

Abstract
Abnormal dendritic cell (DC) differentiation and accumulation of immature myeloid suppressor cells (ImC) is one of the major mechanisms of tumor escape. We tested the possibility of pharmacological regulation of myeloid cell differentiation using all-trans-retinoic acid (ATRA). Eighteen patients with metastatic renal cell carcinoma (RCC) were treated with ATRA followed by subcutaneous IL-2. Eight healthy individuals comprised a control group. As expected, the cancer patients had substantially elevated levels of ImC. We observed that ATRA dramatically reduced the number of ImC. This effect was observed only in patients with high plasma concentration of ATRA (>150 ng/ml), but not in patients with lower ATRA concentrations (<135 ng/ml). Effects of ATRA on the proportions of different DC populations were minor. However, ATRA significantly improved myeloid/lymphoid DC ratio and the ability of patients’ mononuclear cells to stimulate allogeneic T cells. This effect was associated with significant improvement of tetanus-toxoid (T-T) specific T-cell response. During the IL-2 treatment, the ATRA effect was completely eliminated. To assess the role of IL-2, specimens from 15 patients with metastatic RCC who had been treated with intravenous IL-2 alone were analyzed. In this group also, IL-2 significantly reduced the number and function of DCs as well as T-cell function. These data indicate that ATRA at effective concentrations eliminated ImC, improved myeloid/lymphoid DC ratio, DC function, and antigen-specific T-cell response. ATRA treatment did not result in significant toxicity and it could be tested in therapeutic combination with cancer vaccines.
Introduction
Tumor-induced defects in the host immune system play a critical role in impeding tumor-specific immune responses and limiting the effect of cancer immunotherapy (1). Abnormal differentiation of myeloid cells is one of the major mechanisms of these defects. It manifests in decreased presence of mature, functionally competent dendritic cells (DC), accumulation of immature DC, and accumulation of immature myeloid suppressive cells (ImC) (2). In mice, transplantable tumor (3–6) or spontaneous development of tumors in transgenic mice with tissue-restricted expression of oncogenes (7) resulted in marked systemic expansion of these cells. The ImC may contribute to the failure of immune therapy in patients with advanced cancer and in tumor-bearing mice.
Recent data from a number of groups have demonstrated that ImC accumulated in tumor-bearing hosts play an important role in tumor non-responsiveness by suppressing antigen-specific T cell responses (2, 5, 7–10). Earlier experiments from H. Schreiber’s group (11) and from V. Bronte and N. Restifo (4) demonstrated that depletion of murine Gr-1+ cells significantly improved CD8+ T cell immune response and allowed for eradication of tumor. More recent work of J. Berzofsky’s group demonstrated that depleting Gr-1+ myeloid cells in vivo prevented tumor recurrence (12). ImC suppress immune response via number of different mechanisms including loss or significant decrease of the expression of the T cell receptor ξ chain (CD3ξ), which is the principal part of the T-cell receptor (TCR) complex (13); inhibition of CD3/CD28-induced T cell activation/proliferation by production of reactive nitrogen and oxygen intermediates (5), inhibition interferon-γ (IFN-γ) production by CD8+ T cells in response to the specific peptide presented by MHC class I molecules (6); prevention the development of cytotoxic T lymphocytes (CTL) in vitro (14, 15), and induction of antigen-specific CD8+ T-cell tolerance in vivo (16). When cultured in vitro in the presence of appropriate growth factors ImC could be differentiated into DC or macrophages (17–19). However, their differentiation is blocked when cells are transferred into tumor-bearing mice, (18) indicating that tumor-derived factors play critical role in preventing differentiation of these cells. The role of these factors was further supported by the fact that surgical resection of the murine tumor decreased the number of ImC (20).
In cancer patients, ImC are defined as cells that express the common myeloid marker CD33 but lack expression of markers of mature myeloid and lymphoid cells and of the MHC class II molecule HLA-DR (8). An accumulation of phenotypic ImC was associated with the decreased number of DCs in the peripheral blood of patients with head and neck, lung or breast cancer (21). In functional testing, ImC isolated from peripheral blood of HLA-A2-positive cancer patients inhibited production of IFN-γ by CD8+ T cells re-stimulated with specific peptide-pulsed DCs (8). Cells with similar phenotype were shown to suppress T-cell function in patients with advanced cancer patients (22). Thus, accumulation of ImC could be one of the mechanisms by which a growing tumor may induce an antigen specific CD8+ T cell unresponsiveness. It seems logical that elimination of those immune suppressive cells may help to enhance the immune antitumor mechanisms in patients.
