

Abstract
EGF and Ras mitogenic signal transduction pathways are frequently activated in breast carcinoma and inhibit mammary differentiation and apoptosis. HC11 mouse mammary epithelial cells, which differentiate and synthesize β-casein following growth to confluency and stimulation with lactogenic hormones, were used to study EGF-dependent signaling during differentiation. Blocking Mek-Erk or phosphotidylinositol-3-kinase (PI-3 kinase) signaling with specific chemical inhibitors enhanced β-casein promotor-driven luciferase activity. Because EGF stimulation of HC11 cells resulted in the activation of Ras, the effect of activated Ras (RasV12) or dominant negative (DNRasN17) on lactogen induced differentiation was examined. HC11 cell lines expressing RasV12 or DNRasN17 under the control of a tetracycline(tet)-responsive promotor were constructed. Activated RasV12 expression resulted in reduced tyrosine phosphorylation of Stat5 and a delay in β-casein expression in response to prolactin. However, the expression of tet-regulated DNRasN17 and adenovirus-encoded DNRasN17 enhanced Stat5 tyrosine phosphorylation, Stat5 DNA binding and β-casein transcription. The expression of DNRasN17 blocked the activation of the Mek-Erk pathway by EGF but did not prevent the phosphorylation of AKT, a measure of activation of the PI-3-kinase pathway. Moreover, the expression of DNRasN17 prevented the block to lactogenic differentiation induced by EGF. Stimulation of HC11 cells with prolactin resulted in the association of the SHP2 phosphatase with Stat5, and this association was prevented by DNRasN17 expression. These results demonstrate that in HC11 cells DNRas inhibits the Mek-Erk pathway and enhances lactogenic hormone-induced differentiation. This occurs, in part, by inhibiting the association of the SHP2 phosphatase with Stat5.
Keywords: Dominant negative Ras, Mek, HC11, lactogenic differentiation, mammary, breast
INTRODUCTION
Mammary epithelial cells undergo periodic cycles of growth, differentiation and apoptosis during pregnancy and lactation. A complex series of signals that include mammotrophic hormones, locally derived growth factors and stroma initiate and regulate these processes. In this study we address the problem of inhibition of mammary cell differentiation by mitogenic growth factors, including epidermal growth factor (EGF), that are found locally in the mammary gland. Because elevated levels of different growth factors in the EGF family such as transforming growth factor α (TGFα) and amphiregulin have been reported in breast tumors (Dotzlaw et al., 1990; Mizukami et al., 1991; Salomon et al., 1999) this study addresses an important issue in both normal development and neoplasia.
The HC11 mouse mammary epithelial cell line used in this study was derived from the COMMA-1D cell line, which was established from a midpregnant BALB/c mouse mammary gland (Danielson et al., 1984). This cell line has been employed as a model system for the study of regulation of mammary differentiation both in vitro and in vivo. HC11 cells introduced into the mammary fat pad differentiate into ductal structures (Danielson et al., 1984). In culture, HC11 mouse mammary epithelial cells differentiate and synthesize β-casein following growth to confluency and stimulation with the lactogenic hormone mixture, DIP (dexamethasone, insulin, prolactin) (Ball et al., 1988; Danielson et al., 1984). β-casein expression in HC11 cells has been used as marker of differentiation, and its regulation in HC11 cells in culture reflects the in vivo regulation of expression of this protein in the mammary gland during pregnancy by prolactin (Ball et al., 1988; Danielson et al., 1984; Peterson and Haldosen, 1998). Production of β-casein in cell culture is dependent upon both the presence of specific mitogens during the growth of the HC11 cells, cell-cell contact, deposition of extracellular matrix, and the subsequent prolactin-dependent activation of Stat5 a and b when the cells have reached confluency (Marte et al., 1995; Merlo et al., 1996; Taverna et al., 1991). Prolactin stimulation results in Jak2-mediated tyrosine phosphorylation of Stat5 a and b and nuclear translocation of these factors (Ali, 1998; Gouilleux et al., 1994; Han et al., 1997; Marte et al., 1995). In HC11 cells the activation of Stat5 by prolactin is not dependent on the Ras-Erk pathway (Wartmann et al., 1996). However, the hormone-induced expression of β-casein can be blocked by the activation of different tyrosine kinase signaling pathways at the time of prolactin addition (Hynes et al., 1990; Marte et al., 1995; Merlo et al., 1996; Peterson and Haldosen, 1998). Previous studies have demonstrated that EGF prevents HC11 differentiation in response to lactogenic hormones. However, several signal transduction pathways have been implicated as responsible for the inhibition of β-casein synthesis. One study reported that the EGF-dependent inhibition of β-casein expression occured through a Ras- and phosphoinositol-3′-kinase (PI-3 kinase)-dependent mechanism, not a Ras-Erk pathway (DeSantis et al., 1997; Salomon et al., 1999). More recently PTP-PEST, a phosphatase that can act on Jak2, was implicated as an EGF-induced protein contributing to this inhibition (Horsch et al., 2001).
Receptor tyrosine kinase (RTK) activation through different growth factor receptors leads to activation of Ras by guanine nucleotide exchange factors. The ErbB family of RTKs use this mechanism to stimulate signal transduction in the Ras pathway (Janes et al., 1994). Signal transduction that is downstream of Ras depends on the association of Ras GTPase with its effector proteins. Several proteins have been identified which associate with Ras in a GTP-dependent manner. These include Raf-1, RasGAP, p110 subunit of PI-3-kinase, AF6, Rin-1, Mek kinase 1, protein kinase C zeta, and RalGDS (Akasaka et al., 1996; Kikuchi et al., 1994; Moodie et al., 1993; Rodriguez-Vicania et al., 1994; Van Aelst et al., 1993; Warne et al., 1993). Activation of Ras initiates a signaling cascade via activation of the Raf-1 and Mek-1 kinases resulting ultimately in the activation of Erk kinases (Moodie et al., 1993; Shibuya et al., 1992). The results of several studies have indicated that the activation of the Ras-Erk kinase pathway can either induce or enhance the differentiation of breast cancer cell lines (Bacus et al., 1992; Giani et al., 1998; Lessor et al., 1998). However, the activation of the Ras-Raf-Mek-Erk pathway by EGF inhibits hormone-induced differentiation in HC11 cells (Hynes et al., 1990), and the expression of v-Raf, which also activates Erk signaling, has a similar effect (Happ et al., 1993; Jehn et al., 1992).
In the present study we have addressed the mechanism of EGF inhibition of HC11 lactogenic hormone-induced differentiation by examining the involvement of specific signal transduction pathways on differentiation. These studies indicated that the Ras-Mek-Erk pathway and, to a lessor degree, the PI-3 kinase pathway contribute to this inhibition by EGF. Moreover, the expression of DN Ras prevented the EGF-dependent disruption of HC11 differentiation indicating that Ras-signaling is central to this process. DN Ras expression blocked EGF-induced activation of the Mek-Erk signaling but not PI-3-kinase signal transduction, indicating that stimulation of the Mek-Erk pathway is the primary mechanism blocking lactogenic differention in HC11 cells.
MATERIALS and METHODS
Cell culture
Mouse mammary epithelial cell lines, HC11 and HC11-luci, kindly provided by Dr. Nancy Hynes, were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum, 5μg/ml Insulin, 10 mM Hepes and 10ng/ml epidermal growth factor (EGF) as described (Hynes et al., 1990; Marte et al., 1995).
Lactogenic Hormone-Induced Differentiation
The HC11 cells were grown to confluence and maintained for 3–5 days in RPMI 1640 medium supplemented with 10% fetal calf serum, 5μg/ml Insulin, 10 mM Hepes, 10ng/ml epidermal growth factor (EGF) to establish competence (Ball et al., 1988; Taverna et al., 1991). To induce lactogenic hormone-induced differentiation EGF-containing media was removed, the cells were rinsed twice and then incubated in differentiation media, i.e. serum free- or serum containing-RPMI with dexamethazone (10−6 M), insulin (5 μg/ml) and prolactin (5 μg/ml) referred to as DIP. The cells were harvested at the stated times after the addition of DIP.
