

Abstract
Background
Deregulation of c-myc plays a major role in the carcinogenesis of human malignancies. We investigated the amplification of the c-myc gene in a surgical series of Barrett cancers.
Methods
Primary resected esophageal (Barrett) adenocarcinomas (n = 84) were investigated for c-myc amplification using chromogene in situ hybridization. Tumor samples were assembled in a tissue microarray. c-myc gene dosage was correlated with clinicopathologic parameters, including the survival and gene expression of cyclo-oxygenases (COX-1 and COX-2) and proangiogenic growth factors (VEGF-A and VEGF-C).
Results
The majority (70 of 84; 83.3%) exhibited amplification of the c-myc gene. There were low-level amplifications in 63 (75.0%) cases and high-level amplifications in 7 (8.3%) cases. No amplification was found in 14(16.7%) cases. Tumors without c-myc amplification had lower VEGF-A, VEGF-C, and COX-2 expression levels than tumors with low-level and high-level c-myc amplification (statistically significant for VEGF-A; P = .0348). c-myc amplification was not correlated with clinicopathological parameters or survival. Only diffuse and mixed-type tumors, according to Lauren classification, exhibited c-myc amplifications more frequently (P = .0466).
Conclusions
Amplifications of the c-myc gene are frequent in Barrett cancer. c-myc may be involved in the regulation of angiogenesis.
Keywords: c-myc, esophageal adenocarcinoma, Barrett cancer, angiogenesis, VEGF
Introduction
Barrett cancers comprise an entity of increasing clinical importance due to a vastly unexplained rapid rise in incidence in recent decades [1,2]. Chances for cure are still limited to surgical resection at an early stage of the disease [3,4]. Histologically, these esophageal tumors are adenocarcinomas arising in the distal esophagus within the precancerous Barrett esophagus, which is defined as a metaplastic change of the normal esophageal squamous mucosa [2]. Malignant progression is driven by the chronically damaging effect of gastroesophageal reflux, which promotes a characteristic histopathologic sequence, from specialized intestinal metaplasia to low-grade and high-grade intraepithelial neoplasia to invasive adenocarcinoma [2]. Despite intensive research, knowledge about molecular mechanisms underlying Barrett cancer is still rather limited [5].
The proto-oncogene c-myc (“cellular myomatosis oncogene”) encodes a transcription factor that is regarded essential for progression in human malignancies. It is located at the chromosomal region 8q23-24 [6]. The c-myc gene has also been implicated in Barrett carcinogenesis. Amplification of the gene c-myc [7,8], as well as overexpression of the c-myc oncoprotein [9], has been demonstrated in invasive Barrett adenocarcinomas, but with great variability (Table 1). Investigations of precursor lesions (high-grade intraepithelial neoplasia, which has been previously addressed as “dysplasia”) [10] have suggested that c-myc dysregulation by amplification is an early event during Barrett carcinogenesis [7–9] because it is frequently found also in precursor lesions of esophageal adenocarcinoma, especially of high-grade intraepithelial neoplasia.
Table 1.
Frequency of c-myc Amplification/Overexpression in Barrett Adenocarcinomas Based on Available Literature.
| Publication | n | Method | Frequency of c-myc Amplification/Overexpression |
| Investigation of the amplification of the c-myc gene in the 8q23 chromosomal region | |||
| Miller et al. [8] | 87 | Southern blot analysis | 4 of 87 (4.6%) |
| Sarbia et al. [7] | 43 | Differential PCR | 17 of 39 (43.6%) |
| Arnould et al. [18] | 15* | Comparative genomic hybridization (CGH) | Gain of 8q24 in 8 of 15 cases (53.3%) |
| Bhargava et al. [19] | 28† | CGH | Gain of 8q in 79% recurrent high-level amplifications of 8q23-24.1 in > 10% |
| Walch et al. [6] | 30 | CGH | High-level amplification of 8q23-24 in 24 of 30 cases (80%) |
| Current investigation | 84 | CISH | 70 of 84 (83.3%) |
| Investigation of c-myc protein overexpression | |||
| Tselepis et al. [9] | 20 | Western blot analysis | 18 of 20 (90%) |
Adenocarcinomas of the esophagus or gastroesophageal junction. No further information is given in the paper. Thus, this paper does not deal with pure Barrett cancer collective data.
