

Abstract
The t(16:16) and inv(16) are associated with FAB M4Eo myeloid leukemias and result in fusion of the CBFB gene to the MYH11 gene (encoding smooth muscle myosin heavy chain [SMMHC]). Knockout of CBFβ causes embryonic lethality due to lack of definitive hematopoiesis. Although knock-in of CBFB-MYH11 is not sufficient to cause disease, expression increases the incidence of leukemia when combined with cooperating events. Although mouse models are valuable tools in the study of leukemogenesis, little is known about the contribution of CBFβ-SMMHC to human hematopoietic stem and progenitor cell self-renewal. We introduced the CBFβ-MYH11 cDNA into human CD34+ cells via retroviral transduction. Transduced cells displayed an initial repression of progenitor activity but eventually dominated the culture, resulting in the proliferation of clonal populations for up to 7 months. Long-term cultures displayed a myelomonocytic morphology while retaining multilineage progenitor activity and engraftment in NOD/SCID-B2M-/- mice. Progenitor cells from long-term cultures showed altered expression of genes defining inv(16) identified in microarray studies of human patient samples. This system will be useful in examining the effects of CBFβ-SMMHC on gene expression in the human preleukemic cell, in characterizing the effect of this oncogene on human stem cell biology, and in defining its contribution to the development of leukemia.
Introduction
The CBF transcription factor is a heterodimeric complex consisting of the AML1 (RUNX1) protein and the CBFβ protein, both of which are common targets of chromosomal translocations in acute myeloid leukemia (AML). The AML1 gene is involved in numerous genetic aberrations, most commonly the 8:21 translocation found in 40% of AML of the FAB M2 subtype.1 The gene encoding the CBFβ subunit is altered by inv(16) and t(16:16), fusing the CBFB gene with the MYH11 gene for smooth muscle myosin heavy chain (SMMHC).2 This genomic alteration is associated with FAB M4Eo consisting of a myelomonocytic blast cell phenotype with eosinophilia in patient bone marrow.3
Several studies have examined the effects of altered CBFβ and AML1 expression in mice. Knockout of CBFβ gives a lethal phenotype around day E12.5 with mice showing a lack of definitive fetal liver hematopoiesis.4-6 The phenotype is nearly identical to knockout of AML1, suggesting that both proteins are required for function of the CBF complex.7,8 Additionally, mice heterozygous for a knock-in of CBFβ-MYH11 (CM) into one of the endogenous CBFβ genes also show the same phenotype, indicating that CM acts as a dominant repressor of CBF function.9 Chimeric mice generated with embryonic stem cells heterozygous for CM did not develop leukemia. However, leukemia was observed on treatment with the DNA alkylating agent, ethylnitrosourea, at doses insufficient to cause disease in wild-type mice, indicating an involvement of CM in leukemogenesis in cooperation with additional genetic hits.10 Neonatal exposure of these chimeric mice to replication-competent retrovirus also induced AML that mimicked the human disease.11 Analysis of the integration sites in the leukemic samples identified the Plag1 and Plagl2 genes, suggesting cooperation between CM and these genes in leukemogenesis. Subsequent studies determined that overexpression of these transcription factors, both of which accelerated entry into S phase, cooperates with CM in leukemia induction.12 Importantly, both transcription factors were found to be overexpressed in human leukemia samples, with Plagl2 being specifically associated with inv(16) samples.12 In addition, expression of CM in murine hematopoietic stem and progenitor cells (HSPCs) was shown to cooperate with either double knockout of p16INK4a and p19ARF or coexpression of HPV-E7 in the development of leukemia in recipient mice, although the leukemia was of a lymphoid phenotype.13 Another group has recently reported that CM cooperates with p19ARF haploinsufficiency to induce AML and that CM expression in wild-type bone marrow cells induces AML, but with a lower penetrance and latency than in p19ARF-deficient cells.14 Recently, a conditional knock-in mouse model demonstrated that CBFβ-SMMHC expression promotes myeloid leukemia with a median latency of 5 months.15 Interestingly, none of the mouse models have replicated the bone marrow eosinophilia that is associated with the inv(16) human disease.
We and others have examined AML1-ETO function using a model system based on transduction of human cord blood (CB) CD34+ cells.16-19 AML1-ETO (AE) expression resulted in long-term growth and increased self-renewal of progenitor cells. These long-term cultures should further our understanding of the molecular consequences of AE expression in a relevant preleukemic cell. Recently, we have used this system to identify TrkA as an induced target gene of AE and found that its up-regulation confers a proliferative advantage in the presence of nerve growth factor.20 We show here that retroviral transduction of CM into human CD34+ hematopoietic cells mimics a preleukemic condition that could occur on inv(16) manifestation in vivo. Transduced cells proliferate in vitro for up to 7 months, retain CD34 expression, and are able to engraft NOD/SCID-B2M-/- mice. Nevertheless, cells display a strong myelomonocytic bias and altered differentiation in vitro and in vivo and a pronounced tendency to differentiate to eosinophils. These data indicate that expression of CM in human CD34+ HSPCs promotes the expansion of a primitive progenitor cell and that long-term cultures recapitulate key aspects of the disease associated with inv(16) in humans.