Construction of Cell Lines
The HC11 cell line was transfected with pTetOff plasmid (Clontech) using Lipofectamine 2000 (Life Technologies). The cells were incubated in G418 (200–500μg/ml) selection media for 10 days, individual colonies were picked with cloning cylinders and expanded in 24 well plates. The colonies were screened for the regulation of the Tet-promoter by transfection with a Tet-promoter-luciferase construct and incubation in medium with and without doxycyclin (0-0.5–2.0μg/ml). The promoter activity was assessed using a luciferase assay system (Promega) with the light emission measured in a luminometer and expressed as light intensity/μg cell protein. Two cell lines exhibited up to 40 fold increase in a Tet-responsive promoter in response to the removal of doxycycline from the cultures. These HC11 tet-off cell lines were used for the construction of cell lines expressing specific genes under the control of the TRE.
The HC11 Tet-off cell lines were infected with retroviral vectors expressing Tet-regulated Ki-Ras genes. pREV-TRE (Clontech), a retroviral vector that expresses a gene of interest from Tet-responsive element (TRE), was derived from pLNCX, a Moloney murine leukemia virus (MoMuLV)-derived retroviral vector. The TRE contains seven direct repeats of the 42-bp tetO operator sequence, which can be bound by tTA transactivators, upstream of a minimal CMV promoter. The 5′ viral LTR regulates expression of the transcript that contains the viral packaging signal and the hygromycin resistance (Hygr) gene. The TRE is an internal promotor in this vector. pREV-TRE was used to inducibly express the Ki-Ras genes in response to removal of doxycyclin (Dox).
pREV-TRE-RasV12 (active K-Ras 2B-V12) and pREV-TRE-DNRasN17 (dominant negative K-Ras 2B-N17) plasmids were constructed by introduction of K-Ras cDNA into pREV-TRE plasmid and selection on hygromycin. For the production of retroviral vector stocks 1.5 × 105 PA317 packaging cells were transfected with 1 μg of recombinant retroviral vector DNA and Lipofectamine 2000 in a 35mm well. 24 hours post transfection the PA317 cells were split and selected in hygromycin containing media (100μg/ml) for 10 days. Mass cultures were prepared from approximately 50–100 colonies and used to produce retroviral vectors stocks. At this point, viral titers were high enough to use for retroviral infection of HC11 Tet-off cells. The HC11-Tet-off cell line was infected with pREV-TRE, pREV-TRE-RasV12 and pREV-TRE-DNRasN17 vector stocks. Cells were selected in hygromycin (100μg/ml) and doxycyclin (2μg/ml) for ten days. Six colonies from each HC11 Tet-off infected cell line were isolated and expanded into cell lines. The clonal cell lines were tested for expression of vector encoded Ras RNA by northern blot following the removal of doxycyclin.
Adenovirus infection
HC11 and HC11 –luci cells were infected with replication defective adenoviruses. A control vector encoding on β-galactosidase (pAd-CMV-β-gal) or a vector encoding Ha-Ras N17, kindly provided by Dr. Craig Logsdon, were used for these experiments (Nicke et al., 1999). Cells were infected with 10–50 MOI of cesium chloride gradient-purified adenovirus by incubation of cells in a low volume of virus-containing media for 5–6 hours. The virus was removed and media was added to the cells for 24 hours prior to additional treatment of the cells.
Luciferase assays
HC11 luci cells were induced to differentiate in DIP-induction media with and without EGF (10ng/ml). Inhibitors were added at the time of DIP-induction. Inhibitors were added at optimal concentrations (PD98059-20μM, LY294002-10μM, wortmannin-100nM, SB203580-10μM and tyrphostin A25-100μM) determined by dose-response curves (data not shown). Cell lysates were harvested 48 hours after transfer to DIP-induction media and luciferase activity was determined using a commercial kit (Luciferase Assay System, Promega) and a luminometer (Thermolab Systems Ascent FL). The total cell protein was determined by BCA assay (Pierce) and luciferase activity was normalized to protein for all the samples. Results are presented as fold induction calculated from the mean of six determinations.
EMSA
Electrophoretic Mobility Shift Assay. HC11 cells were grown to confluency in media containing 10% fetal calf serum, 10 ng/ml EGF, and 5 μg/ml insulin then maintained for 3 days without EGF. The cells were then starved for 24 h in serum-free media prior to induction for 15 min and 48 h with DIP as described above. Nuclear extracts were prepared according to a previously published protocol with little modification (Wartmann et al., 1996). Briefly, harvested cells were suspended in CEB (10 mM KCl, 20 mM Hepes, pH 7.0, 1 mM MgCl2, 0.1% Triton X-100, 20% glycerol, 0.1 mM EGTA, 0.5 mM dithiothreitol, 2 mM sodium orthovanadate, 50 μM β-glycerophosphate, 50 mM sodium fluoride, 2 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin) and sheared with 20 strokes using a Dounce homogenizer (Wheaton, pestle B). Nuclei were pelleted by centrifugation at 800 × g for 5 min and then extracted with NEB (CEB + 300 mM NaCl) by incubating for 30 min on ice. Extracts were clarified by centrifugation for 5 min at 16,000 × g. EMSAs were performed by incubating 10 μg of nuclear protein with the Stat5 DNA binding site from the bovine β-casein promoter (5′-AGATTTCTAGGAATTCAATCC-3′) or Sp1-binding oligonucleotide, end-labeled with32 P-γ-ATP, for 30 min on ice in 16 μl of EMSA buffer (10 mM Hepes, pH 7.6, 2 mM NaH2PO4, 0.25 mM EDTA, 1 mM dithiothreitol, 5 mM MgCl2, 80 mM KCl, 2% glycerol, and 100 μg/ml poly[dI-dC]). Specific binding was analyzed on 6% DNA retardation gel and pre-run for 2 h at 200 V in 0.25 × TBE (22.5 mM Tris borate, pH 8.0, 0.5 mM EDTA) at 4 C°. The samples were loaded and electrophoresed for 2 h at 200 V, the gels were dried and autoradiographed. For antibody supershift assays, nuclear extracts were pre-incubated with Stat 5b C17 antibody (Santa Cruz) for 20 min prior to the addition of the labeled probe.
Northern Blots
Total RNA was extracted using TriPure reagent (Roche). Northern blots were prepared using 7.5 or 10μg of total RNA separated on 1% agarose-formaldehyde gel and transferred to a nylon filter. Blots were hybridized as described previously (Masuelli et al., 1999). The probes used included: mouse β-casein, human KiRas2B, and mouse actin. Mouse β-casein probe is a 601 bp fragment (nucleotide 3-603) from the mouse β-casein cDNA, (accession number X04490.1); it was obtained by RT-PCR and TA-cloning into PCR2.1 and sequence verified. The Ki-Ras probe is a 650 bp fragment representing the human Ki-Ras 2b cDNA, and the actin probe was obtained from Clontech. Mouse Socs-3 probe consisted of nucleotides 467-1006 (accession number NM_007707.2) and mouse Cis-1 was nucleotides 526-1046 (accession number NM_009895.2).
MTT Assay
The rate of replication of HC11-TRE and HC11-DNRasN17(12) cell lines was determined by proliferation assay using MTT dye (CellTiter96 Assay by Promega). The cells were propagated for 96 hours in the absence of doxycycline. The viable cells were counted by 0.4% trypan blue dye exclusion test and the cell count was adjusted of 1× 106 cells/ml in RPMI with 0.5% FBS. Cells were plated at density of 1.5 × 103 per well in quadruplicate wells in 96 well plate with or without EGF (10ng/ml) incubated at 37° C for 24, 48, or 72 hours. For analysis of proliferation 15μl of MTT dye solution was added to each well and the culture plate was incubate at 37° C in CO2 incubator for 4 hours. After 4 hours 100μl of solubilization-stop solution was added to each well. Following one hour incubation at 37° C the samples were mixed by pipeting and the optical density was measured at 570 nm. The mean and standard deviation of the absorbance values for the quadruplicate wells were calculated.