Only 11 adenocarcinomas of the distal esophagus. The remaining 17 tumors under investigation were adenocarcinomas located at a lower level of the esophagogastric junction (not Barrett cancers).
c-myc has been implicated in various cellular processes, including cell growth, proliferation, loss of differentiation, apoptosis [11,12], and regulation of angiogenesis [13]. Angiogenesis is regarded as an essential feature of malignant tumors. Only recently, we have demonstrated the importance of proangiogenic factors (VEGF-A and VEGF-C) and their regulation by the prostaglandin biosynthetic pathway (COX-1 and COX-2) in Barrett cancer [14].
It has been estimated that c-myc is associated with poor prognosis and has been, therefore, suggested as a clinically relevant marker [12]. This has been shown for c-myc expression in colorectal carcinomas [15]. Moreover, inactivation of c-myc, or downstream targets of c-myc, might provide important therapeutic targets [16].
Study Aims
We have investigated the frequency of c-myc amplification in a large series of primary resected Barrett cancers using chromogene in situ hybridization (CISH). We aimed at the following:
To clarify controversial results on the frequency of c-myc amplifications, with previous studies reporting different results in this respect [7,8], using a highly sensitive CISH method
To elucidate whether c-myc amplification is correlated with the expression of genes involved in angiogenesis, which we have investigated previously in a subset of tumors included in the current study [14]
To analyze the correlation of c-myc amplifications with clinicopathological parameters, including survival.
Materials and Methods
Patients
One hundred thirty-seven primary resected esophageal adenocarcinomas arising in association with Barrett epithelium were investigated. Due to technical reasons associated with CISH (described below), only 84 cases were eligible for the final analysis. Patient and tumor characteristics, such as age, gender, tumor size, pT/pN/pL category, Union Internationale Contre le Cancer (UICC) stage [17], and Lauren classification and grading [10], are included in Table 2.
Table 2.
Level of c-myc Amplification in the Whole Population (n = 84 Eligible Primary Resected Barrett Cancer Cases Investigated with CISH) and in Subgroups.
| n | No Amplification [n (%)] | Low-Level Amplification [n (%)] | High-Level Amplification [n (%)] | P (Chi-Square Analysis) | |
| Whole population | 84 | 14 (16.7) | 63 (75.0) | 7 (8.3) | |
| Subgroups Age (years) | |||||
| < 64 | 45 | 6 (13.3) | 34 (75.6) | 5 (11.1) | ns |
| > 64 | 39 | 8 (20.5) | 29 (74.4) | 2 (5.1) | |
| Sex | |||||
| Male | 78 | 14 (18.0) | 57 (73.1) | 7 (9.0) | ns |
| Female | 6 | 0 | 6 (100.0) | 0 | |
| Tumor size* (mm) | |||||
| < 50 | 52 | 7 (13.5) | 38 (75.0) | 6 (11.5) | ns |
| > 50 | 32 | 6 (18.8) | 25 (78.1) | 1 (3.1) | |
| Depth of invasion (pT category) | |||||
| pT1/2 | 47 | 8 (9.5) | 34 (40.5) | 5 (6.0) | ns |
| pT3/4 | 37 | 6 (7.1) | 29 (34.5) | 2 (2.4) | |
| Lymph node involvement (pN category) | |||||
| pN0 | 40 | 8 (20.0) | 28 (70.0) | 4 (10.0) | ns |
| pN1 | 44 | 6 (13.6) | 35 (79.6) | 3 (6.8) | |
| Lymphatic vessel invasion (L category) | |||||
| L0 | 59 | 11 (18.6) | 45 (76.3) | 3(5.1) | ns |
| L1 | 25 | 3 (12.0) | 18 (72.0) | 4 (16.0) | |
| UICC stage | |||||
| I/II | 55 | 9 (16.4) | 40 (72.7) | 6 (10.9) | ns |
| III/IV | 29 | 5 (17.2) | 23 (79.3) | 1 (3.5) | |
| Differentiation (grade) | |||||
| G1/2 | 41 | 6 (14.6) | 33 (80.5) | 2 (4.9) | ns |
| G3/4 | 43 | 8 (18.6) | 30 (69.8) | 5 (11.6) | |
| Lauren classification | |||||
| Diffuse | 2 | 0 | 1 (50.0) | 1 (50.0) | .0466 |
| Intestinal | 74 | 14 (18.9) | 56 (75.7) | 4 (5.4) | |
| Mixed | 8 | 0 | 6 (75.0) | 2 (25.0) | |
Size range: 5 to 130 mm.
