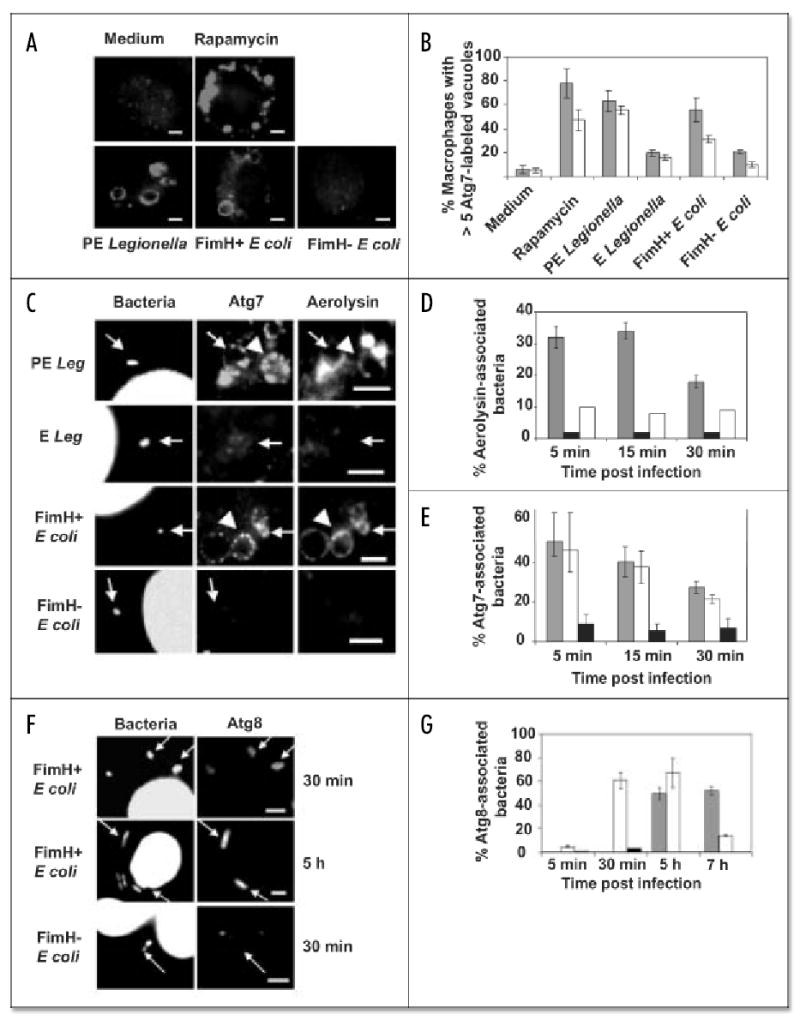
L. pneumophila and uropathogenic E. coli phagosomes colocalize with markers of the lipid raft and autophagy pathways. (A) Atg7 redistributed to vacuoles within 15 min of incubating macrophages with the autophagy stimulator rapamycin or with virulent PE L. pneumophila or FimH+ E. coli but not with medium or FimH− E. coli. (B) The percent macrophages with > 5 Atg7-labeled vacuoles at 5 min (grey bars) and 15 min (white bars) after treatments described in (A); avirulent E L. pneumophila served as an additional negative control. Values are the mean percent of at least three independent experiments ± standard deviation. (C) PE L. pneumophila or FimH+ E. coli stimulated formation of vacuoles (arrowheads) and phagosomes (arrows) rich in Atg7 and GPI-molecules (aerolysin), but E L. pneumophila or FimH− E. coli did not. (D) Phagosomes containing PE L. pneumophila (grey bars) retained Alexa 594-aerolysin, unlike those with E wild-type (black) or PE dotA mutants (white bars). Values are the mean percent of at least two independent experiments ± standard deviation. (E) Atg7 accumulated on phagosomes containing PE L. pneumophila (grey bars) or FimH+ E. coli (white bars), but not FimH− E. coli (black bars). (F) FimH+ E. coli persisted in vacuoles that colocalized with Atg8, unlike FimH− E. coli. Values are the mean percent of at least two independent experiments ± standard deviation. (G) Atg8 accumulated on phagosomes of PE L. pneumophila (grey bars) or FimH+ E. coli (white bars), but not FimH− E. coli (black bars). Values are the mean percent of at least two independent experiments ± standard deviation. Scale bar, 10 μm.