Immunoprecipitations and Western Blots
HC11 cell lysates were prepared in triton-glycerol buffer (1% triton ×100, 10% glycerol, 25 mM Hepes, 150 mM NaCl, 2mM EDTA), NP40 buffer (1% NP40, 25 mM Hepes, 150 mM NaCl) or high salt buffer (Wyszomierski et al., 1999). All lysis buffers contained AEBSF (20μg/ml), aprotinin, (5μg/ml), leupeptin, (5μg/ml), β-glycerol phosphate (100μM) and NaVaO4 (1mM). Immunoprecipitates were prepared by incubation of 0.5 or 1 μg of primary antibody with equal amounts of protein (400μg) and collected by binding to Protein A agarose (In Vitrogen). Antibodies include anti Stat5, sc-835 (SantaCruz), anti phosphoStat5 (Cell Signaling). For western blots equal amount of protein were separated by SDS-PAGE and transferred to PVDF filters. Filters were blocked with 2.5% nonfat dried milk (Blotto) in TBS-T for 1 hour, then incubated with the appropriate dilution of antibody for 1 hour at room temperature or 16 hours at 4 degrees with agitation. Following the incubation with HRP-labeled secondary antibodies, blots were washed and reactivity was detected using ECL (Amersham). Blots were either exposed to Kodak XAR film or results were quantified using a CCD camera (Fuji). Films were quantitated by densitometry. Antibodies included anti Stat5, sc-835 (SantaCruz), anti phosphoStat5 (Cell Signaling), anti phospho Erk, V8031 (Promega), anti pan Erk (Transduction), anti AKT and anti phosphoAKT-ser 473 (Cell Signaling), anti Mek1,2 (Transduction). Anti PTP-PEST was supplied by Dr. Michael Schaller. Antibodies purchased from Santa Cruz Biotechnology were used at 1 μg/ml, and the antibodies from other suppliers were used at the dilution suggested by the manufacturer.
RESULTS
EGF blocks hormone-induced HC11 differentiation through Mek and PI-3-kinase-dependent pathways
Previous studies demonstrated that EGF blocked lactogenic hormone-induced differentiation of HC11 cells (Hynes et al., 1990), and recent data suggested that this block required Ras and PI-3-kinase activity (DeSantis et al., 1997). In the present study specific chemical inhibitors of signal transduction pathways were used to further analyze the contribution of individual signaling pathways to the block of HC11 differentiation by EGF. Because lactogenic hormone-induced differentiation of HC11 cells is characterized by the initiation of β-casein transcription, the HC11-luci cell line, which contains a β-casein promotor linked to the luciferase gene, was used to provide a rapid readout of the differentiation process.
The HC11-luci cells were induced to differentiate with DIP in the absence and presence of EGF. Specific inhibitors of Mek, and PI-3-kinase were added to cells at the time of induction of differentiation. As expected there was a significant inhibition of β-casein driven luciferase activity in the EGF-treated samples compared to the DIP control. However, several compounds (PD98059, LY294002 and wortmannin) restored the β-casein promotor driven luciferase activity that was blocked by EGF (Figure 1A). The results demonstrated that the inhibition of Mek-Erk signaling by PD98059 and PI-3-kinase signaling by LY294002 and wortmannin disrupted the EGF signaling that inhibited lactogenic hormone-induced differentiation, as measured by the activation of β-casein promotor driven luciferase expression.
Figure 1.

A. The effect of signal transduction inhibitors on EGF disruption of differentiation. HC11-luci cells were grown to confluence in EGF-containing media then induced to differentiate in DIP-induction media with serum in the presence or the absence of EGF (10ng/ml). Inhibitors were added at the time of DIP induction at previously determined optimal concentrations (PD98059-20μM, LY294002-10μM, wortmannin-100nM). The luciferase activity in lysates was determined at 48 hours post induction. Luciferase activity was normalized to cell protein. The results, presented as luciferase activity in relative units and represent the mean of six determinations. *These values represent statistically significant difference (p-value .001) from the DIP + EGF condition.
B. The effect of signal transduction inhibitors on EGF disruption of β-casein transcription in HC11 cells. The HC11 cells were induced to differentiate in DIP-induction media with and without EGF (10ng/ml). Inhibitors were added at the time of induction at slightly lower than optimal concentrations to avoid toxicity (PD98059-10μM, LY294002-5μM, wortmannin-50nM).. Total cell RNA was harvested at 48 or 72 hours after transfer to DIP-induction media. β-casein induction was determined via northern blot. For quantitation β-casein expression at 48 hours was normalized to β-actin. The level of expression in DIP-treated cells was set as 1.
The effect of chemical inhibitors of signal transduction pathways on the synthesis of β-casein RNA was examined (Figure 1B). The results confirmed that exposure of HC11 cells to DIP activated β-casein expression and that EGF reduced the expression. The inclusion of PI-3-kinase or Mek1 inhibitors in the induction media with EGF reversed the EGF-induced inhibition of the endogenous β-casein promotor activity in the HC11-luci cells.
Previous studies demonstrated that the treatment of HC11 cells with DIP resulted in increased Stat5 DNA binding and that the DNA binding activity of Stat5 was reduced by the simultaneous addition of EGF and lactogenic hormones (Marte et al., 1995). Therefore, EMSA was performed to examine the ability of the signal transduction inhibitors to alter Stat5 DNA binding. Nuclear extracts were prepared from HC11 cells induced to differentiate in the presence of Jak2, Mek1 or PI-3-kinase inhibitors. The results of this reproducible experiment indicated that DIP stimulation in the presence of the Mek1 (PD98059) and PI-3-kinase (wortmannin) inhibitors enhanced Stat5 binding to DNA compared to the binding detected with DIP alone (Figure 2A). In contrast, exposure of the HC11 cells to DIP plus AG490, an inhibitor of Jak2 tyrosine phosphorylation, inhibited Stat5 DNA binding (Figure 2A, lanes 4 and 8). The results in figure 1 indicated that Mek1 and PI-3-kinase inhibitors restored the DIP-induced Stat5 promotor activity inhibited by EGF, and the same Mek and PI-3-kinase inhibitors enhanced Stat5 DNA binding. Blocking the Mek-Erk and PI-3-kinase pathways with specific inhibitors both enhanced HC11 markers of differentiation and prevented the EGF-dependent disruption of HC11 differentiation.
Figure 2.
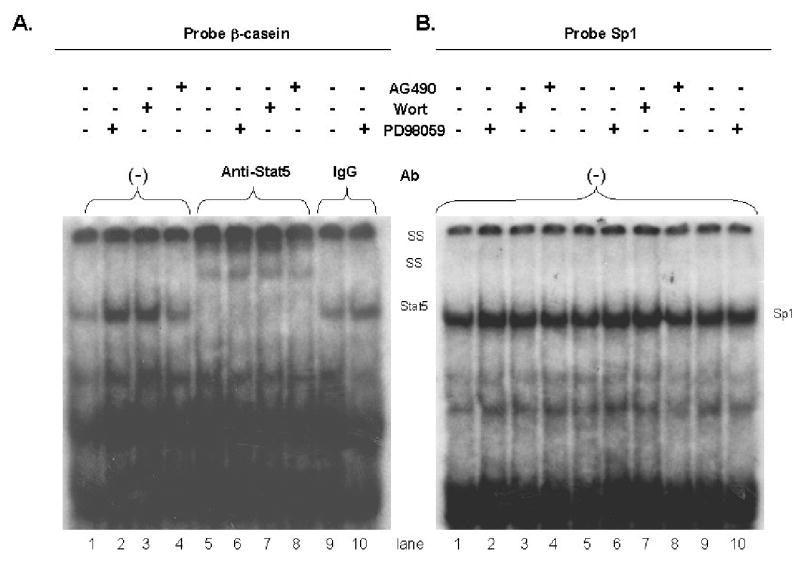
The effect of inhibitors on Stat5 DNA binding by EMSA. A. HC11 cells were grown to confluence in EGF-containing media, then incubated in EGF-free media for three days and serum-free media for 1 day. HC11 cells were pretreated with specific kinase inhibitors for 2 hours prior to DIP-induced differentiation for 15 minutes in the presence of the inhibitors. Nuclear lysates were prepared and used for Stat5 binding to the β-casein GAS element in the presence or absence of anti-Stat5 antibody. Lanes 1 and 5, control (DIP alone); lanes 2 and 6, PD 98059 (20 μM) plus DIP; lanes 3 and 7, wortmannin (20 nM) plus DIP; lanes 4 and 8, AG 490 (20 μM) plus DIP. Lane 5, 6, 7, 8 the binding was performed in the presence of anti-Stat 5 antibody for supershift. Lane 9, 10 the samples were the same as lanes 1, 2 but rabbit IgG was added. B. Gel shift (control) using Sp1 oligos as a loading control. The same protein lysates were used as in Part A, but the binding was to an Sp1 oligonucleotide. SS: supershift of Stat5.
HC11 cells expressing dominant negative RasN17 exhibit an enhanced lactogenic differentiation response
Because Ras activation regulates the activation of the Erk pathway by EGF and may contribute to the activation of PI-3-kinase, the role of Ras activation in the disruption of HC11 differentiation by EGF was examined. HC11 cell clones expressing either activated Ki-Ras(RasV12) or dominant negative Ki-Ras (DNRasN17) were constructed as described in Materials and Methods. The HC11 cell lines constructed contained the Ras cDNAs under the control of a Tet-responsive promotor in a Tet-off system. Hence, the expression of Ras increased following the removal of doxycycline from the culture media. Several independent clones containing each vector were isolated and characterized for the inducibility of Ras gene expression following the removal of doxycycline from the cultures. As expected, the inducibility varied for the individual RasV12 and DNRasN17 clones. The results obtained with three independent clones derived from each vector are shown in Figure 3.