Materials and methods
Retroviral production and transduction
The retroviral constructs, pMSCV-IRES-GFP (MIG) and pMSCV-HA-AE-IRES-GFP, and the retroviral transduction protocol have been described previously.16 The CM cDNA was cloned into pMSCV-IRES-GFP using EcoRI restriction sites. Human CD34+ umbilical CB and adult mobilized peripheral blood progenitor cells (PBPCs) were obtained from the Translational Research Development and Support Laboratory of Cincinnati Children's Hospital under an approved Institutional Review Board protocol. Cells were prestimulated for 2 days in IMDM containing 1× BIT serum substitute (Stem Cell Technologies, Vancouver, BC, Canada), 100 U/mL penicillin/streptomycin, 100 μM 2-mercaptoethanol, and human cytokines (100 ng/mL each of stem cell factor [SCF], megakaryocyte growth and development factor, Flt3 ligand, IL-6, and 20 ng/mL IL-3 [CB media]). Transduction was carried out on plates coated with Retronectin (Fisher Scientific, Pittsburgh, PA) preloaded with virus. Medium was replaced with fresh viral supernatant 6 to 8 hours later and again the next day.
Cell culture
Sorted cultures. After transduction, cells were grown in the described serum-free media with 20 ng/mL each of SCF (Kit ligand), megakaryocyte growth and development factor (thrombopoietin), Flt3 ligand, and IL-6 and 10 ng/mL IL-3 (KTF36 media) for the duration of the experiments. Three days after transduction, cells were sorted for GFP expression on a FACSVantage (Becton Dickinson, San Jose, CA). Positive cells were stained with the indicated antibodies to assess lineage or were plated in methylcellulose assays. Three days after sorting, positive cells were assessed for apoptosis and cell cycle using the annexin V-PE kit (Becton Dickinson) and BrdU-APC kit (Becton Dickinson) according to the manufacturer's directions. For BrdU analysis, cells were labeled for 1 hour with 10 μM BrdU.
Nonsorted cultures. After transduction, cells were grown in KTF36 media and monitored by weekly viable cell counts (trypan blue exclusion). Cytospins were stained with Wright-Giemsa stain for eosinophil quantification. Approximately 200 cells were counted in random fields for each sample. Cytospins were photographed using a SPOT RT color camera (Diagnostic Instruments) connected to an OPTIPHOT-2 (Nikon) microscope at × 40 magnification (40×/0.7 numeric aperture [NA] Planar objective) with SPOT4.0.6 (Diagnostic Instruments) software. Pictures of the cell cultures were taken with a DMIRB fluorescence microscope (Leica, Bannockburn, IL) at × 10 magnification (16×/0.36 NA Planar objective) with an ORCA-ER C4742-95 camera (Hamamatsu, Bridgewater, NJ) equipped with a deconvolution system (Leica) and Openlab 4.0.3 software (Improvision, Lexington, MA). For many experiments, CD34+ cells were selected using the EasySep human CD34+ selection kit according to the manufacturer's recommendations (Stem Cell Technologies). CD34+ preparations were verified to be more than 90% CD34+ by flow cytometry. Flow cytometry was performed using a FACSCalibur (Becton Dickinson). Antibodies were from Becton Dickinson and all are PE conjugates except CD15 (APC), CD33 (APC), CD36 (APC), and CD71 (biotin, used with streptavidin-APC). The lineage stain cocktail included CD2, CD8, CD10, CD11b, CD14, CD16, CD19, CD56, and CD235a antibodies (all PE conjugates; Becton Dickinson). All flow cytometry data were analyzed with FlowJo software (Tree Star, Ashland, OR).
CFU assays
Two thousand GFP+ (sorted cultures, Figure 1) or 5000 CD34+ (long-term cultures, Figure 5) were seeded per 35-mm plate in MethoCult H4100 medium (Stem Cell Technologies) supplemented with 20% BIT, 100 μM 2-mercaptoethanol, 2 mM l-glutamine, 100 U/mL penicillin/streptomycin, and the human cytokines erythropoietin (EPO; 6 U/mL), granulocyte colony-stimulating factor (G-CSF; 10 ng/mL), IL-6 (20 ng/mL), IL-3 (20 ng/mL), and SCF (20 ng/mL). Cultures were plated in triplicate except for CM GFP+-sorted cultures (Figure 1) for which there were 10 plates. Colonies of more than 50 cells were scored after 14 days of incubation. Methylcellulose assays were also performed under conditions favoring red cell differentiation (Figure 5C-D). CD34+ cells were cultured as described with the cytokine mix (SCF [100 ng/mL], IL-3 [20 ng/mL], and EPO [6 U/mL]). Red cell colonies were photographed after 14 days. Red cell lineage was determined by staining for expression of CD235a, CD71hi, and CD36. Red colonies plucked and pooled from parallel plates were subjected to the same staining to confirm colony identification.
Figure 1.

CBFβ-SMMHC expression inhibits human HSPC differentiation and negatively affects progenitor cell activity. (A) Human CD34+ cells were transduced and sorted for GFP expression. On the day of sorting, GFP+ cells were analyzed by flow cytometry for the indicated surface markers. (B) Two thousand GFP+ cells were plated in serum-free methylcellulose culture. Total colony numbers were scored after 14 days. Average total colonies (top) or frequency (bottom) are shown with SDs for a representative experiment. *P < .05. (C) After expansion of GFP+ cells for 3 days, apoptosis and cell cycle analysis by annexin V staining and BrdU incorporation, respectively.
Figure 5.