A promising, clinically relevant approach to reduce the proportion of ImC in tumor-bearing hosts may be use of agents that promote the differentiation of myeloid progenitors. Retinoic acids, ligands of the retinoic acid receptors (RAR, RXR) are among the compounds which may stimulate differentiation of myeloid progenitors into myeloid DCs (6, 23). Mice with vitamin A deficiency (24) and mice treated with a pan-RAR antagonist (25) show accumulation of CD11b+Gr-1+ myeloid cells similar to the ImC that accumulate in cancer patients. Physiologic concentrations of all-trans retinoic acid (ATRA) induced in vitro differentiation of ImC from tumor-bearing mice into CD11c+MHC class II+ myeloid DC (6). In vivo, parenteral or oral administration of ATRA significantly reduced the presence of ImC in two different tested tumor models (26), independent of a direct anti-tumor effect of ATRA or decreased production of growth factors by tumor cells. Experiments with adoptive transfer demonstrated that ATRA differentiated ImC in vivo into mature DC, macrophages, and granulocytes. The decreased presence of ImC in tumor-bearing mice noticeably improved CD4- and CD8-mediated tumor-specific immune responses. The combination of ATRA with two different types of cancer vaccines, in two different tumor models significantly prolonged the anti-tumor effect of the treatment (26). These data suggest that elimination of ImC with ATRA could be a promising approach to improvement of immune response in cancer.
However, all these studies were performed in vitro or in vivo in mice. The critical questions are if and how to use ATRA therapeutically to eliminate ImC in cancer patients. For the first time, in this study we evaluate the effect of ATRA treatment on phenotype and function of DCs, the presence of ImC, and antigen-specific immune response in metastatic, clear-cell type, kidney cancer patients.
Material and Methods
Patient selection and treatment
Competent adults (>18) with metastatic kidney cancer were eligible to participate in this study, which was approved by the University of South Florida Institutional Review Board. All patients in the ATRA treated group had histologically confirmed renal carcinoma with “clear cell component”, identifiable metastatic disease, ECOG performance status ≤2, adequate renal and liver function, and no signs of anemia and leukopenia. Primary tumor was removed via nephrectomy or partial nephrectomy at least 90 days before start of treatment; other tumor debulking surgery at least 30 days before start; radiation therapy completed at least 7 days before start, and no required steroid treatment. Main exclusion criteria were the presence of not-resected primary tumor, brain metastasis with <90 days of stability after end of active treatment; 3 or more days of steroids at greater than physiologic replacement dose within the last 90 days; HIV; pregnancy (ATRA is a known teratogen); seizure medication that was contraindicated with ATRA; or other medical contraindication such as non-healing wound, peripheral vascular disease, or congestive heart failure worse than NYHA class I, or myocardial infarction or hypertension within 1 year.
ATRA (Vesanoid™) was obtained from commercial supply and provided to the subjects. A range of ATRA dose based on the experience in treatment of promyelocytic leukemia patients was selected: 50 mg/m2/day, 100 mg/m2/day, 150 mg/m2/day. The ATRA treatment dose was divided in 3 portions using 10 mg Vesanoid™ capsules, with instructions to take three times a day, with at least 6 hours between each dose; missed doses were not made up, for 21 doses. One week after the finish of ATRA, therapy with IL-2 started. The IL-2 (Proleukin™) treatment included 30 planned doses, given as 5 per week with 2-day breaks (weekends), over 6 weeks. The first five doses were 250,000 IU per kg, and for the next 25 dose 125,000 IU per kg (27).
Observed toxicity was graded using CTCAE3 criteria and tabulated. Management of toxicity was based on dose omission (ATRA, IL-2) or dose reduction. Following the completion of the IL-2 there was a 2–3 week break, with clinical and radiologic evaluation of the treatment response. Eight healthy donors comprised a control group.
Fifteen subjects receiving intravenous, high dose IL-2 for therapy of kidney cancer or melanoma among patients on a separate trial testing a novel schedule using courses of 5 doses of 600,000 units/kg/dose intravenous interleukin-2 at 8 hour intervals, repeated on 4 consecutive weeks; (clinical results to be reported separately). The specimens collected at baseline and at the start of the 3rd week (after planned 10 doses of 600,000 units/kg IL-2) comprised the specimens used for an “IL-2 only” treatment group. These are at a 14-day interval, corresponding for the purpose of assessing IL-2 effect, to the day 14 and day 28 specimens from the ATRA-treated subjects.
