

Abstract
The synthesis and molecular structures of three iron(II) porphyrinates with only CO as the axial ligand(s) are reported. Two five-coordinate [Fe(OEP)(CO)] derivatives have Fe–C = 1.7077 (13) and 1.7140 (10) Å, much shorter than those of six-coordinate [Fe(OEP)(Im)(CO)], although νC-O is 1944–1948 cm−1. The six-coordinate species [Fe(OEP)(CO)2] has also been studied. The competition for π-back-bonding of two CO ligands leads to Fe–C distances of 1.8558 (10) Å and νC-O is increased to 2021 cm−1. The Mössbauer spectrum has a quadrupole splitting constant of 0 mm/s at 4.2 K, indicating high electronic symmetry.
Many heme-based sensing proteins that use/detect the diatomic ligands O2, CO, or NO are known.1–4 These sensing proteins rely on variations in coordination number and the character of their axial ligands to induce conformational changes leading to protein activation.1 Effects involving the trans ligand (or lack thereof) on binding diatomic molecules are important in elucidating the mechanism of small-molecule-sensing proteins. In order to achieve a better understanding of the heme interactions involved with diatomic ligand sensing as well as broadly extending our understanding of their electronic and molecular structure, we have been investigating heme/diatomic ligand interactions.
We report for the first time the solid-state syntheses and structural characterization of unambiguously five-coordinate and six-coordinate heme CO complexes: [Fe(OEP)(CO)], [Fe(OEP)(CO)].C6H6, and [Fe(OEP)(CO)2].5,6 Although these species have been previously reported in solution and CO binding constants determined for three different porphyrins,7 we now demonstrate the structural effects of changing coordination environments. We compare the five-coordinate structures with those of several other diatomic ligand heme complexes, examine effects of the addition of a sixth ligand and note structural differences.
These adducts may elucidate important coordination chemistry features involving ligand loss, ligand switching, and ligand photolability. CO photolysis of six-coordinate heme carbonyl derivatives has been a mainstay in biophysical investigations,8 and is very efficient with quantum yields nearing unity. The suggestion that five-coordinate heme carbonyls are less photolabile than their six-coordinate counterparts9 generates important questions about structural differences.
We first consider the vibrational properties of five-coordinate [Fe(OEP)(CO)]. νC-O in unperturbed imidazole-ligated hemes is typically near 1970 cm−1, but solid-state environmental factors can lead to variation in νC-O in the range of 1926 to 2000 cm−1.10,11 Thus, it is perhaps surprising to find that νC-O in [Fe(OEP)(CO)] and [Fe(OEP)(CO)].C6H6 is well within this range at 1944 cm−1 and 1948 cm−1, respectively (Nujol mull). These values might suggest that the iron center is involved in a significant intermolecular interaction to form a pseudo six-coordinate complex, but this is not correct (vide infra).
The molecular structures of the two crystalline [Fe(OEP)(CO)] complexes are illustrated in Figure 1 (and Fig. S1). There are strong similarities as well as differences in their structures: the iron out-of-plane displacements are similar at 0.20 Å or 0.22 Å, the off-axis tilt of the Fe–C vector is 3.8° or 2.4°, and the Fe–C–O bond angle is 177.20(8)° in both. Both forms show some ring–ring interactions although these lead to some strong differences as well. Figure 2a shows the pairwise interactions in [Fe(OEP)(CO)], while the equivalent packing in the benzene solvate is shown in Figure 2b. In both cases, there is inversion symmetry; metrical information is given in the figure caption. The inter-ring packing pattern in the unsolvated form of [Fe(OEP)(CO)] (Figure 2a) is similar to that of both crystalline forms of [Fe(OEP)(NO)] for which little por-phyrin overlap is observed.12 The closest intermolecular contact to the iron in [Fe(OEP)(CO)] is 3.12 Å whereas in [Fe(OEP)(CO)].C6H6 the closest contact to iron is 3.58 Å. In the two [Fe(OEP)(NO)]+ structures (isoelectronic to the CO's), however, there is a large porphyrin ring overlap with the π-system of an adjacent molecule acting as a pseudo sixth ligand.13 The tighter interactions in [Fe(OEP)(CO)].C6H6 (Figure 2b) are comparable although slightly larger than in the isoelectronic [Fe(OEP)(NO)]+ complexes. The porphyrin core conformation in [Fe(OEP)(CO)] is nearly planar whereas that in [Fe(OEP)(CO)].C6H6 displays modest core ruffling. The average equatorial Fe–Np distances are 1.988 (2) Å and 1.984(3) Å for the unsolvated and solvated forms. The very short Fe–Np bond distances reflect the strong bonding interaction and low-spin state of iron. Significantly, the axial ligand-induced equatorial (Fe–Np) bond distance differences observed in five-coordinate [Fe(por)(NO)] derivatives12 are not observed; individual Fe–Np distances are tightly clustered.