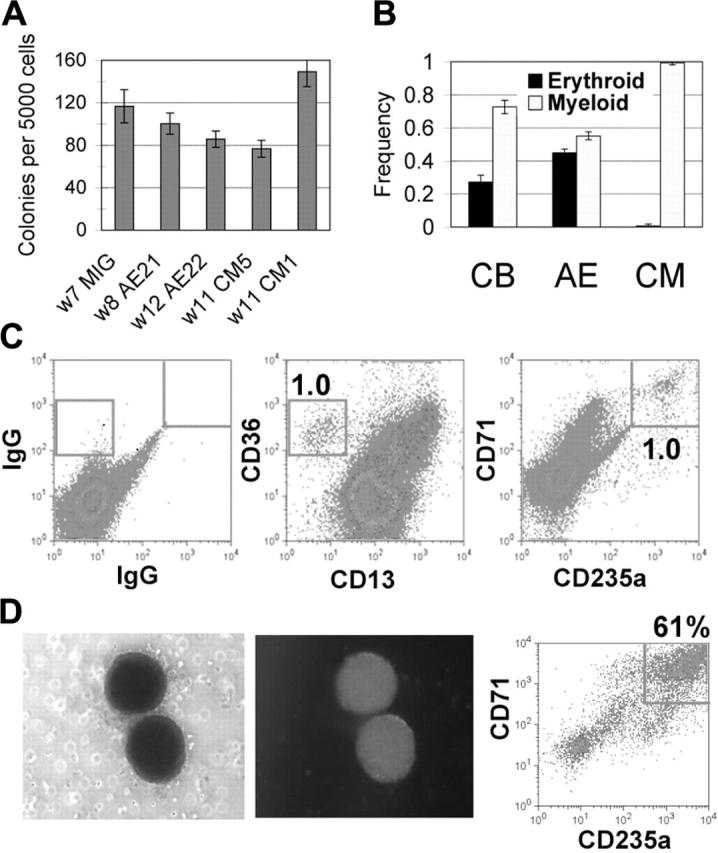
CM-expressing CD34+ cells retain progenitor activity but are inefficient in erythroid differentiation. (A-B) Five thousand magnetically selected CD34+ cells were plated to methylcellulose. (A) Total colony number was determined after 14 days. W indicates weeks in culture at the start of the assay. (B) Colony type frequency was scored as myeloid or erythroid for CD34+ cells from independent CB (n = 2), AE (n = 4), and CM (n = 4) cultures. Averages and SDs are shown. (C-D) Similarly, CD34+ cells from CM cultures were grown in methylcellulose assays with cytokines promoting red cell differentiation. After 14 days, (C) total cells or (D) pooled red cell colonies were stained for erythroid differentiation markers. Photographs show a representative red cell colony expressing GFP that was plucked and pooled for the CD71/CD235a stain in panel D.
Western blotting
Cells were lysed in protein lysis buffer (50 mM HEPES, 1% Triton X-100, 5 mM EDTA, 50 mM NaCl, 10 mM sodium pyrophosphate, 50 mM sodium fluoride, HALT protease inhibitor cocktail [Pierce, Rockford, IL]) followed by incubation on ice for 10 minutes. After brief sonication and centrifugation for 5 minutes, BCA assays (Pierce) were performed. Lysate (25 μg) was boiled in loading buffer (2% SDS, 10% glycerol, 360 mM 2-mercaptoethanol, 100 mM Tris) for 5 to 10 minutes and separated on 8% to 16% Tris-glycine gels followed by transfer to PVDF (Invitrogen, San Diego, CA). After 1 hour of incubation in blocking buffer (Tris-buffered saline/5% BSA/0.1% Tween-20), primary antibodies were added to Tris-buffered saline/2.5% BSA/0.1% Tween-20 solution for 2 hours at room temperature, a secondary HRP-conjugated goat antimouse antibody (Becton Dickinson) was added for 1 hour, and signal was detected using Super Signal substrate (Pierce) on BioLight-1 film (Kodak). Antibodies are monoclonal mouse anti-CBFβ supernatant (a gift of Dr Nancy Speck, Dartmouth) and monoclonal mouse anti-β-actin (Sigma, St Louis, MO).
Southern blotting
gDNA (10 μg) prepared with the QIAamp DNA Blood mini kit (Qiagen, Valencia, CA) was digested with EcoRI overnight. Samples were processed as previously described.16
B-cell differentiation
CD34+ cells from long-term CM cultures (8-15 weeks in culture) were cocultured with a monolayer of MS-5 stroma in B-cell media [MEMα containing 10% FBS and 10 ng/mL each of SCF, Flt3L, and IL-7 [PeproTech, Rocky Hill, NJ]). Cultures were demi-depopulated weekly and tested for expression of B-cell antigens using antibodies for CD10 (PE), CD19 (PE), and CD20 (PerCp; Becton Dickinson).
Xenoengraftment of NOD/SCID-B2M-/- mice
Long-term cultures (12-13 weeks in culture) were injected into the tail veins of 5- to 7-week-old sublethally irradiated (350 rad) NOD/SCID-B2M-/- mice. Mice were killed at 8 to 10 weeks and bone marrow was harvested from the 4 long bones of the legs. Following red cell lysis, human cells were identified by GFP expression and human CD45-APC staining. For positive control, fresh and short-term (< 1 week) cultured CB cells were used, giving engraftment between 0% and 20%.
qPCR
RNA was isolated from magnetically separated CD34+ cells with the RNAeasy kit (Qiagen). RNA was reversed transcribed using MuLV reverse transcriptase (RT) and random hexamers according to the manufacturer's recommendations (Applied Biosystems, Weiterstadt, Germany). The cDNA was then subjected to quantitative polymerase chain reaction (qPCR) using SYBR Green technology (Applied Biosystems). Reactions were run on a 7700 sequence detection system and data were analyzed using SDS version 1.9 and Dissociation Curves 1.0 software (Applied Biosystems). Only samples with the appropriate dissociation curves were considered for analysis. Results were normalized to c-abl expression. qPCR primers are available on request.