L. pneumophila and uropathogenic E. coli phagosomes colocalize with markers of the lipid raft and autophagy pathways
Pathogenic FimH + E. coli or PE L. pneumophila that resided in vacuoles with features of lipid rafts immediately recruited the autophagy machinery, as judged by the coincident fluorescent staining with antibody specific to Atg723,18,43 and either the GPI-anchored protein ligand aerolysin, the GM1 ganglioside ligand cholera toxin B, or the cholesterol-binding protein filipin (Fig. 1C–E; unpublished data; ref. 7). By 30 min after infection, a second conjugating enzyme of the autophagy machinery, Atg8,41,18,43 accumulated on phagosomes that contained FimH+ E. coli; the enzyme decorated the PE L. pneumophila compartments more slowly (Fig. 1F and G), as expected.16 Like maturing autophagosomes17 and L. pneumophila vacuoles,14–16,42 phagosomes that harbored FimH+ E. coli subsequently acquired the fluorescent dye monodansyl-cadaverine and markers of the biosynthetic and the endosomal pathways. At 2 h after infection, 18% (± 2) of the pathogen vacuoles colocalized with the ER lumenal protein BiP (Fig. 2B). In contrast, < 4% (± 3) of the FimH− E. coli did so when examined 15 min to 2 h after infection; instead, the majority were degraded (Fig. 4). The late endosomal and lysosomal protein LAMP-1 was apparent on some vacuoles of FimH+ E. coli 30 min after infection (Fig. 2A); by 3 h, 84% (± 4) were associated with LAMP-1. Association with the autophagy machinery was more frequent and/or more prolonged for pathogenic bacteria that resided within lipid raft-derived vacuoles, since aerolysin, cholera toxin B, Atg7, Atg8, monodansyl-cadaverine, and BiP were rarely observed on phagosomes containing either FimH− E. coli, E wild-type L. pneumophila, or PE dotA mutant L. pneumophila (Fig. 1 and 2; ref. 16), microbes that macrophages efficiently deliver to the endosomal pathway.1,13,37
Figure 2.
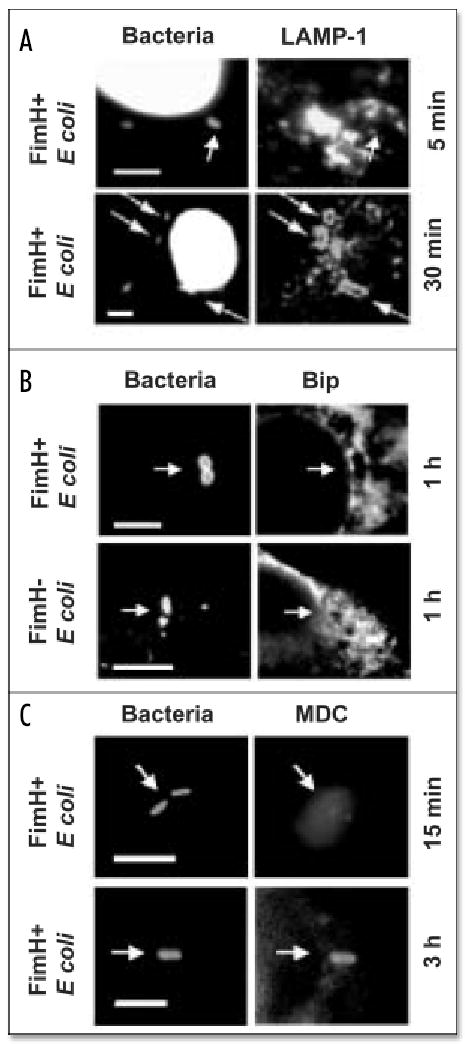
Uropathogenic E. coli phagosomes acquire Bip, LAMP-1 and MDC. (A) DAPI-stained FimH+ E. coli vacuoles required 30 min to accumulate the late endosomal and lysosomal protein LAMP-1. (B) FimH+ but not FimH− E. coli vacuoles colocalized with the ER protein Bip 1 h after infection. (C) The autophagosomal marker MDC accumulated in FimH+ E. coli vacuoles after 3 h of infection. E. coli were stained with NHS-carboxy-fluorescein in (B and C). Scale bar, 10 μm.