All patients had undergone primary surgical resection (radical transthoracic or transhiatal esophagectomy with lymphadenectomy) at the Technical University of Munich between 1991 and 2003. None of these patients had received prior antineoplastic therapy (neither chemotherapy nor radiochemotherapy). Patients' approval was secured according to local arrangements by the ethics committee. This study was performed with patients' consent allowing molecular research to be performed on specimen obtained during surgical resection.
Tissue Array
The tumor tissues of the 137 esophageal adenocarcinomas under analysis were assembled in tissue microarrays. Core needle biopsies were retrieved from original tumor blocks using a manual arrayer (Beecher Instruments, Sun Prairie, WI) and positioned in a recipient paraffin array block. Viable representative areas of tumor specimens were marked by an experienced pathologist (M.S.). At least three tissue cylinders with a diameter of 0.6 mm were obtained from each tumor block.
CISH
Four- to 5-µm-thick sections were cut from microarray paraffin blocks and dewaxed with xylene, and then rehydrated with 100% ethanol and water. Target retrieval and enzyme digestion were achieved using a commercially available tissue pretreatment kit (no. 00-8401; from Zymed Laboratories, distributed through Zytomed, Berlin, Germany). The sections were dehydrated in upgrading ethanol series and air-dried. Fifteen microliters of digoxigenin-labeled c-myc probe (84-1700; from Zymed Laboratories, distributed through Zytomed) was applied on the microarray section, enclosed by a coverslip, sealed, and codenatured on a hot plate (Hybrite; from Vysis, distributed through Abbott, Wiesbaden, Germany) for 5 minutes at 94°C. Hybridization was performed overnight at 37°C. Then, the coverslip was removed by soaking in standard saline citrate (SSC) solution at room temperature and washed in SSC for 5 minutes at 75°C. The remaining hybridized probe linked to digoxigenin was detected by mouse antidigoxigenin antibody followed by polymerized horseradish peroxidase-goat antimouse immunoglobulin. Peroxidase was developed with diaminobenzidine, and nuclei were counterstained with hematoxylin. All detection reagents were provided in commercially available kits (Spotlight CISH Polymer Detection Kit, 84-9246; from Zymed Laboratories, distributed through Zytomed).
The interpretation of CISH results was performed by a senior pathologist (M.S.) with a light microscope using a x40 objective (original magnification, x400; Figure 1, A and B). Signals were seen as nuclear dark brown dots. Fifty to 100 nonoverlapping tumor cell nuclei were evaluated per sample. A gene copy number of one to five copies per nucleus was scored as “no amplification” (Figure 1A). A gene copy number of 6 to 10 copies per nucleus in at least 50% of cancer cells was considered “low-level amplification.” A gene copy number of more than 10 copies per nucleus or the presence of clusters in at least 50% of cancer cells was considered as “high-level amplification” (Figure 1B) [18,19]. Non-neoplastic cells in tissues were always evaluated as intest quality controls.
Figure 1.