Results
CM expression affects CFU activity
Several studies in both mouse and human cells have shown that expression of CM results in inhibition of the cell cycle.21-24 To confirm these results in human progenitor cells, we transduced CD34+ CB cells with viral supernatant generated from the retroviral constructs pMSCV-IRES-GFP (MIG), pMSCV-CBFβ-MYH11-IRES-GFP, or pMSCV-AML1-ETO-IRES-GFP, sorted the cells for expression of GFP, and plated the cells to methylcellulose cultures. We have previously shown that expression of AML1-ETO results in a profound negative effect on the growth of committed progenitors using this assay.16 We used a similar multiplicity of infection to ensure that no experimental artifact was introduced due to differential target cell transduction. Both the MIG and the CM transductions resulted in approximately 15% of cells expressing GFP (Figure 1A). After sorting GFP+ cells, we stained the cells for lineage-specific surface markers. The CM-expressing cells had a marked decrease in erythroid progenitor cells, as demonstrated by loss of the CD36+CD13- population and a decreased CD71hi population, with a concomitant increase in CD13+ cells (Figure 1A). This correlated with a significant decrease in erythroid blast-forming unit (BFU-E) colonies in methylcellulose assays, in addition to an overall decreased clonogenicity (Figure 1B). Remarkably, the transient expression of CM resulted in a skewing of the colony type, with a significantly increased frequency of primitive granulocyte-erythroid-megakaryocyte-macrophage (GEMM) colonies as well as granulocyte-macrophage (GM) colonies (Figure 1B). To determine whether this negative effect on progenitor activity was due to increased apoptosis or cell cycle inhibition, we analyzed cells for annexin V positivity and also compared the incorporation of the base analog bromodeoxyuridine (BrdU) to quantify the number of cells in S phase. CM-expressing cells had an increase in the percentage of cells in the G1 phase of the cell cycle, with fewer annexin V+ cells compared to the MIG control and fewer cells in S phase. These results confirm the findings of others that expression of CM affects the proliferation and differentiation of myeloid progenitor cells and implicates a cell cycle effect rather than cell survival as the mechanism involved. In addition, this effect seems particularly pronounced in cells of the erythroid lineage.
Expression of CM in primary human CD34+ cells leads to long-term proliferation
We have consistently found that the transient inhibitory effect of AE expression on progenitor growth is quickly followed by a period of increased self-renewal in liquid culture that allows for the reproducible establishment of long-term cultures.18,20 To determine whether disruption of the CBF complex by expression of the inv(16)-associated fusion protein CM is sufficient to cause a similar proliferative or survival advantage, we transduced human CB or PBPC CD34+ cells with MIG or MIG-CM. There was a gradual decrease in the percentage of MIG-transduced cells over time in culture, as evidenced by the loss of GFP-expressing cells (Figure 2A). In contrast, CM-expressing cells gradually increased in numbers, beginning at week 4, and eventually dominated the culture (Figure 2A). This selective increase was the result of a continued expansion of primitive CD34+ cells expressing CM (Figure 2B). A large increase in total cell numbers occurred in these experiments, and cells reproducibly grew for approximately 7 months in liquid culture, similar to our findings with AE-expressing CD34+ cells (Figure 2C).18 CM cultures displayed clonal dominance by Southern blot analysis as early as week 6, which is consistent with AE studies (Figure 2D and data not shown). These results demonstrate that the expression of CM is able to promote the expansion of human CD34+ HSPCs in vitro.
Figure 2.
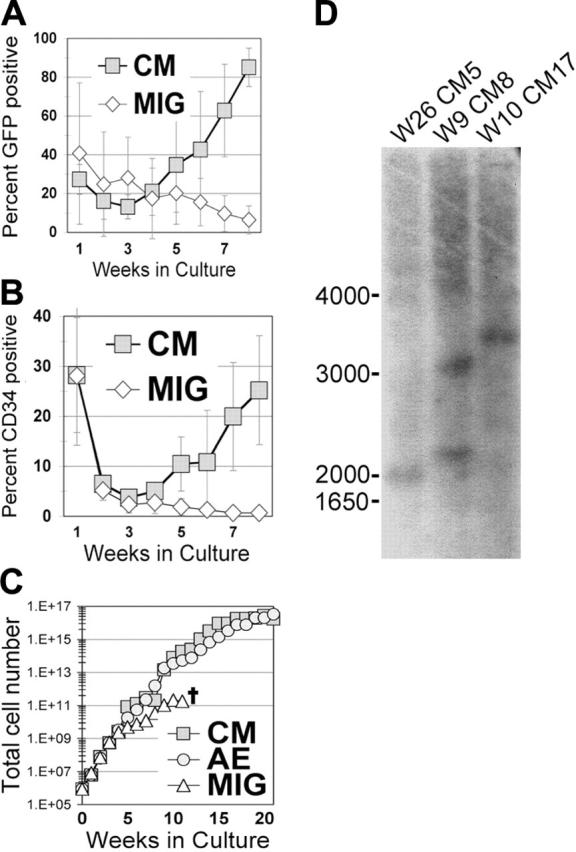
Expression of CBFβ-MYH11 in primary human CD34+ cells leads to long-term proliferation. (A-C) Transduced (nonsorted) human CD34+ cells were expanded in KTF36 media, counted weekly by trypan blue dye exclusion, and stained and analyzed by flow cytometry. (A) GFP expression, (B) CD34 expression, and (C) total cell number are shown. A transduction with the related oncogene AML1-ETO (AE) is shown as a comparison for cell growth during the first 20 weeks. The error bars represent the SD from 3 separate experiments. (D) Southern blot of long-term cultures indicating the clonal nature of the long-term CM cultures. GFP was used as the probe. W indicates weeks in culture.