Collection of blood samples and ATRA plasma level testing
For evaluation of immunological parameters blood was collected prior to start of ATRA treatment, after finish of ATRA treatment (7 days after start), one week after finish of ATRA treatment before start of IL-2 therapy (14 days after start) and then every week for 4 weeks. Mononuclear cells isolated at each of these time points were cryopreserved and stored for future analysis.
Specimens for ATRA pharmacokinetics were collected after the first (day 1), 10th (day 4) and 21st (day 7) doses, at time points “baseline”, 1, 2, 3 and 4 hours after ATRA administration. These time points were selected based on previously published pharmacokinetic of ATRA (28, 29). The measurement of ATRA level in plasma was performed at the Clinical Pharmacology Laboratory at the H. Lee Moffitt Cancer Center & Research Institute. Heparinized plasma samples that had been stored frozen were thawed and processed as described (30, 31).ATRA used to prepare solutions for standards was obtained from Sigma-Aldrich (St. Louis, MO). The internal standard (I.S.) chosen was retinol acetate (Sigma-Aldrich). The chromatography was performed on an Agilent 1100 HPLC system using a Zorbax 5 μm RX-C18 column (Agilent Technologies, Wilmington, DE) with a mobile phase consisting of a gradient of acetic acid (0.1%, v/v): methanol: methyl tert-butyl ether (20:65.5:14.5, v/v/v) that increased in organic content to (10:74:16, v/v/v) over 5 minutes, then held constant for 6 minutes and returned to original conditions in 2 additional minutes. The run time was 16.5 minutes at a flow rate of 0.8 ml/min. The compounds were detected at a wavelength of 354 nm, with a linear range from 10 ng/ml to 500 ng/ml. Quality Control samples were checked at 30, 80, and 250 ng/ml and the limit of quantitation for the assay was 10 ng/ml. Calibration curves were calculated by linear regression with 1/y weighting to determine the slopes, intercepts and correlations coefficients. Unknowns were plotted against each assay’s regression analysis to determine the concentration.
Evaluation of cell phenotype
All samples from one patient were analyzed simultaneously. PBMC were thawed, cultured overnight in complete culture medium (RPMI-1640 and 10% FCS) and then used for the analysis. Cells were labeled with the antibodies for 40 minutes on ice and analyzed the same day by flow cytometry. The cocktail of lineage (Lin) specific antibodies included PE conjugated antibodies against CD3, CD19, CD56 and CD14 (all from BD Pharmingen). In addition we used PerCP conjugated anti-HLA-DR antibody, APC conjugated anti-CD33 and CD11c antibodies, FITC conjugated anti-CD86, CD83, and CD40 antibodies (all from BD Pharmingen). FITC conjugated antibodies against CCR7 and CD123 were obtained from R&D Systems (Minneapolis, MN) and Miltenyi (Auburn, CA) respectively. The phenotype of the cells was evaluated by multi-color flow cytometry using a FACSCalibur flow cytometer (BD Biosciences, Mountain View, CA). 100,000 cells were collected from each parameter in order to obtain reliable data. The following subsets of cells were evaluated: 1) Immature Myeloid Cells: Lin− HLA-DR− CD33+; 2) Dendritic cells: Lin− HLA-DR+; 3) Myeloid dendritic cells (MDC): Lin− HLA-DR+ CD11c+ CD123−; 4) Plasmacytoid dendritic cells (PDC): Lin− HLA-DR+ CD11c− CD123+; 5) Mature dendritic cells: Lin+ HLA-DR+ and either CD86+, CD40+, CD83+, or CCR7+; 6) Regulatory T cells: The presence of T regulatory cells was determined by staining the cells with APC conjugated anti-CD4, FITC conjugated anti-CD25 (both of which were from BD Pharmingen) and PE conjugated anti-GITR (R&D Systems, Minneapolis) antibodies.
Allogeneic mixed leukocyte reaction
T cells were purified from a leukocyte enriched buffy coat from healthy donors using T cell enrichment column (R&D Systems) according to the manufacturer’s instructions. MNC obtained from patients were first irradiated at 25 Gy and then incubated with donors’ T cells in triplicates in round-bottom 96-well plates at T cells: MNC ratios 1:1, 1:2, 1:4 and 1:8. On day 4 3[H]-Thymidine was added (1μCi/well) and the cells were harvested 18 hours later. Thymidine uptake was measured using a liquid scintillation counter (Packard Instrument, Meriden, CT). T cells in culture media alone were used as a background control.