Figure 3.
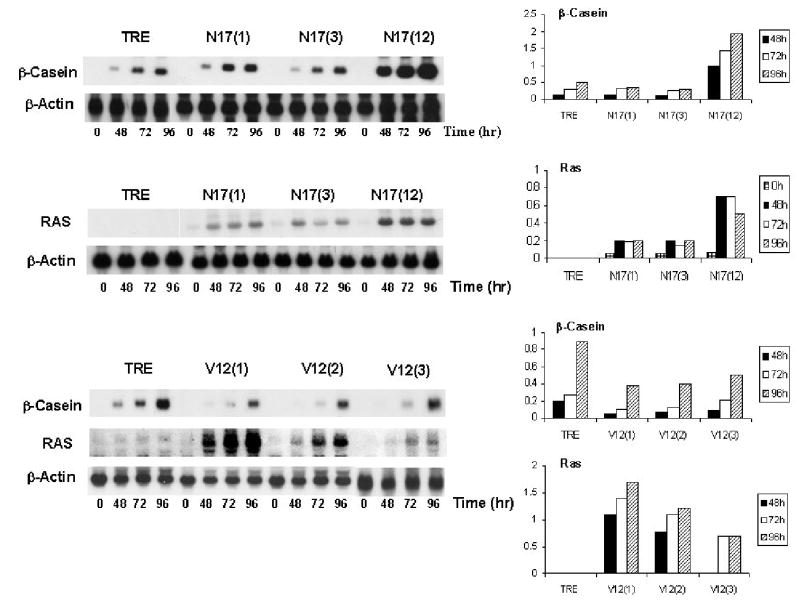
The effect of RasV12 and DNRasN17 on lactogenic differentiation. HC11 cells expressing activated RasV12 and DNRasN17 under the control of the Tet-responsive promotor were utilized to evaluate the effect of Ras-based signal transduction on lactogenic differentiation. Three individual clones of HC11 cells expressing either RasV12 (clones 1,2,3) or RasN17 (clones1,3,12) under the control of the Tet responsive promotor were grown to confluence, incubated in the absence of doxycycline and exposed to DIP differentiation media. The vector control cell line, TRE, was treated in parallel. RNA was harvested from cells at 0, 48, 72 and 96 hours after addition of DIP and used to determine the level of Ras and β-casein expression by northern blotting. The Ras and β-casein expression was quantitated using a beta scanner and were normalized to the actin signal and reported in relative units.
The DNRasN17 and the RasV12 HC11 cell lines were compared to the vector control cell line, REV-TRE, to determine the effect of the Ras gene expression on lactogenic hormone-induced differentiation. HC11 transfectant cell lines expressing DNRasN17 or activated RasV12 along with the vector control cell line were grown for 72 hours in the absence of doxycycline at which point the confluent cultures were incubated in DIP differentiation media. RNA was harvested from cells at 0, 48, 72 and 96 hours post addition of DIP and used to determine the level of Ras and β casein expression by Northern blotting. The results in Figure 3 indicated that RasV12 expression inhibited β-casein expression by approximately 50% compared to the TRE control cell line. In contrast, the expression of DNRasN17 enhanced β-casein induction up to two-fold compared to the control. The results demonstrated that the amount of DNRasN17 expression correlated with the effect on differentiation. The HC11 cell clone expressing the greatest amount of DNRasN17 (clone 12) exhibited the greatest level of β-casein expression. In contrast, all clones of expressing RasV12 inhibited β-casein expression.
In parallel experiments the effect of Ras expression on the prolactin-induced tyrosine phosphorylation of Stat5 was examined. HC11 TRE vector control cells as well as the RasV12 (clone 1) and DNRasN17 (clone 12) cells were stimulated with DIP, and the phosphorylation status of the Stat5 protein was determined by immunoprecipitation and western blotting using anti-Stat5 tyrosine 694 (Y694) phosphorylation site-specific antibodies. The results, seen in Figure 4A, indicated that the tyrosine phosphorylation of Stat5 was enhanced and sustained in the DNRasN17 HC11 cell line compared to the TRE vector control cell line. However, the tyrosine 694 phosphorylation was of a shorter duration in the cell lines expressing activated RasV12 than in the TRE control cells. These results suggested that Ras-dependent signal transduction can modulate Stat5 phosphorylation in HC11 cells in response to DIP. The Stat5 EMSA results supported this conclusion (Figure 4B). Enhanced Stat5 DNA binding in response to DIP stimulation was observed in the DNRasN17 HC11 cell lysates as compared to the vector control. In contrast, the Stat5 DNA binding activity was reduced in cells expressing activated RasV12. In conclusion, an increase in HC11 cell lactogenic hormone-induced differentiation is observed following blockade of the Ras signaling pathway. Moreover, in the HC11 cells that have Ras activity blocked, the enhancement of hormone-induced differentiation appeared to be attributable to an increase in Stat5 tyrosine phosphorylation and to an increase in Stat5 DNA binding resulting in enhanced transcription of β-casein, a Stat5-regulated gene.
Figure 4.

The effect of RasV12 and DNRasN17 expression on Stat5 phosphorylation and DNA binding. A. HC11 TRE vector control cells and HC11 cell lines expressing activated RasV12 (clone 1) or DNRasN17 (clone 12) were grown to confluence in EGF-containing media without doxycycline to induce the expression of Ras. The cells were stimulated with DIP, and nuclear extracts were prepared from cells at 0, 15 minutes, 1 hour and 24 hours post stimulation. Total Stat5 was immuno-precipitated and analyzed by western blotting with antibodies for phospho-Stat5 or total Stat5. The amount of phospho-Stat5 and total Stat5 on the western blots was quantitated, and the amount of phospho-Stat5 was normalized to the total Stat5 and reported in relative units. B. EMSA. HC11 TRE vector control, RasV12(1), and RasN17(12) cells were grown to confluence in EGF-containing media, then incubated in EGF-free media for three days and serum-free media for 1 day. Treated cells were exposed to differentiation media for 15 minutes and control cells (T=0) were not exposed to DIP. (left panel) Nuclear lysates were prepared and used for Stat5 binding to the β-casein GAS element in the presence or absence of anti-Stat5 antibody as indicated. Lanes 1, 4, 7, TRE control; lanes 2, 5, 8, RasV12(1); lanes 3, 6, 9, RasN17(12). (right panel) Sp1 binding oligonucleotides were used as a loading control. Lanes 1, 4, 7 TRE control; lanes 2, 5, 8 RasV12(1); lanes 3, 6, 9 RasN17(12). Lanes 1,2,3 contain contol lysate; lanes 4, 5, 6 contain lysate from DIP-treated cells; lanes 7, 8, 9 contain lysate from DIP-treated cells with the addition of 50 X cold Sp1 oligonucleotides. SS: Stat5 supershift.
Infection of HC11 cells with dominant negative Ha-Ras adenovirus enhances lactogenic differentiation
Infection of cells with replication defective adenovirus encoding dominant negative Ha-RasN17 (DNRasN17) was used as another mechanism to examine the influence of the Ras pathway on lactogenic differentiation. HC11 cells and HC11-luci cells were infected with 10 MOI of either replication defective control adenovirus or adenovirus encoding DNRasN17. At 48 hours post infection the cells were examined for the effect of DNRasN17 on Stat5 phosphorylation, β casein promotor activity and β casein RNA levels. As demonstrated in Figure 5A HC11-luci cells infected with control virus or DNRasN17 virus were stimulated with DIP and the level of Stat5 tyrosine 694phosphorylation was determined. The results indicated that the expression of DNRasN17 increased the level of Stat 5 phosphorylation in response to DIP compared to either uninfected or vector control-infected cells. HC11-luci cells infected with either replication defective control adenovirus or adenovirus encoding DNRasN17 were tested for activation of β-casein promotor-driven luciferase activity (Figure 5B). There was a five-fold increase in the activation of luciferase activity in the DNRasN17 cells compared to the uninfected cells or the control adenovirus infected cells. In addition, there was some activation of luciferase activity in cells infected with the DNRasN17 virus without DIP exposure. This result was reproducible and is not seen when uninfected cells or vector infected cells were exposed to DIP. Finally, HC11 cells infected with either replication defective control adenovirus or adenovirus encoding DNRasN17 were examined for expression of the endogenous β-casein gene following exposure to DIP for 24 or 48 hours. The results of northern blots (Figure 5C) indicated that the infection with DNRasN17 virus resulted in a two-fold increase in β-casein RNA compared to the uninfected or vector infected cells exposed to DIP.