Long-term cultures expressing CM exhibit a myelomonocytic phenotype with eosinophilia
Following the establishment of several independent long-term cultures, samples were analyzed by Western blot for expression of CBFβ-SMMHC. CBFβ-SMMHC protein was observed using antibodies to either CBFβ or SMMHC (Figure 3A and data not shown). Importantly, levels of CBFβ-SMMHC appeared similar to those found in the human inv(16)+ AML cell line, ME-1, indicating these in vitro effects occur at physiologically relevant doses. The surface profile of the CM cultures was consistent with an in vitro myelopoiesis culture, demonstrating the presence of both primitive CD34+ cells as well as some differentiated myeloid (CD11b and CD16) and monocytic (CD14) cells (Figure 3B). Most of the proliferative potential in the CM-expressing cells was detected in the CD34+ fraction, implying that some normal level of hematopoietic hierarchy is maintained in the long-term cultures expressing CBFβ-SMMHC (data not shown). CM cultures displayed a clearly distinct morphology when compared with the control cultures, consistent with the presence of immature myelomonocytic cells at multiple stages of differentiation (Figure 3C). In contrast, control cultures primarily differentiated to macrophage, as shown by both light microscopic and Wright-Giemsa staining of long-term MIG cultures (Figure 3C-D). Strikingly, Wright-Giemsa staining demonstrated that many of the CM-expressing cells were immature or maturing eosinophils, which are often found in excess amounts in patients with AML with inv(16) (Figure 3D). In 4 different paired cultures, a significant increase in eosinophils was detected in CM cultures compared to control MIG cultures (Figure 3E). This increase was maintained throughout the culture period (Figure 1E).
Figure 3.

Long-term cultures expressing CM exhibit a myelomonocytic phenotype with eosinophilia. (A) Long-term cultures expressing CM or AE, and leukemia cell lines from patients with inv(16) (ME-1) or t(8;21) (Kasumi-1) were analyzed by Western blot using an anti-CBFβ antibody. (B) Surface marker expression of long-term CM cultures expressed as averages of 4 independent cultures with SDs. (C) Photographs of typical cultures transduced with MIG or CM and grown for 8 weeks. (D) Wright-Giemsa-stained cytospins of week 10 MIG and CM cultures, with eosinophils indicated by the arrows. (E) Average eosinophil counts determined by Wright-Giemsa staining of 4 independent MIG- and CM-matched cultures (5-10 weeks in culture) and 3 independent longer-term (week 15-20) CM cultures. Error bars represent SD.
Cells expressing CM have aberrant differentiation potential
CD34+ cells were selected from long-term CM-expressing and control cultures and analyzed for expression of immature and lineage-specific markers over a 4-week period. After 4 weeks, the majority of cells in the normal CB cultures expressed CD11b while losing nearly all CD34 expression (Figure 4A-B). In contrast, the CM-expressing cultures retained a population of cells that continued to express CD34, and the vast majority of cells failed to up-regulate any of the lineage-specific molecules that typically indicate terminal myelomonocytic differentiation (Figure 4A-B). After 4 weeks, 65% of control CB cells expressed mature lineage markers, whereas fewer than 10% of the CM-expressing cells were positive for these differentiation molecules (Figure 4C). All cells were negative for lymphoid markers CD8, CD10, CD19, and CD56, consistent with an almost 100% staining for the myeloid marker CD13 (data not shown). These results indicate a dramatic slowing in the normal myeloid differentiation program of human CD34+ cells on expression of CM.
Figure 4.

Cells expressing CM retain CD34 expression and have aberrant differentiation potential. (A-C) Proliferating cells from normal CB samples (3-5 weeks in culture) or from long-term CM cultures (3 different clones, weeks 13-16 in vitro) were magnetically purified for CD34+ cells. Weekly thereafter cells were stained for (A) differentiation markers, (B) CD34, or (C) a lineage cocktail (week 4 only). Error bars indicate the SD of the 3 independent experiments.
The CD34+ cells expressing CM had a clonogenic potential of between 0.5% and 5.0%, similar to what we find using CD34+ cells from long-term AE cultures (Figure 5A). CD34+ cells from control cultures grown in vitro for 5 or more weeks also show a similar clonogenicity, implying that the greatly expanded proliferative capacity of the CBF-expressing cultures may be due to the continued expansion of CD34+ cells coupled with their inability to differentiate normally. CM-expressing cells predominantly formed myeloid colonies, with very few erythroid colonies detected (Figure 5B). This was in contrast to the colony types from AE-expressing or control CD34+ cells, which were a more equal mix of myeloid and erythroid cells (Figure 5B). Surface staining of cells collected from methylcellulose confirmed that CD235a+ erythroid cells were present at very low numbers in the CM samples, with virtually all of the cells retaining expression of the CD13 myeloid marker (data not shown). To determine whether the long-term CM cultures possessed any erythroid potential at all, CD34+ cells from long-term CM cultures were plated in methylcellulose in a cytokine cocktail that promotes red blood cell survival and differentiation. A small percentage of colonies were of the erythroid phenotype, which was confirmed by detection of a population of CD36+CD13- cells and a corresponding number of CD71+CD235a+ cells (Figure 5C). These erythroid colonies were GFP+, and when plucked and assayed for surface expression, showed a clear enrichment for the erythroid phenotype (Figure 5D). Thus, similar to the finding of a myeloid bias at the expense of erythroid differentiation on transient expression of CM, we detected a similar bias in the long-term CM cultures not seen in AE cultures, demonstrating a particular effect of CM on erythroid potential.