Evaluation of T-cell function
MNC were cultured in triplicates in round-bottom 96-well plates (105 MNC/well) for 4 days with either 0.1 μg/ml tetanus toxoid (TT) (List Biological Labs, Campbell, CA) or 5 μg/ml phytohemagglutinin PHA (Sigma). These concentrations of TT and PHA were selected after initial testing. 3[H]-thymidine (1μCi/well) was added 18 hr prior to cell harvest. Thymidine uptake was measured using a liquid scintillation counter (Packard Instrument, Meriden, CT). MNC in culture media without any antigen were used as background controls. To measure T-cell response to stimulation with immobilized anti-CD3 antibody round-bottom 96-wells plates were coated overnight at 4°C with 1μg/ml anti-CD3 antibody (BD Pharmingen) diluted in 1 × DPBS (Dulbecco’s phosphate buffered saline). Excess of unbound anti-CD3 antibody was washed off with DPBS. Then 1 × 105 MNC in 100 μl were added in triplicates and incubated for 4 days. T-cell proliferation was measured as described above.
Cytokine Bead Array Assay
Irradiated patients’ MNC were cultured with donors’ T cells at 1:1 ratio for 48 hr. The supernatants were collected and stored at −80°C, until the analysis. The profile of cytokines secreted by T cells after stimulation with patient’s MNC was evaluated using the BD Cytometric Bead Array (CBA) kit (BD Biosciences). Each sample tube was analyzed on the BD FACSCalibur flow cytometer using the BD FACSComp Software.
Statistical analysis
Patients were assigned to one of three ATRA dose levels, at a 1:1:1 ratio, using a randomly permuted list assignments, with the assignment generally being made on the initial day of treatment. The present report describes the experience in treatment of all 18 subjects; the original protocol allowed treatment of up to 36 subjects. The purpose of the randomization was to provide an unbiased assignment, not to compare the arms. Data were analyzed using 2-sided Wilcoxon-Mann-Witney test or Student’s paired T test.
Results
Clinical results and ATRA pharmacokinetics
Between September 2004 and November 2005, 18 patients with metastatic kidney cancer were treated with ATRA followed by course of IL-2. There were 11 men and 7 women, with an age range 45–79 (median 66), with performance status range 0–1. Disease features included metastatic spread to lungs (16), liver (7), bone (8), brain (1), lymph nodes (7), and other (9). Among blood test identified to be prognostic, the hemoglobin range was 9.2–16.0, median (13.0), the serum calcium 8.4–10.0 (median 9.0) and LDH 279–978 (median 430).
Among the side effects observed during the ATRA portion of the treatment, were dry skin, myalgia, and headache, all mild-moderate grades; these typically remitted within a day or two of end of ATRA administration. Per-patient observed best clinical responses, using RECIST criteria, were evaluated after 11–12 weeks of treatment. Among 18 treated subjects, there were 1 complete response (lung, adrenal, lymph nodes) ongoing at 9+ months; no partial responses, 11 stable diseases, 3 progression. Three patients could not tolerate IL-2 treatment and it was discontinued before finishing planned treatment. Two of these three patients had progressive disease and one stable disease.
Consistent with previous reports (32, 33) the peak of the plasma ATRA concentration was observed 2–3 hours after oral administration. Importantly, when assayed on day 4 or 7 of ATRA treatment a smaller peak was observed (Fig. 1A), in the same time frame. ATRA concentration in patients’ plasma varied depending on dose of the drug (Fig. 1B). However, based on the day 1 peak ATRA level, the subjects were easily separated in to two groups. Nine patients had peak of ATRA concentration below 135 ng/ml and eight patients had ATRA concentration above 150 ng/ml (Fig. 1C; one patient specimen not collected). These two groups were comprised of patients who received different doses of ATRA (Fig. 1B, C). Subsequent analysis suggests a critical role of plasma ATRA concentration in the biological effects of the drug. In further discussion we refer to the patients with lower ATRA concentrations (<135 ng/ml) as LC patients and patients with higher ATRA concentrations (>150 ng/ml) as HC patients.
Figure 1. ATRA pharmacokinetics.