Figure 4.
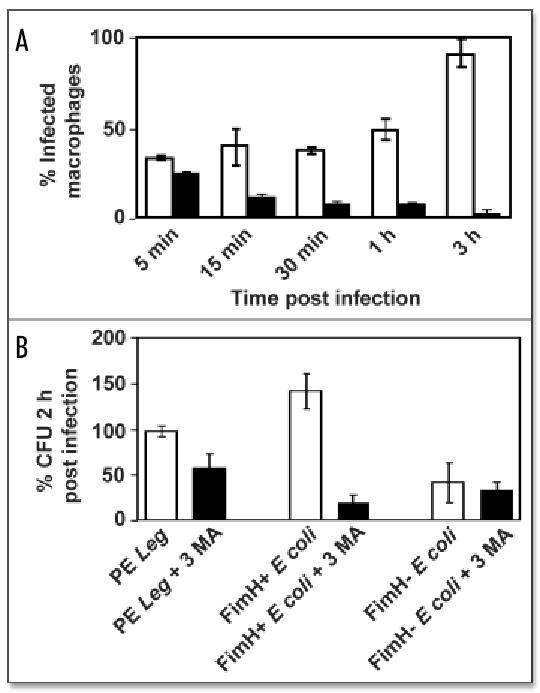
Viability of pathogens in autophagosomes. (A) During a 3 h incubation period, FimH+ E. coli infected additional cells (white bars), whereas FimH− E. coli were cleared (black bars), as judged microscopically by the percent of macrophages that contained intact bacteria. (B) Compared to control cultures (white bars), macrophages pretreated for 1 h with 3MA to inhibit autophagy (black bars) decreased the colony forming units (CFU) of pathogenic PE L. pneumophila and FimH+ E. coli, whereas FimH− E. coli were rapidly killed by both treated and untreated macrophages. Percent CFU 2 h post-infection was calculated as [(intracellular CFU 2 h post-infection)/(intracellular CFU 5 min post-infection)] x 100. Values are the mean percent of at least two independent experiments ± standard deviation.
Pathogen internalization and stimulation of autophagy are cholesterol-sensitive
To investigate by an independent approach the link between vacuole composition and accumulation of autophagy enzymes, lipid rafts were perturbed pharmacologically,7 then the capacity of the bacteria to enter macrophages quickly and to stimulate autophagosome formation was quantified. Pretreatment of macrophages for 1 h with methyl-β-cyclodextrin or filipin had the most pronounced effect during a 15 min period of internalization of infectious PE L. pneumophila, an intermediate effect on uptake of PE dotA mutant L. pneumophila, and the least impact on phagocytosis of nonvirulent DH5α E. coli (Fig. 3A and B). Thus, as previously reported for FimH+ E. coli1 and predicted for L. pneumophila,13 macrophage internalization of certain pathogens appears to be more sensitive to the cholesterol content of the plasma membrane than is uptake of avirulent E. coli.
Figure 3.