Figure 1.
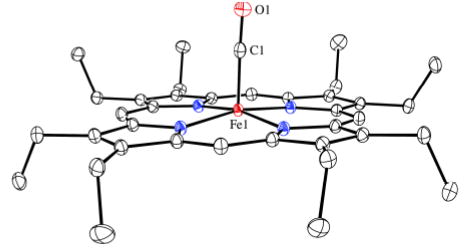
Thermal ellipsoid plot of [Fe(OEP)(CO)] (50% probability ellipsoids). Hydrogen atoms are omitted for clarity.
Figure 2.
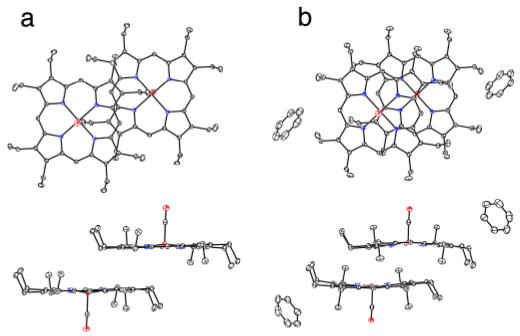
ORTEP plots (50% probability ellipsoids) of [Fe(OEP)(CO)] (a) and [Fe(OEP)(CO)].C6H6 (b) displaying the pairwise interactions. In (a) the 24-atom mean plane separation is 3.42 Å and the lateral shift of the two ring centers is 6.76 Å. The Fe…Fe distance is 7.58 Å. The corresponding distances in (b) are 3.46, 3.88, and 5.20 Å, respectively.
The short axial Fe–C distance of 1.7140(11) Å in [Fe(OEP)(CO)] is shorter than those of the six-coordinate imidazole adducts by ~0.03–0.06 Å.10,14 This might suggest stronger Fe→C π-back-donation, but the relatively normal νC-O value suggests that the Fe π-donation into the CO π* orbitals is similar to that of the six-coordinate species. This would then suggest that it is the σ-bonding component that leads to the shorter Fe–C bond distance. The distance in [Fe(OEP)(CO)].C6H6 is 1.7077 (13) Å; the possibly slightly shorter Fe–C distance is not consistent with the 4 cm−1 increase in νC-O, again suggesting the importance of σ-bonding. We thus conclude that differences in the σ-bonding component of Fe–C have no or modest effects on νC-O. As we have noted for a different series of six-coordinate carbonyl derivatives,10 there is a strong correlation between νC-O and the Fe–C/C–O bond lengths, this question is being examined in more detail for OEP derivatives.