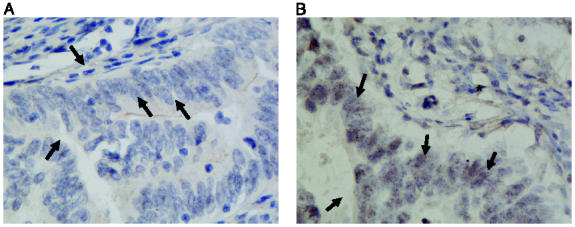
(A) Cells of a moderately differentiated adenocarcinoma (center and bottom) with a maximum of five nuclear brown dots (arrows indicate nuclei) indicating no evidence of c-myc gene amplification (original magnification, x600). (B) In contrast, another moderately differentiated adenocarcinoma (bottom left) with more than 10 dots per nucleus (arrows indicate nuclei) indicating high-level amplification of the c-myc gene (original magnification, x600). Adjacent stromal cells show no evidence of c-myc gene amplification (asterisk).
Due to various reasons (i.e., loss of tissue cores during hybridization procedures, lack of hybridization, or lack of viable tumor tissues in tissue cores), only 84 of the initial 137 esophageal adenocarcinoma cases could finally be assessed for c-myc gene dosage.
Results
c-myc gene amplification was assessed using tissue arrays that included 137 esophageal adenocarcinomas. Due to various technical reasons (i.e., loss of tissue cores during hybridization procedures, lack of hybridization, or lack of viable tumor tissue in tissue cores), only 84 of the initial 137 esophageal adenocarcinoma cases could be finally assessed for c-myc gene dosage. The majority of cases (70 of 84; 83.3%) exhibited amplification of the c-myc gene. These were low-level amplifications in 63 (75.0%) cases and highlevel amplifications in 7 (8.3%) cases. No amplification was found in 14 (16.7%) cases (Table 2).
Subgroup analysis according to clinicopathological parameters (Table 2) did not show any statistically significant differences with respect to c-myc amplification (patient age, sex, tumor size, pT/pN category, lymphatic vessel invasion, UICC stage, and tumor grade), except for Lauren classification (P = .0466). Thus, none of the diffuse or mixed-type tumors according to Lauren classification lacked c-myc amplifications. Six (75.0%) of eight mixed-type tumors under investigation and one (50.0%) of two diffuse-type tumors exhibited low-level amplifications of the c-myc gene. Furthermore, two (25.0%) of eight mixed-type tumors and one (50.0%) of two diffuse-type tumors showed high-level amplifications of c-myc.
Survival analysis according to the univariate Kaplan-Meier method did not show any significant impact of c-myc amplification on patients' survival (Figure 2).
Figure 2.

c-myc amplifications did not significantly affect outcome after the resection of Barrett cancer (univariate survival analysis according to the Kaplan-Meier method).
In a subset of tumors under analysis (n = 51), data on the mRNA expression of four genes [i.e. cyclooxygenases (COX-1 and COX-2) and proangiogenetic growth factors (VEGF-A and VEGF-C), which were determined in a previous investigation] [14] were available. Correlation analysis showed that tumors without c-myc amplification tended to have lower COX-2, VEGF-A, and VEGF-C mRNA expression levels than tumors with low-level amplification, which had lower expression levels compared to cases with high-level amplification (Table 3). This trend toward increased gene expression with increasing level of c-myc amplification was statistically significant for VEGF-A (P = .0466; chi-square analysis).
Table 3.
Correlation of c-myc Amplification Data with Relative mRNA Expression Levels of COX-1, COX-2, VEGF-A, and VEGF-C, as Determined by Quantitative Reverse Transcription PCR (TaqMan).