Figure 5.

The effect of dominant negative Ha-Ras (N17) adenovirus expression on lactogenic differentiation in HC11 cells. A. The effect of DNRasN17 adenovirus on Stat5 phosphorylation in response to lactogenic hormone was determined. Uninfected HC11 cells, HC11 cells infected with a control adenovirus vector and HC11 cells infected with adenovirus encoding DNRas(N17) (at M.O.I. = 10) were incubated for 24 hours; the cells were then serum-starved overnight and stimulated with DIP for 7.5 minutes. Total Stat5 was immunoprecipitated and analyzed by western blotting with antibodies for phospho-Stat5 or total Stat5. The amount of phospho-Stat5 and total Stat5 was quantitated using a CCD camera, and the amount of phospho-Stat5 was normalized to the total Stat5 and reported in relative units. B. HC11-luci cells infected with adenovirus vector control or adenovirus encoding dominant negative RasN17 were used to determine the effect of DNRasN17 on β-casein driven luciferase activity. The cells were infected with the viruses described above and incubated for a period of 24 hours in media without EGF. The cells were then either stimulated with DIP for 24 hours or incubated in media without EGF for an additional 24 hours. The luciferase activity in lysates was determined and normalized to cell protein; the results, presented as luciferase activity in relative units, represent the mean of four determinations. C. The effect of DNRasN17 adenovirus infection on HC11 expression of β-casein was determined. The HC11 cells were infected with the control or DNRasN17 virus as described above. RNA was isolated at 0, 24 and 48 hours post induction of differentiation and used to determine the amount of β-casein transcription by northern blotting. Hybridization of the blots with an actin probe was used as a control for RNA loading. The expression of the β-casein RNA was quantitated by measurement on a β-scanner, normalized to actin and expressed on a relative scale.
HC11 cells expressing dominant negative Ras exhibit reduced response to EGF
Studies were performed to determine if the DNRasN17 expression could block EGF-induced responses in stable transfectants of HC11 cells. HC11 cells respond mitogenically to EGF. The TRE vector control cells and the DNRasN17 cells were stimulated with EGF and the ability of the cells to proliferate was examined using the MTT assay. Cells were removed from doxycylcine for 96 hours and then grown in reduced serum media in the absence and the presence of EGF. MTT assays were performed over the course of four days to follow cell proliferation. The results in Figure 6A demonstrated that the DNRasN17 cell line was growth inhibited by 40% in both the absence and presence of EGF compared to the vector control cell line. This experiment was repeated using TGFα treatment of HC11 vector control and DNRasN17 cells. Again, the DNRasN17 cells exhibited a significantly lower response to EGF and TGFα than did the vector control cell line (Figure 6B).
Figure 6.

DNRas N17 expression inhibits EGF-induced proliferation and prevents EGF-dependent disruption of lactogenic differentiation. A. HC11 TRE vector control and DNRasN17 (clone 12) cells were grown in absence of doxycycline and then seeded in microtiter plates in 0.5% serum-containing media with and without EGF (10μg/ml). Cell proliferation was determined at 24, 48, 72, 96 hours post addition of EGF using the MTT assay. The results are reported as the mean of four determinations. B. The HC11 TRE vector control and DNRasN17 (clone 12) cells were grown as described above and exposed to EGF (10ng/ml) or TGFa (10ng/ml). Cell proliferation was determined using the MTT assay and the results represent the mean of four determinations. C. HC11 TRE vector control and RasN17 (12) cells were grown to confluence in absence of doxycycline and then exposed to DIP in the presence or absence of EGF (10ng/ml). Total RNA was isolated after 72 hours and used for northern blotting. The blots were hybridized to probes for β-casein and actin. The β-Casein and actin RNA was quantitated using a beta scanner; the β-casein RNA was normalized to the actin RNA. The % reduction of β-casein RNA by the addition of EGF during DIP-induced differentiation was calculated using the values for normalized β-casein expression.
The ability of DNRas to prevent the disruption of lactogenic hormone-induced differentiation by EGF in HC11 cells was examined. The HC11 TRE vector control cells and cells expressing DNRasN17 under the control of a Tet-responsive promotor were grown in the absence of doxycycline for 72 hours. The cells were exposed to lactogenic hormone differentiation media in the presence and absence of EGF for varying lengths of time; RNA was extracted and the level of β-casein mRNA was analyzed by northern blotting. The results in Figure 6C demonstrated that EGF did not inhibit the induction of β-casein transcription in the DNRasN17 cell line and, hence, it appeared that differentiation proceeded in these cells even in the presence of EGF. In contrast, the expression of β-casein was blocked by EGF in the TRE vector control cell line in two separate experiements. These results demonstrated that DNRasN17 expression prevented the disruption of hormone-induced differentiation by EGF in HC11 cells.
HC11 cells expressing dominant negative Ras exhibit reduced Erk activation in response to EGF
HC11 cells expressing DNRasN17 were examined to determine if expression of DN Ras prevented the activation of Mek-Erk or PI-3-kinase signaling in response to EGF. In Figure 7A the stable transfectants were removed from doxycycline and grown to confluence. The cells were starved and then stimulated with EGF for varying amounts of time. Cell lysates were prepared and analyzed by western blot using antibodies that detect phosphorylated forms of different signaling proteins. The results, shown in Figure 7A, revealed that stimulation of HC11 vector control cells with EGF resulted in activation of p44Erk as detected by reactivity with an antibody that recognizes the active phosphorylated forms of Erk1,2. In contrast, in HC11 cells expressing DNRasN17 there was no activation of p44Erk, although the Erk protein levels in the cells were similar to those in the vector control cells. The analysis of other signaling proteins revealed little or no difference in Akt activation between the control HC11 cells and the DNRasN17 HC11 cells following treatment with EGF. This demonstrated that the PI-3-kinase pathway was not significantly blocked by DNRasN17 expression in HC11 cells. Moreover, activation of Jun kinase and p38 kinase by EGF was not deficient in the DNRasN17 HC11 cells (data not shown). These results suggest that the Mek-Erk pathway was most sensitive to inhibition by DNRas17 expression.
Figure 7.

The effect of DNRasN17 expression on signal transduction pathways in HC11 cells. A. The HC11 TRE vector control cells and DNRasN17 (clone 12) cell lines were grown to confluence in EGF-containing media lacking doxycycline. The cells were incubated in media without EGF or media without EGF and serum (*) prior to restimulation with EGF (100ng/ml) for the time indicated. Lysates of cells were harvested and analyzed by western blotting using antibodies specific for phosphorylated and nonphosphorylated forms of the indicated proteins. B. HC11 cells infected with control adenovirus vector or DNRasN17-encoding adenovirus at an MOI of 10 were incubated in serum-containing media for 24 hours and incubated in EGF-free media for 20 hours prior to stimulation with EGF (100ng/ml) for the time indicated. Lysates of cells were harvested and analyzed by western blotting as in part A.
Cells infected with the control adenovirus vector or adenovirus encoding DNRasN17 were examined for the effect of EGF on signal transduction pathways in an analogous fashion. The results in Figure 7B demonstated that DNRasN17 adenovirus also blocked the activation of Erk but not the phosphorylation of AKT on serine 473, used as a measure of PI-3-kinase activity. The results from the DNRasN17 expressing cells indicated that blocking the Ras pathway in this manner in HC11 cells primarily blocked signaling to the Raf-Mek-Erk pathway. Hence, these data support the conclusion that in HC11 cells activated RasV12 inhibits β-casein transcription via Mek-Erk signaling, and that the effect of DNRasN17 expression on β-casein is primarily a result of its inhibition of the Mek-Erk pathway.