A primitive, self-renewing progenitor is responsible for the long-term expansion of CM-expressing cells
We have previously shown that human CD34+ cells expressing the AML1-ETO fusion protein are able to differentiate to B cells in vitro, even after extended culture in myeloid-promoting conditions.18 These AE cultures also retained the ability to engraft immunocompromised mice at low levels. These data implicate a primitive cell as the source of the proliferative potential in vitro. To determine whether the oncogene CBFβ-MYH11 is similarly promoting the long-term growth of a primitive progenitor, we tested the ability of 5 independent CM cultures to differentiate to B-lymphoid cells. Long-term CM cultures (8-15 weeks in myeloid conditions) were plated onto the MS-5 stroma for 2 to 4 weeks and then analyzed for surface expression of B-lymphoid surface molecules. Two of the CM cultures demonstrated GFP-expressing cells that were positive for CD10, CD19, and CD20, demonstrating that at least for these 2 cultures, the clone that promoted long-term growth was most likely a multipotent progenitor with both myeloid and lymphoid potential (Figure 6A). To determine whether the CM long-term cultures retained the ability to engraft into immunocompromised mice, we injected cells from cultures that had been proliferating in vitro for 12 to 13 weeks. We used 10 million cells per mouse, which is a common number used for engraftment of AML patient samples using this model. Eight to 9 weeks later, we analyzed the bone marrow of the mice and found that GFP+ human cells were detectable in all 5 mice tested, at levels ranging from 0.2% to 1% (Figure 6B). These cells were exclusively myeloid, as determined by CD13+ staining and lack of reactivity to the CD19 antibody. High percentages of the cells were positive for the mature myeloid markers CD11b and CD14, indicating that the cells differentiated in vivo (data not shown). Therefore, CM expression is able to promote the extended in vitro expansion of a primitive hematopoietic cell, similar to what we have described using the related leukemia fusion protein AML1-ETO.
Figure 6.

A primitive progenitor is responsible for the long-term expansion of CM-expressing cells. (A) Long-term CD34+ CM cells (week 9-15 in vitro) were cultured on the MS-5 stroma line under B cell-differentiating conditions. After 4 weeks, cells were collected and analyzed for GFP expression (indicating expression of CBFβ-SMMHC) and for expression of the B-cell markers CD10, CD19, and CD20. (B) Ten million cells from a week 12 or week 13 CM culture were injected intravenously into NOD/SCI-B2M-/- mice. Eight or 9 weeks later, mice were killed and bone marrow from the 4 long bones of the leg was obtained and analyzed for GFP expression and for expression of the human blood cell molecule CD45.
Cells expressing CM show altered expression of known inv(16)-associated genes
Several microarray studies have identified genes with altered expression in human leukemia samples with inv(16).25-28 These studies compared inv(16)+ samples to AML samples containing other genetic alterations. Despite the diverse setup of these different studies, several genes appeared in multiple lists generated by analyses of the gene expression profiles. To determine whether the in vitro preleukemia model we established was recapitulating the molecular signature of inv(16)+ patient samples, we performed qPCR to assess the expression levels of 8 genes that appeared in at least 2 of the 4 studies of primary inv(16) patient samples. qPCR analysis of CD34+ cells from CM cultures demonstrated that 4 of these genes were similarly regulated when compared to control CD34+ cells isolated from cultured CB cells (Figure 7A). Three of these genes, MN1, NRP1, and SPARC, were significantly increased in the CM cultures when compared to control cultures, and the RUNX3 gene was significantly decreased. It is also possible that CLIPR59 would become significant if the sample size were increased because 2 CM samples showed a very significant increase in transcript expression but the remaining sample did not, leading to a large standard deviation. In addition, the SDR1 transcript was very difficult to detect in normal CB CD34+ cells, and much of the signal in these samples was generated from diffuse background bands. An analysis of the PCR product by gel electrophoresis indicates that this transcript is also significantly increased in the CM samples compared to the control CD34+ cells (Figure 7B).
Figure 7.

Cells expressing CM show altered expression of known inv(16)-associated transcripts. (A) qPCR analysis of samples from CD34 magnetically purified cells from CM and control nontransduced CB cultures. Error bars represent SD of 2 to 6 samples. (B) Gel electrophoresis of SDR1 and c-abl amplicons after 40 rounds of qPCR amplification. Samples are derived from CD34+ cells from 2 CM cultures (CM17 and CM6) and 3 different long-term control CB cultures (CB952A, CB935A, and CB56).
Discussion
The results presented here demonstrate that expression of CM in primary human hematopoietic progenitor cells greatly enhances the expansion and promotes clonal outgrowth of primitive progenitor cells in vitro. The ability of CM to increase the proliferative potential of human hematopoietic progenitor cells in a similar fashion as described for AE strongly suggests that this phenotype is due to disruption of normal CBF function. In agreement with published studies on this oncogene, CM expression results in a dramatic inhibition of human progenitor cell growth in initial clonogenic assays, similar to our findings with the AE fusion protein. The expression of CM has an inhibitory effect on the differentiation of the human HSPC, with a greatly increased accumulation of eosinophils and a clear bias toward a myelomonocytic phenotype. In addition, there is a pronounced proliferative or differentiation defect for cells of the erythrocytic lineage. Nevertheless, it is evident that the cell that drives the proliferation of the CM cultures is a primitive progenitor because 2 of the cultures are able to differentiate to a CD19+CD20+ B cell, and the cells are able to engraft in NOD/SCID-B2M-/- mice. Finally, we demonstrate that CM-expressing progenitor cells from the long-term cultures recapitulate many of the hallmark changes seen at the molecular level in cells from inv(16) patient samples, indicating that these cultures will serve as a good starting point for studying the events initiated by this oncogene in human HSPCs.
The effect of CM expression on cell cycle progression has been studied in a number of different model systems, including human HSPCs.21,24 In our studies, the inhibitory effect on committed progenitor proliferation seems to be due to an early delay in differentiation, preferentially for the erythroid lineage, given the reproducible decrease in the number of erythroid precursors as defined by surface expression. This delayed differentiation is accompanied by a decrease in cell cycling and an increase in cell survival. Similar to our observations for AE-expressing cells, we find that expression of CM in human HSPCs does not initiate an absolute block in differentiation but rather sets up a condition of delayed differentiation with a skewing toward self-renewal divisions. This effect would explain the extended proliferative ability of the CBF oncogene-expressing human HSPCs.