| n* | No Amplification | Low-Level Amplification | High-Level Amplification | P (Two-Sided Amplification Jonckheere-Terpstra Test) | |
| COX-1 | |||||
| Mean | 51 | 12.18 | 13.65 | 9.36 | ns |
| SD | 16.03 | 23.48 | 4.73 | ||
| Median | 8.13 | 3.85 | 9.51 | ||
| Min | 0.05 | 0.01 | 3.57 | ||
| Max | 49.47 | 108.11 | 15.57 | ||
| COX-2 | |||||
| Mean | 51 | 12.98 | 24.36 | 27.03 | ns |
| SD | 13.96 | 42.87 | 19.77 | ||
| Median | 6.22 | 10.14 | 16.15 | ||
| Min | 0.82 | 0.05 | 7.37 | ||
| Max | 39.25 | 259.63 | 59.32 | ||
| VEGF-A | |||||
| Mean | 51 | 3.22 | 7.93 | 9.841 | .0348 |
| SD | 3.29 | 15.92 | 7.92 | ||
| Median | 2.88 | 3.71 | 7.78 | ||
| Min | 0.08 | 0.08 | 1.88 | ||
| Max | 11.99 | 104.57 | 22.80 | ||
| VEGF-C | |||||
| Mean | 51 | 0.06 | 0.08 | 0.15 | ns |
| SD | 0.08 | 0.17 | 0.11 | ||
| Median | 0.01 | 0.00 | 0.14 | ||
| Min | 0.00 | 0.00 | 0.01 | ||
| Max | 0.22 | 1.14 | 0.36 | ||
The number of cases varies according to the eligibility of available information (CISH and gene expression analysis with TaqMan).
Discussion
According to our current investigation, amplification of the c-myc gene is frequent in esophageal adenocarcinomas. However, the majority of cases showed low-level amplifications (75% of the cases), whereas only a minority of the tumors (7.3%) showed high-level amplifications. Only at first glance do these findings contradict existing data in the literature (Table 1). Thus, Miller et al. [8], investigating a series of similar size (87 Barrett cancers) for c-myc amplification, found amplification of c-myc in only 4 (4.6%) cases. However, this discrepancy can be well explained by the fact that Miller et al. used Southern blot analysis, which has a considerably lower sensitivity than CISH. In contrast, comparative gene expression data have also shown that genetic gains are frequent at 8q.23-24, the chromosomal region where c-myc is localized (Table 1). Walch et al. [6] demonstrated high-level amplification of the region in 80% (24 of 30) Barrett cancer cases. Moskaluk et al. [20] found gains of 8q24 in 53.3% (8 of 15) cases, whereas van Dekken et al. [21] found gains of 8q in 79%. However, the results of the latter two studies have to be interpreted with caution because the tumors under investigation were not exclusively Barrett cancers (see Table 1 and footnotes). Adenocarcinomas of the esophagogastric junction, which have been included in these studies, are known to share more similarities with gastric cancers, which are known to exhibit c-myc amplification quite frequently.
A polymerase chain reaction (PCR)-based investigation found c-myc amplifications in 43.6% (17 of 39 Barrett cancer cases under investigation) [7]. Although these results are more similar to the findings of our current investigation, this PCR approach is, again, inferior to CISH. A third study, in which overexpression of the c-myc oncoprotein was studied by Western blot analysis, revealed a high level of tumors overexpressing c-myc (18 of 20 cases; 90%). All these data suggest that c-myc is an important molecular feature of invasive Barrett cancers, and especially low-level amplifications can frequently be found when a sensitive technique is applied.
c-myc amplifications have previously been shown to be less frequent or absent in precursor lesions (non-neoplastic Barrett esophagus and high-grade intraepithelial neoplasia) than in invasive Barrett cancer [7,8]. This might explain the lack of correlation of our results (obtained from invasive Barrett cancer specimens) with clinicopathological parameters such as pT/pN category, UICC stage (Table 2), and survival (Figure 2). Furthermore, this supports the concept that c-myc amplification is an early event during Barrett carcinogenesis, as indicated by previously published investigations [6–8].
The association of c-myc amplification with VEGF-A expression in the current investigation is well in accordance with the current understanding of the function of c-myc. Apart from various other effects by which c-myc contributes to carcinogenesis (i.e., promotion of cell growth, proliferation, loss of differentiation, and apoptosis) [12], the gene is also suspected to be involved in the regulation of angiogenesis [12,13]. c-myc deficiency is a lethal condition that has been shown to be due to the associated profound defects of vasculogenesis [13]. Embryos of c-myc-/- mice lack virtually the whole the vasculature. These defects can partially be addressed by the substitution of VEGF, suggesting that c-myc regulates angiogenesis through VEGF. This result has been substantiated by further studies indicating that Myc activation is a sufficient trigger to increase VEGF expression [12]. Our results in Barrett cancer (significant correlation of gene expression level with level of c-myc amplification) also support this link between c-myc and angiogenesis through the proangiogenic growth factor VEGF-A.