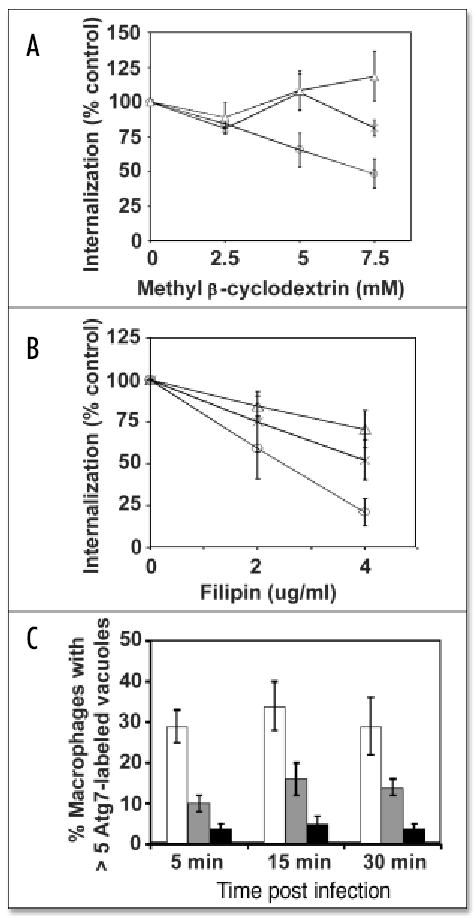
Perturbation of lipid rafts decreased pathogen entry and autophagosome formation. Methyl-β-cyclodextrin (A) or filipin (B) pretreatment for 1 h primarily decreased macrophage internalization of PE L. pneumophila (circles) but had moderate effect on uptake of dotA− L. pneumophila (X) or DH5α E. coli (triangles). Percent internalization was quantified microscopically as: [(# intracellular bacteria 15 min post-infection)treated/(# intracellular bacteria 15 min post-infection)]untreated × 100. (C) Macrophages contained numerous Atg7-decorated vacuoles when infected by PE L. pneumophila (white bars), unless they were pretreated for 1 h with 5 mM methyl-β-cyclodextrin (grey) or 4 μg/ml filipin (black bars). Values are the mean percent of at least three independent experiments ± standard deviation.
The integrity of macrophage lipid rafts also affected formation of autophagosomes in response to PE L. pneumophila. Methyl-β-cyclodextrin or filipin treatment decreased the number of autophagosomes observed in macrophages incubated with L. pneumophila (Fig. 3C). Perturbation of macrophage lipid composition had minimal effect when autophagosome formation was induced by rapamycin or starvation (unpublished data). Accordingly, components of lipid rafts do not appear to be essential for assembly of autophagosomes, but instead may affect a signal transduction pathway dedicated to host recognition of microbes.
Pathogens that reside in autophagosomes avoid immediate killing
By entering the lipid raft pathway, a variety of microbes avoids immediate digestion in lysosomes of cultured phagocytes.1–4 For L. pneumophila, uptake into GPI-rich vacuoles correlates with its ability to evade the endosomal pathway.13 Likewise, uropathogenic FimH+ E. coli persisted in macrophage cultures for at least 3 h, whereas FimH− E. coli were efficiently cleared, as expected (Fig. 4A; ref. 1). To assess the impact of autophagy on survival of virulent L. pneumophila and FimH+ E. coli, autophagosome formation was inhibited pharmacologically, then viability of the intracellular bacteria was quantified. During a 2 h infection of untreated macrophage cultures, the number of intracellular PE L. pneumophila remained constant, consistent with the several hour lag period this pathogen requires to differentiate to a replicative form,42 whereas the yield of intracellular FimH+ E. coli increased slightly (Fig. 4B). In contrast, when autophagy was inhibited with 10 mM 3-methyladenine (3MA), which blocks class III phosphatidylinositol 3’ kinase activity,17 the macrophages killed ~50% of the intracellular PE L. pneumophila, as expected,16 and ~80% of the intracellular FimH+ E. coli (Fig. 4B). Therefore, in the A/J mouse model of the encounter between microbes and macrophages of a host that lacks opsonic antibodies, the autophagy pathway did not rapidly kill the pathogens.
DISCUSSION
Like the intracellular respiratory pathogen L. pneumophila and the uropathogen FimH+ E. coli analyzed here, a variety of other particles that engage components of lipid rafts may also be targets of the macrophage autophagy machinery. Brucella spp.,4,6,22,44 T. gondii,9 SV40,5,8 cholera toxin B,10 and latex beads11,12 are each first internalized within cholesterol-rich vacuoles before associating with ER. Moreover, a diverse array of pathogens have been reported to interact with components of the autophagy pathway, namely B. abortus, Coxiella burnetii, Porphyromonas gingivalis, Salmonella enterica, Chlamydia trachomatis, Listeria monocytogenes, Group A Streptococcus, Mycobacterium tuberculosis, Leishmania mexicana, poliovirus, herpes simplex virus and coronavirus.22–34,45,46 Accordingly, we postulate that autophagy is not only a surveillance system to detect cytosolic microbes28,34 but also a mechanism for macrophages to capture, document, and digest microbes that engage components of lipid rafts.