The structures of other [Fe(OEP)(XY)]+, 0 adducts (XY = NO or CS) are available for comparison. Structural parameters for five- and six-coordinate species are listed in Table 1. The iron out-of-plane displacement of [Fe(OEP)-(CO)] is seen to be at the low end of the range. In all cases, the addition of a sixth ligand leads to a decreased iron atom displacement and in most cases to an increase in the length of the Fe–X(XY) bond. Although this increase is never large, the COs are the system that experiences the largest increase in Fe–C bond distance (from 1.7140(11) Å to 1.7733(12) Å in [Fe(OEP)(CO)(1-MeIm)].14 Interestingly, the Fe–C bond would appear to have changed minimally when the trans ligand is the weakly coordinating tetrahydrofuran ligand in [Fe(Deut)(CO)(THF)]. Unfortunately, the relatively low precision of this structure (from ~25 years ago) is not adequate to comment on further, but clearly merits further attention.
Table 1.
Notable bonding parameters for [Fe(OEP)(CO)] and related compounds.
| Complex | Fe–XYa | X–Ya | Fe–X–Yb | Fe–Npa | Fe–Laxa | ΔFed | νX–Yc | ref |
|---|---|---|---|---|---|---|---|---|
| [Fe(OEP)(CO)] | 1.7140(11) | 1.1463(12) | 177.20(8) | 1.988(2) | – | 0.20 | 1944e | tw |
| [Fe(OEP)(CO)].C6H6 | 1.7077(13) | 1.1259(16) | 177.20(11) | 1.984(3) | – | 0.22 | 1948e | tw |
| [Fe(OEP)(NO)] | 1.722(2) | 1.167(3) | 144.4(2) | 2.004(15) | – | 0.29 | 1666e | 12 |
| [Fe(OEP)(NO)] | 1.7307(7) | 1.1677(11) | 142.74(8) | 2.010(13) | – | 0.27 | 1673e | 12 |
| [Fe(OEP)(NO)]+ | 1.6528(13) | 1.140(2) | 173.19(13) | 1.994(5) | – | 0.32 | 1838e | 13 |
| [Fe(OEP)(NO)]+ | 1.644(3) | 1.112(4) | 176.9(3) | 1.994(1) | – | 0.29 | 1868e | 16 |
| [Fe(OEP)(CS)] | 1.662(3) | 1.559(3) | 176.3(2) | 1.982(5) | – | 0.23 | 1292e | 17 |
| [Fe(Deut)(CO)(THF)] | 1.706(5) | 1.144(5) | 178.3(14) | 1.98(3) | 2.127(4) | 0.10 | 1955f | 18 |
| [Fe(OEP)(CO)(1-MeIm)] | 1.744(5) | 1.158(5) | 175.1(4) | 2.000(3) | 2.077(3) | 0.00 | 1965g | 19 |
| [Fe(OEP)(CO)(1-MeIm)] | 1.7733(12) | 1.1413(15) | 175.67(11) | 2.010(4) | 2.0544(9) | 0.02 | 1980e | 20 |
| [Fe(OEP)(NO)(1-MeIm)]+ | 1.6465(17) | 1.135(2) | 177.28(17) | 2.003(5) | 1.9889(16) | 0.02 | 1921e | 21 |
| [Fe(TPP)(NO)(1-MeIm)] | 1.750(2) | 1.182(3) | 137.7(2) | 2.008(13) | 2.173(2) | 0.04 | 1628e | 22 |
| [Fe(OEP)(CS)(1-MeIm)] | 1.703(4) | 1.563(4) | 172.2(2) | 2.001(4) | 2.112(3) | 0.10 | 1272e | 23 |
| [Fe(OEP)(CO)2] | 1.8558(10) | 1.1216(13) | 173.95(9) | 2.0133(7) | 1.558(10) | 0.00 | 2021e | tw |
Å
degrees.
cm−1
Displacement from 24-atom meanplane.
Nujol mull.
THF.
CD2Cl2 solution.