Expression of dominant negative Ras prevents the prolactin-induced association of SHP2 with STAT5
Our results demonstrated that the expression of DNRasN17 resulted in enhanced DIP-induced activation of STAT5 as measured by tyrosine phosphorylation, DNA binding and activation of the β-casein promotor. To determine the mechanism by which this occurs, the functionality of several STAT5 regulatory pathways in DNRasN17 cells was examined. The increased activity of STAT5 likely resulted from the higher level of STAT5 tyrosine 694 phosphorylation; hence, regulation of STAT5 tyrosine phosphorylation was examined by looking for the association of tyrosine phosphatases with Jak2 and STAT5. Previous reports demonstrated that the phosphatase SHP2 interacted with STAT5; SHP2 associated with STAT5 following stimulation of the STAT5 pathway by prolactin as detected by co-immunoprecipitation of SHP2 with STAT5 (Chughtai et al., 2002). To determine if dominant negative Ras expression affected the prolactin-induced association of SHP2 with STAT5, HC11-TRE and DNRasN17 cells were stimulated with prolactin. Cell lysates were prepared, STAT5 was immunoprecipitated, and the amount of SHP2 associated with the STAT5 was determined by western blotting. The results in figure 8A demonstrated that the level of SHP2 protein expression was not reduced in HC11-DNRasN17 cells. However, in the vector control cell line, prolactin stimulation for 30 minutes resulted in significant association of SHP2 with STAT5, but very little SHP2 was associated with STAT5 in prolactin stimulated DNRasN17 cells (Figure 8A). Accordingly, the degree of STAT5 phosphorylation at Y694 was significantly greater in the DNRasN17 cells than in the TRE cells.
Figure 8.

Prolactin-induced binding of SHP2 to Stat5 and prolactin-induced expression of SOCS-3 and CIS-1. HC11-TRE and HC11-DNRasN17 (clone 12) cells were grown to confluency in EGF-containing media then incubated in EGF-free media for 24 hours; the cells stimulated with prolactin (5μg/ml) for the indicated time. A. Lysates were prepared and Stat5 was immunoprecipitated. The lysates and the immunoprecipitates were analyzed by western blotting for Stat5 and SHP2. B. RNA was extracted from cells and analyzed by northern blotting for expression of SOCS-3 and CIS-1. β-actin was hybridized to the blots as a loading control.
In previous reports PTP-PEST was identified as a phosphatase that was activated by EGF and prolactin and associated with Jak2 (Horsch et al., 2001). These studies indicated that PTP-PEST was co-immunoprecipitated with JAK-2 from HC11 cells following prolactin stimulation. However, we did not observe this association in either the HC11-TRE or HC11-DNRasN17 cell lines (data not shown).
A previous report indicated that both EGF and prolactin stimulation induced the expression of the inhibitors of cytokine signaling, SOCS-3 and CIS-1, in HC11 cells (Tonko-Geymayer et al., 2002). To determine if changes in the regulation of SOCS-3 and CIS-1 expression was affected by dominant negative Ras expression, the level of SOCS-3 and CIS-1 expression was examined by northern blotting at various times after the addition of prolactin to HC11-TRE and DNRasN17 cell lines. The results, shown in Figure 8B, indicated that the expression of CIS-1 and SOCS-3 was stimulated by prolactin and that the expression was similarly regulated in the HC11- DNRasN17 cells and the HC11-TRE control cell line.
These results indicated that the association of SHP2 tyrosine phosphatase activity with STAT5 was blocked by dominant negative Ras expression. The resulting block to STAT5 dephosphorylation constitutes a likely mechanism for the enhancement of STAT5 activation by DNRasN17 expression.
DISCUSSION
Members of the EGF family of peptide growth factors are found in the mammary gland and appear to play a role in growth and differentiation in that tissue (Jhappan et al., 1990). EGF and amphiregulin are expressed in the ductal epithelial cells and TGFα is expressed in cap stem cells in the terminal end buds (Kenney et al., 1995; Snedeker et al., 1992). EGF and TGFα, bind to EGF receptor (ERB1) and can stimulate the proliferation of mammary epithelial cells and enhance lobular-aveolar development in the mammary gland of virgin mice (Vonderhaar, 1987). These growth factors can also prevent milk protein expression in HC11 cells and inhibit apoptosis of secretory alveolar epithelial cells in the involuting mammary gland (Smith et al., 1995). Hence, these factors play a dual role in mammary differentiation.
Growth factors of the EGF family have been detected in human breast tissue and elevated levels have been associated with breast tumors (Dotzlaw et al., 1990; Mizukami et al., 1991). The stimulation of mammary cells in culture by these growth factors activates signal transduction pathways that lead to cell survival and mitosis, and the activation of the EGF-R (ErbB1) correlates with aggressive behavior of breast tumors (Arteaga et al., 1988; Umekita et al., 1992). One of the signaling molecules activated by EGF family growth factors in breast tumors is the Ras GTPase (von Lintig et al., 2000). Previous studies have demonstrated that EGF and activated Ras inhibit differentiation in HC11 cells. Both stimulation of mammary epithelial cells with EGF and the expression of activated Ras initiate signaling through the Mek-Erk pathway. While EGF stimulation also leads to activation of the PI-3-kinase pathway, the influence of Ras on this pathway in HC11 cells has not been examined. Therefore, the present study analyzed the effects of EGF on the Ras, Erk and PI-3-kinase pathways in HC11 cells and the contribution of those pathways to lactogenic hormone-induced differentiation.
The results confirmed the findings of several previous studies by demonstrating that EGF can block lactogenic hormone-induced differentiation in HC11 cells (Hynes et al., 1990). Chemical inhibitor studies indicated that the inhibition of β-casein promotor activity by EGF required both the Mek-Erk and PI-3-kinase pathways. While a previous study found that activation of the Erk pathway was not required for lactogenic differentiation (Wartmann et al., 1996), the contribution of Erk to the inhibition of lactogenic hormone-induced differentiation by EGF was less clear. Merlo et al. correlated the inhibition of lactogenic hormone-induced differentiation by growth factors with the ability of different growth factors to induce a high level of Erk activation (Merlo et al., 1996). Also, expression of v-Raf, an activator of Mek-Erk signaling, inhibited lactogenic hormone-induced differentiation of HC11 cells (Happ et al., 1993). However, a previous study by DeSantis et al. (DeSantis et al., 1997) demonstrated that inhibition of Ras and PI-3-kinase blocked the inhibitory effects of EGF on β-casein synthesis. Our study extends this previous study and demonstrates that the inhibition of the Erk pathway strongly correlates with an increase in β-casein promotor activation. Moreover, in our study the stable expression of dominant negative Ki-RasN17 or the infection of HC11 cells with dominant negative Ha-RasN17 adenovirus effectively enhanced β-casein synthesis in response to lactogenic hormones, and these cells exhibited inhibition of the Mek-Erk pathway but not the PI-3-kinase signaling pathway. Hence, it appears that the Erk pathway is critical in the negative regulation of lactogenic hormone-induced differentiation by DNRas. This appears be a function of its effect on Stat5 tyrosine phosphorylation and activation. Our results are in agreement with those of Gao et al, which suggest that Erk activation alters prolactin-induced expression at a step prior to Stat5 DNA binding (Gao and Horseman, 1999). The HC11 cells expressing dominant negative Ras, which were defective in Erk activation, exhibited both an increase in Stat5 tyrosine phosphorylation and an increase in Stat5 DNA binding. The SH2 protein tyrosine phosphatase, SHP2, has been identified in a complex with Stat5 and a role for this phosphatase in regulation of Stat5 activity has been proposed (Berchtold et al., 1998; Chughtai et al., 2002). Our results indicated that DNRas expression blocked the association of SHP2 with Stat5. While EGF stimulation of HC11 cells has been linked to the activation of PTP-PEST and dephosphorylation of Jak2 (Horsch et al., 2001), no association of PTP-Pest with Jak2 was detected following prolactin stimulation in the HC11-TRE or HC11-DNRasN17 cell lines. In addition, although expression of SOCS-3 and CIS-1 has been demonstrated in HC11 cells following exposure to prolactin (Tonko-Geymayer et al., 2002), DNRasN17 expression did not alter the transcriptional activation of Socs-3 or Cis-1 following prolactin stimulation in our experiments. Collectively these results suggested that DNRasN17 expression enhanced Stat5 tyrosine phosphorylation primarily by blocking the association of the SHP2 phosphatase with Stat5.
The data presented here demonstrate that the addition of EGF to HC11 cells stimulates the PI-3-kinase pathway resulting in the phosphorylation of Akt and its downstream signaling pathway. The data also demonstrate that inhibition of the PI-3-kinase pathway increases β-casein promotor activity. The expression of dominant negative Ki-Ras did not prevent the activation of PI-3-kinase-Akt pathway, indicating that the activation of PI-3-kinase was primarily a consequence of the binding of the p85 subunit to the EGF receptor rather than the direct activation of p110 by activated Ras (Rodriguez-Vicania et al., 1994). These results suggest that the PI-3-kinase pathway influences a stage in Jak-Stat signaling that occurs prior to or at the level of DNA binding. A recent study has demonstrated that PI-3-kinase inhibition enhanced Stat5 activation by thrombopoitin in part by preventing nuclear export of Stat5 (Kirito et al., 2002).