Whether virulent and avirulent microbes follow distinct routes in macrophages, or instead traffic on the same pathway but at quite different rates cannot be distinguished by the microscopy methods employed here. At a given time, the fraction of vacuoles observed to colocalize with a particular marker is affected by several factors, including the half-life of the interaction, the synchrony of vacuole biogenesis, and the strength of the fluorescence signal. Therefore, we cannot ascertain whether perturbation of lipid rafts alters which host proteins and organelles productively interact with the phagosome or instead affects their rate of maturation. The decreased number of autophagosomes observed after lipid raft disruption is likely not accounted for solely by the concomitant decrease in ingestion of the pathogen, based on three previous observations: Cytochalasin D treatment dramatically decreases phagocytosis of L. pneumophila yet has minimal effect on autophagy stimulation by the bacteria; sterile supernatants obtained from L. pneumophila cultures stimulate autophagosome formation independently of phagocytosis; and autophagosomal vacuoles are frequently observed in macrophages that do not contain a bacterium.16 Accordingly, our present model is that internalization of pathogens and autophagosome formation are concurrent but distinct activities that are each promoted by components of lipid rafts.
A number of testable predictions concerning the composition and function of macrophage autophagosomes follow from the present and published data. As a mechanism that captures cytosolic material for lysosomal degradation,17 autophagy would be another host barrier that exerts selective pressure on microbes; consequently, some pathogens are predicted to manipulate the autophagy machinery as a strategy to establish a persistent infection. Indeed, in macrophages derived from A/J mice, the only strain known to be susceptible to infection by L. pneumophila,38,39 autophagosomes that harbor the intracellullar pathogen mature more slowly than those stimulated by either rapamycin or the extracellular pathogen FimH+ E. coli (refs. 16, 42 and unpublished data). Moreover, autophagosomes of A/J mouse macrophages mature more slowly than those of resistant C57BL/6J mouse macrophages, whether stimulated by L. pneumophila, rapamycin, or starvation.16,42 Taken together, available data support the model that L. pneumophila exploit Type IV secretion and other mechanisms to deliver virulence factors that delay maturation of its autophagosomal vacuole.15,16,37,42 Consistent with this model, virulent B. abortus persist in an ER-derived vacuole, whereas mutants that lack Type IV secretion transit through an ER compartment en route to the lysosomes.44
As a mechanism to assemble mixed phagosome-ER compartments.12,15,16,42 autophagy may orchestrate not only recognition by certain Toll-like receptors47 but also proteasome- and TAP-dependent antigen cross-presentation by macrophages.19–21 The survival of L. pneumophila and FimH+ E. coli in the autophagy pathway (Fig. 4B) raises the possibility that biogenesis of lipid raft-rich autophagosomes does not trigger the rapid assembly of active NADPH-oxidase, an enzyme detrimental not only to microbes but also to antigen structure. According to this reasoning, in immune hosts, Fc-mediated phagocytosis accomplishes a rapid killing and digestion of pathogens; whereas, in naïve hosts, autophagy of lipid raft-derived phagosomes promotes presentation of native antigens prior to their disposal in lysosomes. The impact of macrophage autophagy on innate and acquired immunity can now be examined directly by exploiting recently obtained knowledge of the broadly conserved enzymatic cascade that governs autophagosome biogenesis.17,18,43
Acknowledgments
For generous gifts of reagents that were essential for this study, we thank H.L.T. Mobley for the pair of phase-locked uropathogenic E. coli strains, W. A. Dunn for anti-Atg7 antibody, and T. Yoshimori for anti-Atg8 antibody.
We thank K. Kirkegaard, A. Molofsky, and JD Sauer for insightful discussions, and J.A. Swanson and B. Tsai for perceptive comments on the manuscript.