Although solutions of Fe(OEP) and CO will always be mixtures of mono- and bis-CO species, crystallization experiments (see SI) at low temperature reproducibly afford crystals of [Fe(OEP)(CO)2]. Analysis of crystals of [Fe(OEP)(CO)2] gave the structure displayed in Figure 3; the complex has a required inversion center. As might be expected, the competition from two COs for π-backbonding from the central iron leads to increased Fe–C bond distances of 1.8558 (10) Å, which is also consistent with the observed asymmetric stretch of 2021 cm−1. The CO ligands are tilted off-axis as shown with other metrical information given in Figure 3. Perhaps as a consequence of the competition for bonding with two CO ligands, the equatorial Fe–Np bonds are at the very long end of values expected for low-spin iron(II).15 The porphyrin core is planar; core diagrams for all three complexes are given in Figure S2.
Figure 3.
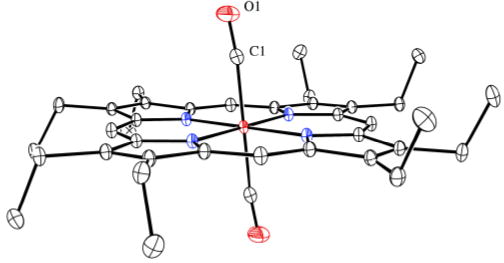
Thermal ellipsoid plot of [Fe(OEP)(CO)2]. The Fe–C vector is tilted from the heme normal by 5.9°; the C and O atoms are shifted laterally by 0.14 and 0.56 Å, respectively.
Mössbauer spectra for [Fe(OEP)(CO)] are consistent with an isolated five-coordinate iron center. The quadrupole splitting is much larger (1.84 mm/s, 4.2 K) than that observed for six-coordinate carbonyls (typically less than 0.7 mm/s), strongly indicative of a d-orbital asymmetry consistent with five-coordination. The isomer shift value of 0.27 mm/s is similar to that of the six-coordinate derivatives; the relatively low value is consistent with strong and effectively equivalent covalency among the occupied Fe 3d orbitals in all derivatives. The six-coordinate derivative is much more symmetric with a quadrupole splitting of 0 or near 0 at 4.2 K and isomer shift of 0.31 mm/s, this increases to a QS of 0.176 mm/s and an isomer shift of 0.18 mm/s at 298 K. Spectra in applied magnetic field for this complex confirmed that it is a diamagnetic species. Complete data are in Table S1.
Five-coordinate coordinate carbonyl hemes have been structurally characterized and compared with related five- and six-coordinated diatomic complexes. Although the biological importance of five-coordinate carbonyl hemes is not known, proteins have been described that have spectroscopic properties that may be associated with this adduct.9,24 Additional spectroscopic and photophysical studies on these carbonyl complexes are in prospect.
Supplementary Material



Acknowledgments
We thank the NIH for support (GM-38401, W.R.S.) and the NSF for X-ray instrumentation (CHE-0443233). We thank Prof. Timothy Sage for useful discussions.
Footnotes
Supporting Information Available: Syntheses, figures and crystallographic data are available (including CIF files). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gilles-Gonzalez MA, Gonzalez G. J Inorg Biochem. 2005;99:1. doi: 10.1016/j.jinorgbio.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Rodgers KR, Lukat-Rodgers GS. J Inorg Biochem. 2005;99:963. doi: 10.1016/j.jinorgbio.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 3.Roberts GP, Kerby RL, Youn H, Conrad M. J Inorg Biochem. 2005;99:280. doi: 10.1016/j.jinorgbio.2004.10.032. (a) [DOI] [PubMed] [Google Scholar]; Aono S, Nakajima H. Coord Chem Rev. 1999;190–192:267. (b) [Google Scholar]
- 4.Boon EM, Marletta MA. J Inorg Biochem. 2005;99:892. doi: 10.1016/j.jinorgbio.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 5.The following abbreviations are used in this paper: Por, generalized porphyrin dianion; OEP, dianion of octaethylporphyrin; Deut, dianion of deuteroporphyrin; TPP, dianion of tetraphenylporphyrin.
- 6.Complete synthetic procedures are given in the Supplemental Information.