There have been several studies in other tissues demonstrating that regulation of Ras-dependent signal transduction contributes to differentiation. For example, there is evidence from both in vivo systems and tissue culture systems that the Ras-Raf-Mek-Erk pathway is required for neuronal differentiation (Cowly et al., 1994; Halegoua et al., 1991; Marshall, 1995; Thomas et al., 1992; Wood et al., 1992). Also, the activation of the Mek-Erk pathway may contribute to the differentiation status of some breast cancer cell lines. For example, differentiation-linked Erk activation in breast cancer cells occurs following ligand-induced activation of receptor tyrosine kinases, including stimulation with heregulin (NDF-Neu differentiation factor) and subsequent activation of HER-3 (Lessor et al., 1998), or following transfection and overexpression of c-erbB-2 (Giani et al., 1998). In both systems activation of the Ras-Erk pathway resulted in increased expression of p21CIP and enhanced differentiation. ErbB4 signaling has also been linked to prolactin-induced Stat5 activation (Jones et al., 1999). Hence, because of dual nature of Mek-Erk signaling in differentiation, it is important to understand the role of the Ras pathway in lactogenic hormone-induced differentiation. The results of this study clearly focus on signaling through the Mek-Erk pathway as a Ras regulated disruptor of lactogenic hormone-induced differentiation. Moreover, by identifying the Mek-Erk pathway as the pathway that is inhibited by DNRasN17, these studies suggest the mechanism by which EGF disrupts differentiation in this cell line.
Acknowledgments
This work was funded by grants DAMD 17-01-1-0264 from the Congressionally Directed Medical Research Program and R01CA90908 to M. L. Cutler.
References
- Akasaka K, Tamada M, Wang F, Kariya K, Shima F, Kikuchi A, Yamamoto M, Shirouzu M, Yokoyama S, Katoaka T. Differential structural requirements for interaction of Ras protein with its distinct downstream effectors. The Journal of Biological Chemistry. 1996;271:5353–5359. doi: 10.1074/jbc.271.10.5353. [DOI] [PubMed] [Google Scholar]
- Ali S. Prolactin receptor regulates Stat5 tyrosine phosphporylation and nuclear translocation by two separate pathways. Journal of Biological Chemistry. 1998;273(13):7709–7716. doi: 10.1074/jbc.273.13.7709. [DOI] [PubMed] [Google Scholar]
- Arteaga C, Hanauske A, Clark G, Osborne C, et al. Immunoreacitve alpha transforming growth factor in effusions from cancer patients as a marker of tumor burden and patient’s prognosis. Cancer Research. 1988;48:5023–5028. [PubMed] [Google Scholar]
- Bacus S, Huberman E, Chin D, Kiguchi K, Simpson S, Lippman M, Lupu R. A ligand for the ErbB2 oncogene product (gp30) induces differentiation of human breast cancer cells. Cell Growth and Differentiation. 1992;3:401–411. [PubMed] [Google Scholar]
- Ball R, Friis R, Schonenberger C, Doppler W, Groner B. Prolactin regulation of a β-casein gene expression and a cytosolic 120kD protein in a cloned mouse mammary epithelial cell line. The EMBO Journal. 1988;7:2089–2095. doi: 10.1002/j.1460-2075.1988.tb03048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold S, Volarevic S, Morrigl R, Mercep M, Groner B. Dominant-negative variants of SHP-2 tyrosine phosphaptase inhibit prolactin activation of Jak2 and induction of Stat5-dependent transcription. Molecular Endocrinology. 1998;12:556–567. doi: 10.1210/mend.12.4.0086. [DOI] [PubMed] [Google Scholar]
- Chughtai N, Schimchowitsh S, Lebrun J, Ali S. Prolactin induces SHP-2 association with Stat5, nuclear translocation, and binding to the beta-casein gene promotor. Journal of Biological Chemistry. 2002;277(34):31107–31114. doi: 10.1074/jbc.M200156200. [DOI] [PubMed] [Google Scholar]
- Cowly S, Paterson H, Kemp P, Marshall C. Activation of Map kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Danielson K, Oborn C, Durbam E, Butel J, Medina D. Epithelial mouse mammary cell line exhibiting normal morphogenesis in vivo and functional differentiation in vitro. Proceedings of the National Academy of Sciences. 1984;81:3756–3760. doi: 10.1073/pnas.81.12.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis M, Kannan S, Smith G, Seno M, Bianca C, Kim N, Martinez-Lacaci I, Wallace-Jones B, Salomon D. Cripto-1 inhibits β-casein expression in mammary epithelial cells through a p21Ras- and phosphatidylinositol 3′-kinase-dependent pathway. Cell Growth and Differentiation. 1997;8:1257–1266. [PubMed] [Google Scholar]
- Dotzlaw H, Miller T, Karvelas J, Murphy L. Epidermal growth factor gene expression in human breast biopsies samples: relationship to estrogen and progesterone receptor gene expression. Cancer Research. 1990;50:4204–4208. [PubMed] [Google Scholar]
- Gao J, Horseman N. Prolactin-independent modulation of the beta-casein response element by Erk2 Map kinase. Cell Signaling. 1999;11:205–210. doi: 10.1016/s0898-6568(98)00067-9. [DOI] [PubMed] [Google Scholar]
- Giani C, Casalini P, Pupa S, DeVecchi R, Ardini E, Colnaghi M, Giordano A, Menard S. Increased expression of c-erbB-2 in hormone-dependent breast cancer cells inhibits cell growth and induces differentiation. Oncogene. 1998;17:425–432. doi: 10.1038/sj.onc.1201954. [DOI] [PubMed] [Google Scholar]
- Gouilleux F, Wakeo H, Mundt M, Groner B. Prolactin induces phosphorylation of Tyr 694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO Journal. 1994;13:4361–4369. doi: 10.1002/j.1460-2075.1994.tb06756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halegoua S, Armstrong R, Kremer N. Dissecting the mode of action of a neuronal growth factor. Current Topics in Microbiology and Immunology. 1991;165:120–170. doi: 10.1007/978-3-642-75747-1_7. [DOI] [PubMed] [Google Scholar]
- Han Y, Watling D, Rogers N, Stark G. Jak2 and Stat5, but not Jak1 and Stat1, are required for prolactin-induced beta-lactoglobulin transcription. Molecular Endocrinology. 1997;11:1180–1188. doi: 10.1210/mend.11.8.9952. [DOI] [PubMed] [Google Scholar]
- Happ B, Hynes N, Groner B. Ha-Ras and v-Raf oncogenes, but not int-2 and c-myc, interfere with lactogenic hormone dependent activation of the mammary gland specific transcription factor. Cell Growth and Differentiation. 1993;4(1):9–15. [PubMed] [Google Scholar]
- Horsch K, Schaller M, Hynes N. The protein tyrosine phosphatase-PEST is implicated in the negative regulation of epidermal growth factor on PRL signaling in mammary epithelial cells. Molecular Endocrinology. 2001;15(12):2182–2196. doi: 10.1210/mend.15.12.0743. [DOI] [PubMed] [Google Scholar]
- Hynes N, Taverna D, Harwerth I, Ciardiello F, Salomon D, Yamamoto T, Groner B. Epidermal growth factor, but not c-erb-2, activation prevents lactogenic hormone induction of β-casein gene in mouse mammary epithelial cell line. Molecular and Cellular Biology. 1990;10:4027–4034. doi: 10.1128/mcb.10.8.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes P, Daly R, DeFazio A, Sutherland R. Activation of the Ras signaling pathway in human breast cancer cells overexpressing erbB-2. Oncogene. 1994;9:3601–3608. [PubMed] [Google Scholar]
- Jehn B, Costello E, Marti A, Keon N, Deane R, Li F, Friis R, Burri P, Martin F, Jaggi R. Overexpression of Mos, Ras, Src, and Fos inhibits mouse mammary epithelial cell differentiation. Molecular and Cellular Biology. 1992;12(9):3890–3892. doi: 10.1128/mcb.12.9.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhappan C, Stahle C, Harkins R, Fausto N, Smith G, Merlino G. TGFalpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and the pancreas. Cell. 1990;61:1137–1146. doi: 10.1016/0092-8674(90)90076-q. [DOI] [PubMed] [Google Scholar]