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institute of Health, 2 R01 AI040694.
References
- 1.Baorto DM, Gao Z, Malaviya R, Dustin ML, van der Merwe A, Lublin DM, Abraham SN. Survival of FimH-expressing enterobacteria in macrophages relies on glycolipid traffic. Nature. 1997;389:636–9. doi: 10.1038/39376. [DOI] [PubMed] [Google Scholar]
- 2.Gatfield J, Pieters J. Essential role for cholesterol in entry of Mycobacteria into macrophages. Science. 2000;288:1647–50. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- 3.Manes S, del Real G, Martinez AC. Pathogens: Raft hijackers. Nat Rev Immunol. 2003;3:557–68. doi: 10.1038/nri1129. [DOI] [PubMed] [Google Scholar]
- 4.Naroeni A, Porte F. Role of cholesterol and the ganglioside GM1 in entry and short term survival of Brucella suis in murine macrophages. Infect and Immun. 2002;70:1640–4. doi: 10.1128/IAI.70.3.1640-1644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson HA, Chen Y, Norkin LC. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol Biol Cell. 1996;7:1825–34. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watarai M, Makino S, Fujii Y, Okamoto K, Shirahata T. Modulation of Brucella-induced macropinocytosis by lipid rafts mediates intracellular replication. Cell Microbiol. 2002;4:341–55. doi: 10.1046/j.1462-5822.2002.00195.x. [DOI] [PubMed] [Google Scholar]
- 7.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 8.Norkin LC, Anderson HA, Wolfrom SA, Oppenheim A. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J Virol. 2002;76:5156–66. doi: 10.1128/JVI.76.10.5156-5166.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mordue DG, Desai N, Dustin M, Sibley LD. Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J Exp Med. 1999;190:1783–92. doi: 10.1084/jem.190.12.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujinaga Y, Wolf A, Rodighiero C, Wheeler H, Tsai B, Allen L, Jobling M, Rapoport T, Holmes R, Lencer W. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to ER. Mol Biol Cell. 2003;14:4783–93. doi: 10.1091/mbc.E03-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dermine JF, Duclos S, Garin J, St-Louis F, Rea S, Parton RG, Desjardins M. Flotillin-1-enriched lipid raft domains accumulate on maturing phagosomes. J Biol Chem. 2001;276:18507–12. doi: 10.1074/jbc.M101113200. [DOI] [PubMed] [Google Scholar]
- 12.Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, Paiement J, Bergeron JJ, Desjardins M. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110:119–31. doi: 10.1016/s0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- 13.Watarai M, Derre I, Kirby J, Growney JD, Dietrich WF, Isberg RR. Legionella pneu-mophila is internalized by a macropinocytotic uptake pathway controlled by the Dot/Icm system and the mouse Lgn1 locus. J Exp Med. 2001;194:1081–96. doi: 10.1084/jem.194.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63:3609–20. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol. 2002;4:945–54. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- 16.Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 2005 doi: 10.1111/j.1462-5822.2005.00509.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogier-Denis E, Codogno P. Autophagy: A barrier or an adaptive response to cancer. Biochim Biophys Acta. 2003;1603:113–28. doi: 10.1016/s0304-419x(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 18.Ohsumi Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–6. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 19.Ackerman AL, Kyritsis C, Tampe R, Cresswell P. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc Natl Acad Sci. 2003;100:12889–94. doi: 10.1073/pnas.1735556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 2003;425:397–402. doi: 10.1038/nature01911. [DOI] [PubMed] [Google Scholar]
- 21.Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, Princiotta MF, Thibault P, Sacks D, Desjardins M. Phagosomes are competent organelles for antigen cross-presentation. Nature. 2003;425:402–6. doi: 10.1038/nature01912. [DOI] [PubMed] [Google Scholar]
- 22.Pizarro-Cerda J, Meresse S, Parton RG, van der Goot G, Sola-Landa A, Lopez-Goni I, Moreno E, Gorvel JP. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect Immun. 1998;66:5711–24. doi: 10.1128/iai.66.12.5711-5724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorn BR, Dunn WA, Jr, Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun. 2001;69:5698–708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorn BR, Dunn WA, Jr, Progulske-Fox A. Bacterial interactions with the autophagic pathway. Cell Microbiol. 2002;4:1–10. doi: 10.1046/j.1462-5822.2002.00164.x. [DOI] [PubMed] [Google Scholar]