- 7.Rougee M, Brault D. Biochemistry. 1975;73:4100. (a) [Google Scholar]; Wayland BB, Mehne LF, Swartz J. J Am Chem Soc. 1978;100:2379. (b) [Google Scholar]; Strauss SH, Holm RH. Inorg Chem. 1982;21:863. (c) [Google Scholar]
- 8.Sage JT, Champion PM. Small Substrate Recognition in Heme Proteins. In: Suslick KS, editor. Comprehensive Supramolecular ChemistryPergamon. Vol. 5. Oxford; UK: 1996. pp. 171–217. [Google Scholar]
- 9.Pal B, Li Z, Ohta T, Takenaka S, Tsuyama S, Kitagawa T. J Inorg Biochem. 2004;98:824. doi: 10.1016/j.jinorgbio.2003.12.007. (a) [DOI] [PubMed] [Google Scholar]; Makino R, Obayashi E, Homma N, Shiro Y, Hori H. J Biol Chem. 2003;278:11130. doi: 10.1074/jbc.M209026200. (b) [DOI] [PubMed] [Google Scholar]
- 10.Silvernail NJ, Roth A, Schulz CE, Noll BC, Scheidt WR. J Am Chem Soc. 2005;127:14422. doi: 10.1021/ja053148x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim K, Ibers JA. J Am Chem Soc. 1991;113:6077. [Google Scholar]
- 12.Ellison MK, Scheidt WR. J Am Chem Soc. 1997;119:7404. (a) [Google Scholar]; Scheidt WR, Duval HF, Neal TJ, Ellison MK. J Am Chem Soc. 2000;122:4651. (b) [Google Scholar]
- 13.Ellison MK, Schulz CE, Scheidt WR. Inorg Chem. 2000;39:5102. doi: 10.1021/ic000789e. [DOI] [PubMed] [Google Scholar]
- 14.Silvernail, N. J.; Noll, B.C.; Scheidt, W. R. unpublished results.
- 15.Scheidt WR. Systematics of the Stereochemistry of Porphyrins and Metalloporphyrins. In: Kadish KM, Smith K, Guilard R, editors. The Porphyrin Handbook . Academic Press; San Diego, CA and Burlington, MA: 2000. p. 3. Chapter 16. [Google Scholar]
- 16.Scheidt WR, Lee YJ, Hatano K. J Am Chem Soc. 1984;106:3191. [Google Scholar]
- 17.Scheidt WR, Geiger DK. Inorg Chem. 1982;21:2056. [Google Scholar]
- 18.Scheidt WR, Haller KJ, Fons M, Mashiko T, Reed CA. Biochemistry. 1981;20:3653. doi: 10.1021/bi00515a054. [DOI] [PubMed] [Google Scholar]
- 19.Salzmann R, McMahon MT, Godbout N, Sanders LK, Wojdelski M, Oldfield E. J Am Chem Soc. 1999;121:3818. [Google Scholar]
- 20.Silvernail, N. J.; Noll, B.C.; Scheidt, W. R. unpublished results.
- 21.Ellison MK, Scheidt WR. J Am Chem Soc. 2001;123:5210. doi: 10.1021/ja010276m. [DOI] [PubMed] [Google Scholar]
- 22.Wyllie GRA, Schulz CE, Scheidt WR. Inorg Chem. 2003;42:5722. doi: 10.1021/ic034473t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao C, Dahal S, Shang M, Beatty AM, Hibbs W, Schulz CE, Scheidt WR. Inorg Chem. 2003;42:5202. doi: 10.1021/ic030043r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu GC, Katakura K, Tomita T, Zhang X, Sun D, Sato M, Sasahara M, Kayama T, Ikeda-Saito M, Yoshida T. J Biol Chem. 2000;275:17494. doi: 10.1074/jbc.M000830200. (a) [DOI] [PubMed] [Google Scholar]; Vogel KM, Spiro TG, Shelver D, Thorsteinsson MV, Roberts GP. Biochemistry. 1999;38:2679. doi: 10.1021/bi982375r. (b) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.