Due to a variety of implications in carcinogenesis, c-myc has been suggested as a promising target for molecular therapies [16]. Although these approaches are still in their infancy, promising strategies, including the application of antisense oligodeoxynucleotides, have been suggested to reduce c-myc expression. One other approach, specifically referring to the pathophysiology of Barrett carcinogenesis, relates to the reported modulation of c-myc expression by the contents of gastroesophageal refluxate. In vitro (cell culture) and in vivo (reflux models in animals) experiments have shown that bile acids activate c-myc [22]. Therefore, it was postulated that preventing esophageal epithelium from coming into contact with bile acids is one strategy against Barrett cancer [23]. Prevention of c-myc activation must be considered as one major aspect of such a strategy.
Conclusion
Our results of frequent (low-level and high-level) c-myc gene amplifications in a majority of Barrett cancers support the previously suggested importance of c-myc in this entity, c-myc amplification is likely to be an early event during Barrett carcinogenesis. Furthermore, our data suggest that an effect of the targeting of this gene is, at least in part, antiangiogenic in nature.
Acknowledgements
We thank Angelika Jahn for expert technical assistance in laboratory work, Reinhart Willers (University Düsseldorf) for statistical analyses, and Susanne Mach-Booms for maintaining our Barrett database.
Abbreviations
- SSC
standard saline citratae
Footnotes
This work was supported by Deutsche Krebshilfe (grant no. 70-2789-Si3).
References
- 1.Spechler SJ. Clinical practice. Barrett's esophagus. N Engl J Med. 2002;346:836–842. doi: 10.1056/NEJMcp012118. [DOI] [PubMed] [Google Scholar]
- 2.Devesa SS, Blot WJ, Fraumeni JF. Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer. 1998;83:2049–2053. [PubMed] [Google Scholar]
- 3.Siewert JR, Stein HJ, Feith M, Brücher BLDM, Bartels H, Fink U. Tumor cell type is an independent prognostic parameter in esophageal cancer: lessons learned from more than 1000 consecutive resections at a single institution in the Western world. Ann Surg. 2001;234:360–369. doi: 10.1097/00000658-200109000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein HJ, von Rahden BHA, Siewert JR. Survival after surgery of cancer of the esophagus. Langenbecks Arch Surg. 2004;390:280–285. doi: 10.1007/s00423-004-0504-9. [DOI] [PubMed] [Google Scholar]
- 5.Morales CP, Souza RF, Spechler SJ. Hallmarks of cancer progression in Barrett's oesophagus. Lancet. 2002;360:1587–1589. doi: 10.1016/S0140-6736(02)11569-8. [DOI] [PubMed] [Google Scholar]
- 6.Walch AK, Zitzelsberger HF, Bruch J, Keller G, Angermeier D, Aubele MM, Mueller J, Stein H, Braselmann H, Siewert JR, et al. Chromosomal imbalances in Barrett's adenocarcinoma and the metaplasiadysplasia-carcinoma sequence. Am J Pathol. 2000;156:555–566. doi: 10.1016/S0002-9440(10)64760-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarbia M, Arjumand J, Wolter M, Reifenberger G, Heep H, Gabbert HE. Frequent c-myc amplification in high-grade dysplasia and adenocarcinoma in Barrett esophagus. Am J Clin Pathol. 2001;115:835–840. doi: 10.1309/MXXH-25N3-UAL2-G7XX. [DOI] [PubMed] [Google Scholar]
- 8.Miller CT, Moy JR, Lin L, Schipper M, Normolle D, Brenner DE, Iannettoni MD, Orringer MB, Beer DG. Gene amplification in esophageal adenocarcinomas and Barrett's with high-grade dysplasia. Clin Cancer Res. 2003;9:4819–4825. [PubMed] [Google Scholar]