Genetic translocations are tightly associated with specific subtypes of AML. Whereas the phenotype produced by CM expression in human CD34+ CB cells is similar to that observed on AE expression, clear differences are apparent in the differentiation of the resulting cultures. These differences are consistent with the idea that the fusion proteins direct the morphologic and phenotypic nature of leukemia. We detect a significant increase in accumulating eosinophils in CM-expressing cultures, and the cell morphology and behavior (adherence to plastic, spreading) demonstrate that CM expression affects the differentiation program differently than does expression of AE. These results indicate that similar model systems could be developed for non-CBF oncogenes and that long-term cultures would possess traits consistent with the oncogene-expressing human leukemia cells.
A myeloid bias is observed in CM-expressing progenitor cells plated to methylcellulose assays, with very few erythroid colonies observed compared to control cells. This bias is likely a result of an early block in committed erythroid progenitor cells, as demonstrated by a failure to generate a CD71hi population. It is possible that CM expression may strongly induce progenitor cells to commit to myeloid differentiation. Alternatively, expression of CM may specifically interfere with red cell differentiation. This hypothesis is supported by the observation that CBFβ knockout mice, partially rescued by expression of a CBFβ-GFP knock-in cassette, express CBFβ in all blood lineages except maturing B cells and erythroid cells.29 Down-regulation of CBFβ could be a prerequisite for erythroid (and B-lymphoid) differentiation. Because we express CM from the MSCV promoter, the required regulation would not occur and we therefore observe an interference with red cell formation. The CM knock-in chimeric mice also display an accumulation of immature erythroid cells, with few mature red cells expressing CM in the peripheral blood, and the conditional knock-in model shows a rapid loss of B cells on CM induction.9,15 Together, these data would argue that expression of CM in primitive progenitors interferes with the commitment toward the erythrocytic and B-lymphoid differentiation programs.
Established long-term cultures driven by AE or CM show no significant difference in average population doubling time, cell cycle status, or clonogenicity in methylcellulose assays, indicating that a common mechanism may be initiated by the related oncogenes that is responsible for the proliferative phenotype in human HSPCs (data not shown). In addition, CD34+ cells from control CB cultures at late time points in vitro have a similar clonogenic potential (0.5%-5%) compared to the oncogene-expressing CD34+ cells when tested in methylcellulose assays (eg, week 7; Figure 5A). These data would indicate that the difference between fusion protein-driven cells and normal cells is the ability to maintain this pool of CD34+ cells over time, possibly through the promotion of self-renewal divisions rather than differentiation divisions. Given the clonal outgrowth that occurs in both the CM and AE cultures, it is possible that specific epigenetic changes in the dominant clone, or insertion site effects of the retrovirus, participate in the phenotype that we describe here. Nevertheless, the long-term cultures initiated by CBF fusion protein expression could shed light on the signals that promote this self-renewal decision.
Several groups have used microarray analysis of AMLs to identify genes with altered expression in inv(16) patient samples.26-28,30,31 However, the key downstream targets of oncoprotein-induced disruption of CBF activity in preleukemic cells remain elusive. One benefit to studying oncogene expression in human cells is the ability to relate downstream gene expression changes to that which actually occurs in patient samples. We have found that a number of inv(16)-associated transcripts are likewise altered in CD34+ cells from our cultures when compared to control CD34+ cells, including NRP1, MN1, RUNX3, and SPARC. Two other genes, SDR1 and CLIPR59, almost reach significance, and it is likely that an analysis using a larger number of samples would reach significance (only 2-6 samples of each group were included in these comparisons). The fact that 6 of 8 genes were regulated in the CM-expressing CD34+ samples similar to what was found in leukemic blast cell comparisons indicates that the majority of class-defining genes may be a reflection of direct or indirect effects of oncoprotein expression rather than a measure of the phenotype of the accumulating blast cell. This system, using purified CD34+ progenitor cells (with and without expression of CM) cultured under the same in vitro conditions, represents a highly controlled comparison using primary human cells and will be useful in examining the effects of CM on gene expression in the human preleukemic cell. It is also likely to reveal that certain genes may be more closely tied to the phenotype of the accumulating blast cell type (eg, CD1c in the case of M4Eo) than to the action of the oncogene. We believe the data generated using primary human HSPC-expressing CM will complement the findings from mouse models and will be useful in characterizing the effect of this oncogene on human stem cell biology and in defining its contribution to the development of leukemia.
Acknowledgments
We thank the mouse core at Cincinnati Children's Hospital for help with animal experiments; Kirin Brewery for the cytokines IL-3, IL-6, SCF, and TPO; and Amgen for Flt3L, SCF, and IL-6.
Prepublished online as Blood First Edition Paper, May 2, 2006; DOI 10.1182/blood-2005-12-012773.
Supported by National Institutes of Health grant CA90370 (J.C.M.).