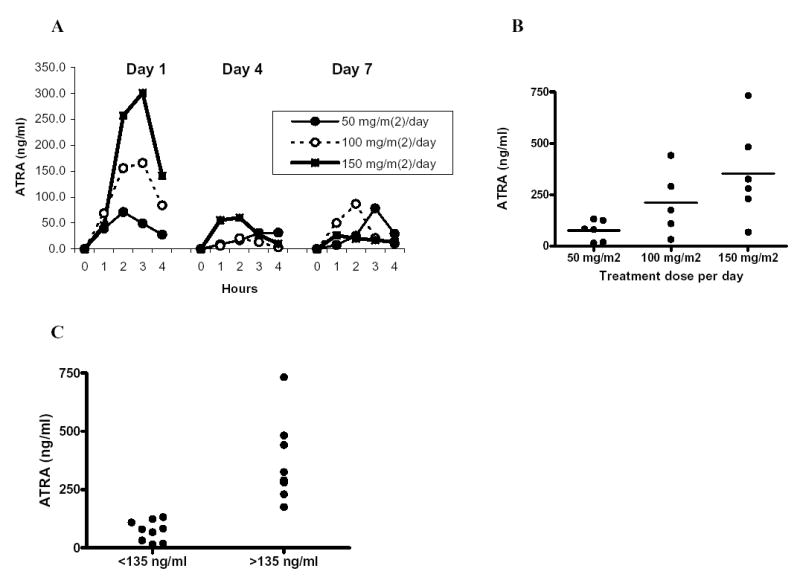
Patients with RCC were treated with indicated doses of ATRA and the level of the compounds was measured in plasma as described in Materials and Methods. Six patients had received 50 mg/m2/day dose, 6 patients – 100 mg/m2/day, and 6 patients – 150 mg/m2/day. ATRA concentrations were measured in 17 out of 18 treated patients. A. Plasma ATRA concentrations at different time points after administration of the single dose of the drug. Measurements were performed on day 1, day 4 and day 8 during the course of the treatment. B. The peak level of ATRA after first dose. Individual results and mean values in the groups receiving different doses are shown. C. The peaks of ATRA concentrations in all patients.
Effect of ATRA on immature myeloid cells and DCs in cancer patients
Blood samples were collected prior to start of ATRA treatment (day 0), at the end of the treatment (day 7), before start of IL-2 therapy (day 14), and then weekly during IL-2 therapy (days 21, 28, and 35).
First, we evaluated the effect of ATRA on the number of white blood cells. Treatment with ATRA did not affect the total number of white blood cells, neutrophils or lymphocytes but induced slight decrease in the number of monocytes, which was restored to pre-treatment level one week after finish of the treatment (Table 1). IL-2 treatment resulted in significant increased in total number of white blood cells, neutrophils lymphocytes but not monocytes (Table 1).
Table 1.
Effect of ATRA and IL-2 treatment on the number of white blood cells
| Parameters | Day 0 Before treatment | Day 7 End of ATRA treatment | Day 14 Before start of IL-2 | Day 28 Two weeks of IL-2 | Day 42 Four weeks of IL-2 |
|---|---|---|---|---|---|
| WBC (× 106/ml) | 6.22 ± 1.55 | 5.41 ± 1.53 p=0.07 | 6.71 ± 1.92 p=0.31 | 15.75 ± 6.32 p=0.01 | 10.01 ± 3.07 p=0.07 |
| Neutrophils (× 106/ml) | 3.99 ± 1.18 | 3.66 ± 1.49 p=0.14 | 4.50 ± 1.60 p=0.18 | 5.51 ± 2.64 p=0.02 | 3.06 ± 1.47 p=0.26 |
| Lymphocytes (× 106/ml) | 1.62 ± 0.52 | 1.47 ± 0.29 p=0.15 | 1.53 ± 0.68 p=0.85 | 4.40 ± 1.73 p=0.005 | 3.50 ± 1.40 p=0.001 |
| Monocytes (× 106/ml) | 0.58 ± 0.19 | 0.40 ± 0.15 p=0.04 | 0.53 ± 0.17 p=0.23 | 0.66 ± 0.52 p=0.73 | 0.57 ± 0.22 p=0.71 |
Eighteen patients were evaluated. Mean ± st. deviation are shown. P values were calculated using 2-sided Student’s t test. Each time point was compared with pre-treatment level (day 0).
ImC were defined as lineage negative HLA-DR negative cells that express myeloid marker CD33. Patients with metastatic RCC had significant increase in this cell population (Fig. 2A). After seven days of ATRA treatment their proportion was significantly reduced and remained at that level seven days later. IL-2 treatment eliminated this effect and increased ImC to the pre-treatment level (Fig. 2A). The effect of ATRA on ImC was dependent on achieved ATRA concentration. Patients with LC and HC had similar background levels of ImC population (Fig. 2B). Only slight decrease in ImC was observed in LC patients, whereas proportion of ImC in HC patients was reduced dramatically and reached the control level. Subsequent IL-2 treatment increased the proportion of ImC in both groups of patients (Fig. 2B).