- 9.Tselepis C, Morris CD, Wakelin D, Hardy R, Perry I, Luong QT, Harper E, Harrison R, Attwood SE, Jankowski JA. Upregulation of the oncogene c-myc in Barrett's adenocarcinoma: induction of c-myc by acidified bile acid in vitro. Gut. 2003;52:174–180. doi: 10.1136/gut.52.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamilton SR. In: Pathology and Genetics of Tumors of the Digestive System. Aaltonen LA, editor. Tumors of Small Intestine, IARC Press: Lyon; 2000. pp. 69–91. [Google Scholar]
- 11.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 12.Pelengaris S, Khan M, Evan GI. Suppression of Mycinduced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109:321–334. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 13.Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16:2530–2543. doi: 10.1101/gad.1024602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Rahden BHA, Stein HJ, Pühringer F, Langer R, Koch I, Siewert JR, Höfler H, Sarbia M. Coexpression of cyclooxygenases (COX-1, COX-2) and vascular endothelial growth factors (VEGF-A, VEGF-C) in esophageal adenocarcinoma. Cancer Res. 2005;65:5038–5044. doi: 10.1158/0008-5472.CAN-04-1107. [DOI] [PubMed] [Google Scholar]
- 15.Smith DR, Goh HS. Overexpression of the c-myc protooncogene in colorectal carcinoma is associated with a reduced mortality that is abrogated by point mutation of the p53 tumor suppressor gene. Clin Cancer Res. 1996;2:1049–1053. [PubMed] [Google Scholar]
- 16.Pelengaris S, Khan M. The c-MYC oncoprotein as a treatment target in cancer and other disorders of cell growth. Expert Opin Ther Targets. 2003;7:623–642. doi: 10.1517/14728222.7.5.623. [DOI] [PubMed] [Google Scholar]
- 17.Sobin LH, Wittekind Ch, editors. TNM Classification of Malignant Tumors. 6th ed. New York: Wiley-Liss; 2002. [Google Scholar]
- 18.Arnould L, Denoux Y, MacGrogan G, Penault-Llorca F, Fiche M, Treilleux I, Mathieu MC, Vincent-Salomon A, Vilain MO, Couturier J. Agreement between chromogenic in situ hybridisation (CISH) and FISH in the determination of HER2 status in breast cancer. Br J Cancer. 2003;88:1587–1591. doi: 10.1038/sj.bjc.6600943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhargava R, Oppenheimer O, Gerald W, Jhanwar SC, Chen B. Identification of MYCN gene amplification in neuroblastoma using chromogenic in situ hybridization (CISH): an alternative and practical method. Diagn Mol Pathol. 2005;14:72–76. doi: 10.1097/01.pas.0000149878.78117.ff. [DOI] [PubMed] [Google Scholar]
- 20.Moskaluk CA, Hu J, Perlman EJ. Comparative genomic hybridization of esophageal and gastroesophageal adenocarcinomas shows consensus areas of DNA gain and loss. Genes Chromosomes Cancer. 1998;22:305–311. [PubMed] [Google Scholar]
- 21.van Dekken H, Geelen E, Dinjens WN, Wijnhoven BP, Tilanus HW, Tanke HJ, Rosenberg C. Comparative genomic hybridization of cancer of the gastroesophageal junction: deletion of 14Q31-32.1 discriminates between esophageal (Barrett's) and gastric cardia adenocarcinomas. Cancer Res. 1999;59:748–752. [PubMed] [Google Scholar]
- 22.Jenkins GJ, Harries K, Doak SH, Wilmes A, Griffiths AP, Baxter JN, Parry JM. The bile acid deoxycholic acid (DCA) at neutral pH activates NF-kappaB and induces IL-8 expression in oesophageal cells in vitro. Carcinogenesis. 2004;25:317–323. doi: 10.1093/carcin/bgh032. [DOI] [PubMed] [Google Scholar]
- 23.Stamp DH. Bile acids aided by acid suppression therapy may be associated with the development of esophageal cancers in westernized societies. Med Hypotheses. 2006;66:154–157. doi: 10.1016/j.mehy.2005.04.045. [DOI] [PubMed] [Google Scholar]