M.W. designed research, performed research, analyzed data, and wrote the paper; O.K. performed research and analyzed data; J.W. performed research and analyzed data; and J.C.M. designed research, performed research, analyzed data, and wrote the paper.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Licht JD. AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene. 2001;20: 5660-5679. [DOI] [PubMed] [Google Scholar]
- 2.Liu P, Tarle SA, Hajra A, et al. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261: 1041-1044. [DOI] [PubMed] [Google Scholar]
- 3.Reilly JT. Pathogenesis of acute myeloid leukaemia and inv(16)(p13;q22): a paradigm for understanding leukaemogenesis? Br J Haematol. 2005;128: 18-34. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki K, Yagi H, Bronson RT, et al. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc Natl Acad Sci U S A. 1996;93: 12359-12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Q, Stacy T, Miller JD, et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996;87: 697-708. [DOI] [PubMed] [Google Scholar]
- 6.Niki M, Okada H, Takano H, et al. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. Proc Natl Acad Sci U S A. 1997;94: 5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84: 321-330. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93: 3444-3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castilla LH, Wijmenga C, Wang Q, et al. Failure of embryonic hematopoiesis and lethal hemorrhages in mouse embryos heterozygous for a knocked-in leukemia gene CBFB-MYH11. Cell. 1996;87: 687-696. [DOI] [PubMed] [Google Scholar]
- 10.Castilla LH, Garrett L, Adya N, et al. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia [letter]. Nat Genet. 1999;23: 144-146. [DOI] [PubMed] [Google Scholar]
- 11.Castilla LH, Perrat P, Martinez NJ, et al. Identification of genes that synergize with Cbfb-MYH11 in the pathogenesis of acute myeloid leukemia. Proc Natl Acad Sci U S A. 2004;101: 4924-4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landrette SF, Kuo YH, Hensen K, et al. Plag1 and Plagl2 are oncogenes that induce acute myeloid leukemia in cooperation with Cbfb-MYH11. Blood. 2005;105: 2900-2907. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Wang W, Cleaves R, et al. Acceleration of G(1) cooperates with core binding factor beta-smooth muscle myosin heavy chain to induce acute leukemia in mice. Cancer Res. 2002;62: 2232-2235. [PubMed] [Google Scholar]
- 14.Moreno-Miralles I, Pan L, Keates-Baleeiro J, et al. The inv(16) cooperates with ARF haploinsufficiency to induce acute myeloid leukemia. J Biol Chem. 2005;280: 40097-40103. [DOI] [PubMed] [Google Scholar]
- 15.Kuo YH, Landrette SF, Heilman SA, et al. Cbf beta-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell. 2006;9: 57-68. [DOI] [PubMed] [Google Scholar]
- 16.Mulloy JC, Cammenga J, MacKenzie KL, Berguido FJ, Moore MA, Nimer SD. The AML1-ETO fusion protein promotes the expansion of human hematopoietic stem cells. Blood. 2002;99: 15-23. [DOI] [PubMed] [Google Scholar]
- 17.Tonks A, Pearn L, Tonks AJ, et al. The AML1-ETO fusion gene promotes extensive self-renewal of human primary erythroid cells. Blood. 2003;101: 624-632. [DOI] [PubMed] [Google Scholar]
- 18.Mulloy JC, Cammenga J, Berguido FJ, et al. Maintaining the self-renewal and differentiation potential of human CD34+ hematopoietic cells using a single genetic element. Blood. 2003;102: 4369-4376. [DOI] [PubMed] [Google Scholar]
- 19.Basecke J, Schwieger M, Griesinger F, et al. AML1/ETO promotes the maintenance of early hematopoietic progenitors in NOD/SCID mice but does not abrogate their lineage specific differentiation. Leuk Lymphoma. 2005;46: 265-272. [DOI] [PubMed] [Google Scholar]
- 20.Mulloy JC, Jankovic V, Wunderlich M, et al. AML1-ETO fusion protein up-regulates TRKA mRNA expression in human CD34+ cells, allowing nerve growth factor-induced expansion. Proc Natl Acad Sci U S A. 2005;102: 4016-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao W, Britos-Bray M, Claxton DF, et al. CBF beta-SMMHC, expressed in M4Eo AML, reduced CBF DNA-binding and inhibited the G1 to S cell cycle transition at the restriction point in myeloid and lymphoid cells. Oncogene. 1997;15: 1315-1327. [DOI] [PubMed] [Google Scholar]
- 22.Cao W, Adya N, Britos-Bray M, Liu PP, Friedman AD. The core binding factor (CBF) alpha interaction domain and the smooth muscle myosin heavy chain (SMMHC) segment of CBFbeta-SMMHC are both required to slow cell proliferation. J Biol Chem. 1998;273: 31534-31540. [DOI] [PubMed] [Google Scholar]
- 23.Kummalue T, Lou J, Friedman AD. Multimerization via its myosin domain facilitates nuclear localization and inhibition of core binding factor (CBF) activities by the CBFbeta-smooth muscle myosin heavy chain myeloid leukemia oncoprotein. Mol Cell Biol. 2002;22: 8278-8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D'Costa J, Chaudhuri S, Civin CI, Friedman AD. CBFbeta-SMMHC slows proliferation of primary murine and human myeloid progenitors. Leukemia. 2005;19: 921-929. [DOI] [PubMed] [Google Scholar]
- 25.Gutierrez NC, Lopez-Perez R, Hernandez JM, et al. Gene expression profile reveals deregulation of genes with relevant functions in the different subclasses of acute myeloid leukemia. Leukemia. 2005;19: 402-409. [DOI] [PubMed] [Google Scholar]
- 26.Ross ME, Mahfouz R, Onciu M, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104: 3679-3687. [DOI] [PubMed] [Google Scholar]
- 27.Valk PJ, Verhaak RG, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350: 1617-1628. [DOI] [PubMed] [Google Scholar]
- 28.Yagi T, Morimoto A, Eguchi M, et al. Identification of a gene expression signature associated with pediatric AML prognosis. Blood. 2003;102: 1849-1856. [DOI] [PubMed] [Google Scholar]
- 29.Kundu M, Chen A, Anderson S, et al. Role of Cbfb in hematopoiesis and perturbations resulting from expression of the leukemogenic fusion gene Cbfb-MYH11. Blood. 2002;100: 2449-2456. [DOI] [PubMed] [Google Scholar]
- 30.Schoch C, Kohlmann A, Schnittger S, et al. Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proc Natl Acad Sci U S A. 2002; 99: 10008-10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bullinger L, Dohner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350: 1605-1616. [DOI] [PubMed] [Google Scholar]