- 25.Suhy DA, Giddings TH, Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J Virol. 2000;74:8953–65. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talloczy Z, Jiang W, Virgin IVth HW, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA. 2002;99:190–5. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beron W, Gutierrez MG, Rabinovitch M, Columbo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002;70:5816–21. doi: 10.1128/IAI.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5:455–68. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 29.Hernandez LD, Pypaert M, Flavell RA, Galan JE. A Salmonella protein causes macrophage cell death by inducing autophagy. J Cell Biol. 2003;163:1123–31. doi: 10.1083/jcb.200309161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Younes HM, Brinkmann V, Meyer TF. Interaction of Chlamydia trachomatis serovar L2 with the host autophagic pathway. Infect Immun. 2004;72:4751–62. doi: 10.1128/IAI.72.8.4751-4762.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279:10136–41. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading group a Streptococcus. Science. 2004;306:1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 33.Gutierrez MG, Master SS, Singh SB, Taylor GA, Columbo MI, Deretic V. Autophagy is a defense mechanism inhibiting Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 34.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–31. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 35.Vogel JP, Andrews HL, Wong SK, Isberg RR. Conjugative transfer by the virulence system of Legionella pneumophila. Science. 1998;279:873–6. doi: 10.1126/science.279.5352.873. [DOI] [PubMed] [Google Scholar]
- 36.Mulvey MA, Schilling JD, Martinez JJ, Hultgren SJ. Bad bugs and beleaguered bladders: Interplay between uropathogenic Escherichia coli and innate host defenses. Proc Natl Acad Sci USA. 2000;97:8829–35. doi: 10.1073/pnas.97.16.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joshi AD, Sturgill-Koszycki S, Swanson MS. Evidence that Dot-dependent and -independent factors isolate the Legionella pneumophila phagosome from the endocytic network in mouse macrophages. Cell Microbiol. 2001;3:99. doi: 10.1046/j.1462-5822.2001.00093.x. [DOI] [PubMed] [Google Scholar]
- 38.Diez E, Lee SH, Gauthier S, Yaraghi Z, Tremblay M, Vidal S, Gros P. Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat Genetics. 2003;33:55–60. doi: 10.1038/ng1065. [DOI] [PubMed] [Google Scholar]
- 39.Wright EK, Goodart SA, Growney JD, Hadinoto V, Endrizzi MG, Long EM, Sadigh K, Abney AL, Bernstein-Hanley I, Dietrich WF. Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr Biol. 2003;13:27–36. doi: 10.1016/s0960-9822(02)01359-3. [DOI] [PubMed] [Google Scholar]
- 40.Gunther IN, Snyder JA, Lockatell V, Blomfield I, Johnson DE, Mobley HL. Assessment of virulence of uropathogenic Escherichia coli type 1 fimbrial mutants in which the invertible element is phase-locked on or off. Infect Immun. 2002;70:3344–54. doi: 10.1128/IAI.70.7.3344-3354.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sturgill-Koszycki S, Swanson MS. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J Exp Med. 2000;192:1261–72. doi: 10.1084/jem.192.9.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klionsky DJ, Cregg JM, Dunn WA, Emr SD, Sakai Y, Sandoval IV, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–45. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 44.Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J Exp Med. 2003;198:545–56. doi: 10.1084/jem.20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaible UE, Schlesinger PH, Steinberg TH, Mangel WF, Kobayashi T, Russell DG. Parasitophorous vacuoles of Leishmania mexicana acquire macromolecules from the host cell cytosol via two independent routes. J Cell Sci. 1999;112:681–93. doi: 10.1242/jcs.112.5.681. [DOI] [PubMed] [Google Scholar]
- 46.Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: Surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–14. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–8. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]