Figure 2. Effect of ATRA treatment on the proportion of ImC and DCs.
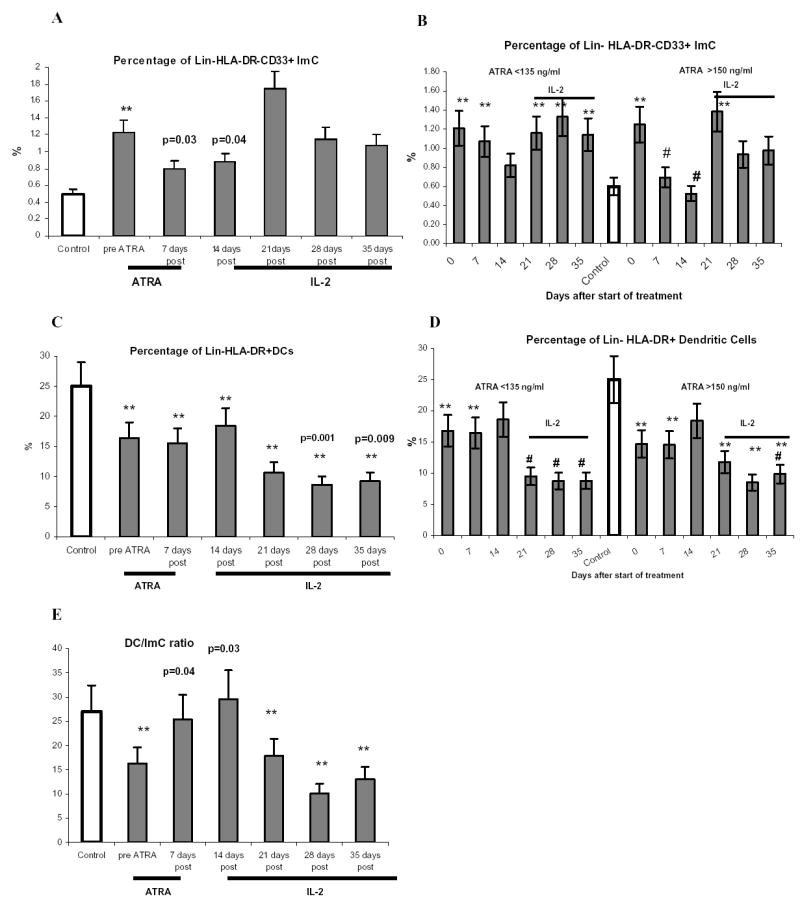
MNC from cancer patients and healthy donors were stored in liquid nitrogen. All samples from the same patient or donor were thawed simultaneously and cells were incubated in complete culture medium overnight at 37°C and 5% CO2. After that time cells were labeled with antibodies and analyzed by multicolor flow cytometry as described in Material and Methods. A, C, E – Proportion of ImC, DCs, and DC/ImC ratio in all 18 patients treated with ATRA. Control represent data from 8 healthy volunteers and donors. Mean and standard error are shown. ** - statistically significant differences from control group (P<0.05). Actual p values are shown in groups that statistically different (p<0.05) from pre-treatment level. B, D. Proportion of ImC and DCs in patients with different ATRA concentration in plasma. Nine patients had lower ATRA concentrations (<135 ng/ml) and 8 patients had higher ATRA concentrations (>150 ng/ml). ** - statistically significant differences from control group (P<0.05). # - statistically significant differenced from pre-treatment level (p<0.05).
The presence of Lin−HLA-DR+ DCs was significantly reduced in the cancer patients. In contrast to the effect on ImC, ATRA did not change the proportion of DCs (Fig. 2C, D). There was a trend for an increased proportion of DCs but it did not reach statistical significance. IL-2 treatment significantly reduced the proportion of these cells (Fig. 2C,D). As a result of the changes in the proportion of ImC and DCs, the cancer patients had significantly reduced DC/ImC ratio. ATRA restored it to the control level (Fig. 2E). Subsequent treatment of patients with IL-2 had just opposite effect by significantly reducing this ratio (Fig. 2E).