- Jones F, Welte T, Fu X-Y, Stern D. ErbB4 signaling in the mammary gland is required for lobuloalveolar development and Stat5 activation during lactation. Journal of Cell Biology. 1999;147:77–87. doi: 10.1083/jcb.147.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney N, Huang R-P, Johnson G, Wu J-X, Okamura D, Matheny W, Kordon E, Gullick W, Plowman G, Smith G, Salomon D, Adamson E. Detection and location of amphiregulin and cripto-1 expression in the developing postnatal mouse mammary gland. Molecular Reproduction and Development. 1995;41:277–286. doi: 10.1002/mrd.1080410302. [DOI] [PubMed] [Google Scholar]
- Kikuchi A, Demo S, Ye Z, Chen Y, Williams L. Ral GDS family members interact with the effector loop of Ras p21. Molecular and Cellular Biology. 1994;14:7483–7491. doi: 10.1128/mcb.14.11.7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirito K, Watanabe T, Sawada K, Endo H, Ozawa K, Komatsu N. Thrombopoitin regulates Bcl-xL gene expression through Stat5 and phophatidylinositol 3-kinase. Journal of Biological Chemistry. 2002;277:8329–8337. doi: 10.1074/jbc.M109824200. [DOI] [PubMed] [Google Scholar]
- Lessor T, Yoo J-Y, Davis M, Hamburger A. Regulation of Heregulinβ1-induced differentiation in a human breast carcinoma cell line by the extracellular-regulated kinase pathway. Journal of Cellular Biochemistry. 1998;70:587–595. doi: 10.1002/(sici)1097-4644(19980915)70:4<587::aid-jcb14>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Marshall C. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Marte B, Jeschke M, Grause-Porta D, Taverna D, Hofer P, Groner B, Yarden Y, Hynes N. Neu differentiaiton factor/heregulin modulates growth and differentitaion of HC11 mammary epithelial cells. Molecular Endocrinology. 1995;9:14–23. doi: 10.1210/mend.9.1.7760847. [DOI] [PubMed] [Google Scholar]
- Masuelli L, Ettenberg S, Vasaturo F, Vestergaard-Sykes K, Cutler M. The Ras suppressor, Rsu-1, enhances NGF-induced differentiation of PC12 cells by induction of p21CIP. Cell Growth and Differentiation. 1999;10:555–564. [PubMed] [Google Scholar]
- Merlo G, Grause-Porta D, Cella N, Marte B, Taverna D, Hynes N. Growth, differentiation and survival of HC11 mammary epithelial cells: diverse effects of receptor tyrosine kinase-activating peptide growth factors. European Journal of Cell Biology. 1996;70:97–105. [PubMed] [Google Scholar]
- Mizukami Y, Nonomura A, Noguchi M, Taniya T, et al. Immunohistochemical study of oncogene product Ras p21, c-myc, and growth factor EGF in breast carcinomas. Anticancer Research. 1991;11:1485–1494. [PubMed] [Google Scholar]
- Moodie S, Willumsen B, Weber M, Wolfman A. Complexes of Ras-GTP with Raf-1 and mitogen activated protein kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- Nicke B, Tseng M-J, Fenrich M, Logsdon C. Adenovirus-mediated gene transfer of Ras N17 inhibits specific CCK actions on pancreatic acinar cells. American Journal of Physiology. 1999;276:G499–G506. doi: 10.1152/ajpgi.1999.276.2.G499. [DOI] [PubMed] [Google Scholar]
- Peterson H, Haldosen L. EGF modulates expression of Stat5 in mammary epithelial cells. Experimental Cell Research. 1998;243:347–358. doi: 10.1006/excr.1998.4160. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Vicania P, Warne P, Dhand R, Vanhaesebroek B, Gout I, Fry M, Waterfield M, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Salomon D, Bianco C, DeSantis M. Cripto: a novel epidermal growth factor (EGF)-related peptide in mammary gland development and neoplasia. BioEssays. 1999;21:61–70. doi: 10.1002/(SICI)1521-1878(199901)21:1<61::AID-BIES8>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Shibuya E, Polverino A, Chang E, Wigler M, Ruderman J. Oncogenic Ras triggers the activation of 42-kDa mitogen-activated protein kinase in extracts of quiscent Xenopus oocytes. Proceedings of the National Academy of Sciences. 1992;89:9831–9835. doi: 10.1073/pnas.89.20.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G, Sharp R, Kordon E, Jhappan C, Merlino G. Transforming growth factor alpha promotes mammary tumorigenesis through selective survival and growth of secretory epithelial cells. American Journal of Pathology. 1995;147:1081–1096. [PMC free article] [PubMed] [Google Scholar]
- Snedeker S, Brown C, DiAugustine R. Expression and functional properties of transforming growth factor alpha and epidermal growth factor during mouse mammary ductal morphogenisis. Proceedings of the National Academy of Sciences. 1992;88:276–280. doi: 10.1073/pnas.88.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna D, Groner B, Hynes N. Epidermal growth factor receptor, platelet-derived growth factor receptor, and c-ErbB receptor activation all promote growth but have distinctive effects upon mouse mammary epithelial cell differentiation. Cell Growth and Differentiation. 1991;2:145–154. [PubMed] [Google Scholar]
- Thomas S, DeMarco M, D’Arcangelo G, Halegoua S, Brugge J. Ras is essential for nerve growth factor- and phorbol ester-induced tyrosine phosphorylation of MAP kinases. Cell. 1992;68:1031–1040. doi: 10.1016/0092-8674(92)90075-n. [DOI] [PubMed] [Google Scholar]
- Tonko-Geymayer S, Goupille O, Tonko M, Soratroi C, Yoshimura A, Streuli C, Ziemiecki A, Kofler R, Doppler W. Regulation and function of the cytokine-inducible SH-2 domain proteins, CIS and SOCS3, in mammary epithelial cells. Mol Endocrinol. 2002;16(7):1680–1695. doi: 10.1210/mend.16.7.0872. [DOI] [PubMed] [Google Scholar]
- Umekita Y, Enokizono N, Sagara Y, Kuriwaki K, et al. Immunohistochemical studies on oncogene products (EGF-R, c-erbB2) and growth factors (EGF, TGFalpha) in human breast cancer: their relationship to oestrogen receptor status, histological grade, mitotic index, and nodal status. Virchows Arch A Pathol Anat Histopathol. 1992;420:345–351. doi: 10.1007/BF01600214. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M. Complex formation between Ras and Raf and other protein kinases. Proceedings of the National Academy of Sciences. 1993;90:6213–6217. doi: 10.1073/pnas.90.13.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lintig F, Dreilinger A, Varki N, Wallace A, Casteel D, Boss G. Ras activation in human breast cancer. Breast Cancer Research and Treatment. 2000;62(1):51–62. doi: 10.1023/a:1006491619920. [DOI] [PubMed] [Google Scholar]
- Vonderhaar B. Local effect of EGF, TGFalpha, and EGF-like growth factors on lobuloalveolar development in the mouse mammary gland in vivo. Journal of Cell Physiology. 1987;132:581–584. doi: 10.1002/jcp.1041320324. [DOI] [PubMed] [Google Scholar]
- Warne P, Viciana P, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364:352–355. doi: 10.1038/364352a0. [DOI] [PubMed] [Google Scholar]
- Wartmann M, Cella N, Hofer P, Groner B, Liu X, Henninghousen L, Hynes N. Lactogenic hormone activation of Stat5 and transcription of the b-casein gene in mammary epithelial cells is independent of p42Erk mitogen-activated protein kinase activity. Journal of Biological Chemistry. 1996;271:31863–31868. doi: 10.1074/jbc.271.50.31863. [DOI] [PubMed] [Google Scholar]
- Wood K, Sarnecki C, Roberts T, Blenis J. ras mediates nerve growth factor receptor modulation of three signal-transducting protein kinases: MAPkinase, Raf-1, and RSK. Cell. 1992;68:1041–1050. doi: 10.1016/0092-8674(92)90076-o. [DOI] [PubMed] [Google Scholar]
- Wyszomierski S, Yeh J, Rosen J. Glucocorticoid receptor/signal transducer and activator of transcription 5 (STAT5) interactions enhance STAT5 activation by prolonging STAT5 DNA binding and tyrosine phosphorylation. Molecular Endocrinology. 1999;13(2):330–343. doi: 10.1210/mend.13.2.0232. [DOI] [PubMed] [Google Scholar]