We evaluated the presence of two major subtypes of DCs: MDCs and PDCs. The proportion of mature DCs was evaluated within the population Lin−HLA-DR+ cells based on the expression of 4 markers typically associated with the phenotype of mature cells: CD86, CD40, CD83, and CCR7. The proportion of MDCs in RCC patients was significantly reduced (Fig 3A), whereas the proportion of PDCs remained at the control level (Fig. 3B). This resulted in significant decrease in MDC/PDC ratio (Fig. 3C). No changes in the levels of these DC populations were observed in patients with ATRA LC (Fig. 3A-C). Patients with ATRA HC demonstrated slight increase in the proportion of MDCs and decrease in the proportion of PDCs. Although these changes were not statistically significant they resulted in a substantial improvement of MDC/PDC ratio (Fig. 3A-C). Subsequent treatment with IL-2 equally reduced the proportion of both these populations and therefore did not change the proportion of MDC/PDC (Fig. 3A-C). The proportion of mature DCs was significantly reduced in patients with RCC Treatment with ATRA resulted in slight increase in their presence. However, it did not reach statistical significance. (Fig. 3D and data not shown).
Figure 3. Effect of ATRA treatment on the proportion of DC populations.
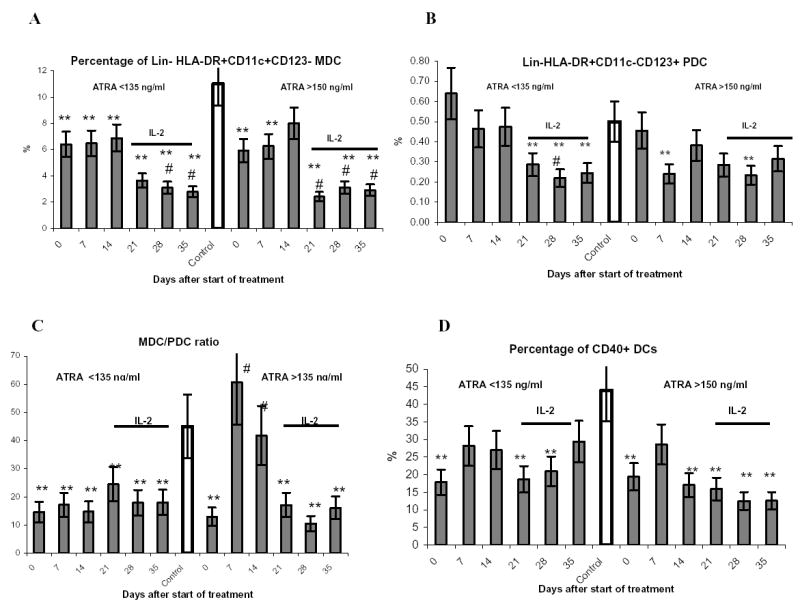
A–D. Results in patients with different levels of ATRA in plasma. All abbreviation as in Figure 2B and D.
ATRA effect on DC function and immune response in cancer patients
Next, we evaluated the function of DCs in allogeneic MLR, a hallmark of DC activity. MNC obtained from cancer patients had significantly reduced ability to stimulate control allogeneic T-cells (Fig. 4A). ATRA substantially improved MNC induced stimulation of allogeneic T cells. This effect was observed only in patients with HC but not in those with LC. IL-2 therapy reversed this effect (Fig. 4A). We studied the type of cytokines produced by T-cells after stimulation with patients’ MNC in allogeneic MLR. MNC from patients treated with ATRA stimulated T-cell production of IFN-γ and IL-2 and decreased the production of Th2-type cytokine (IL-5). Another Th2-type cytokine IL-10 was below the level of detection. These changes resulted in increase of Th1/Th2-type cytokines ratios from 4.6 before ATRA treatment to 51.2 seven days after ATRA treatment (p<0.05) and 69.3 after 14 days of ATRA treatment (p<0.05).
Figure 4. Effect of ATRA treatment on DC function and antigen-specific immune response.
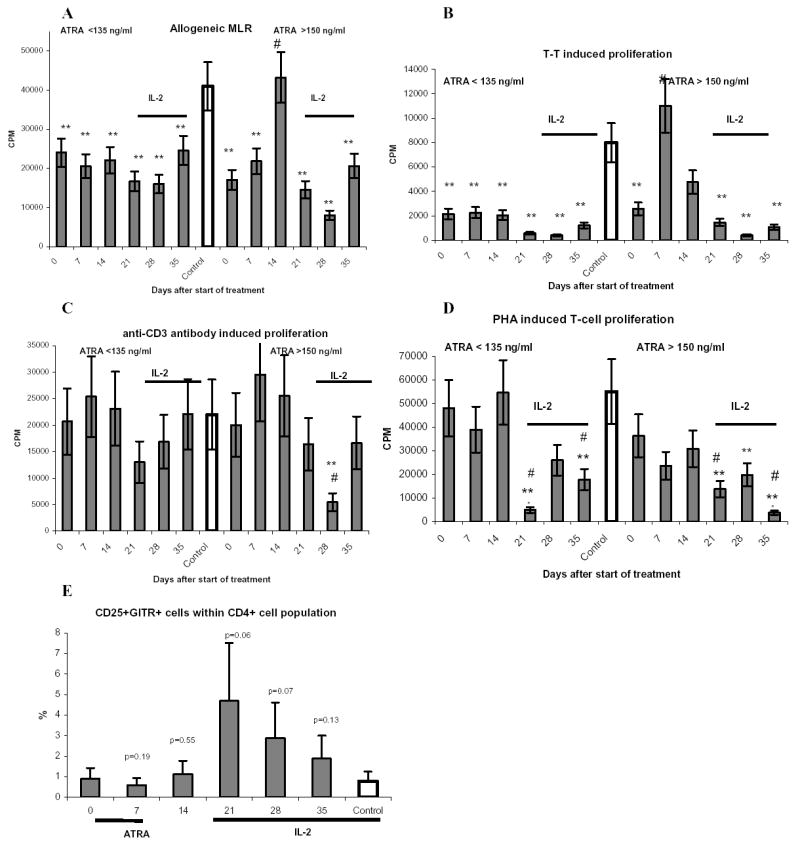
A. MNC isolated from patients and donors were irradiated and cultured for 5 days in triplicates in U-bottom 96-well plates with 105 allogeneic T cells at different ratios. 3[H]-thymidine (1 μCi) was added to each well 18 hr prior cell harvesting. Thymidine uptake was measured in liquid scintillation counter as described in Methods. The background values of T-cell proliferation were subtracted. Only one (1:1) ratio is shown. ** - statistically significant differences from control group (P<0.05). # - statistically significant differenced from pre-treatment level (p<0.05). B – D. MNC (2 × 105/well) were incubated in triplicates with 0.1 μg/ml T-T (B), immobilized anti-CD3 antibody (C), or 5 μg/ml PHA (D) for 4 days. 3[H]-thymidine (1 μCi) was added to each well 18 hr prior cell harvesting. The background values (MNC incubated with medium alone) were subtracted. E. – The proportion of CD4+CD25+GITR+ T cells was evaluated. All abbreviations are as in Figure 2.
To evaluate antigen-specific T-cell response in cancer patients MNC were stimulated with tetanus toxoid (T-T). Consistent with previous observations (rev. in (1)), T-T specific response was significantly reduced in cancer patients (Fig. 4B). In LC group no effect of ATRA on T-T specific T-cell proliferation was found. In HC group ATRA significantly increased this response, which was restored to the control level (Fig. 4B). In contrast, anti-CD3 antibody or PHA-induced T-cell responses were not affected in these patients and treatment with ATRA did not result in significant changes in T-cell proliferation (Figs. 4C and D).
ATRA treatment did not affect the proportion of CD4+CD25+GITR+ regulatory T cells. IL-2 therapy initially increased that proportion albeit the change was not statistically significant (Fig. 4E), then it decreased slightly later during the course of IL-2 treatment.
The effect of IL-2 therapy alone on ImC, DCs and T-cell responses
The data presented above demonstrated that effects of ATRA on ImC, DC subsets and function were eliminated after start of IL-2 therapy. It was not clear whether those changes were indeed the effect of IL-2 therapy or the result of a time-dependent attenuation of the ATRA effect. The original design of the trial did not allow for long observation period between ATRA and IL-2 treatment. Therefore to evaluate potential role of IL-2 on immunological parameters we tested samples of blood obtained from patients enrolled in separate clinical trial.
Fifteen patients with metastatic cancer were treated as described in Material and Methods. They have received several cycles of IL-2 therapy alone. Blood was collected before treatment and after the first 3-week cycle. Treatment with IL-2 alone produced significant decrease in the proportion of DCs and MDCs but not PDCs (Fig. 5). Slight increase in the proportion of ImC was observed. As the result DC/ImC and MDC/PDC ratios were reduced significantly (Fig. 5). IL-2 therapy also significantly decreased the ability of MNC to stimulate allogeneic T cells. The response of T cells to stimulation with T-T or anti-CD3 antibody was reduced. However, those differences did not reach statistically significance (Fig. 5). In addition, IL-2 significantly increased the proportion of regulatory T cells.
Figure 5. Effect of IL-2 on ImC and DCs.
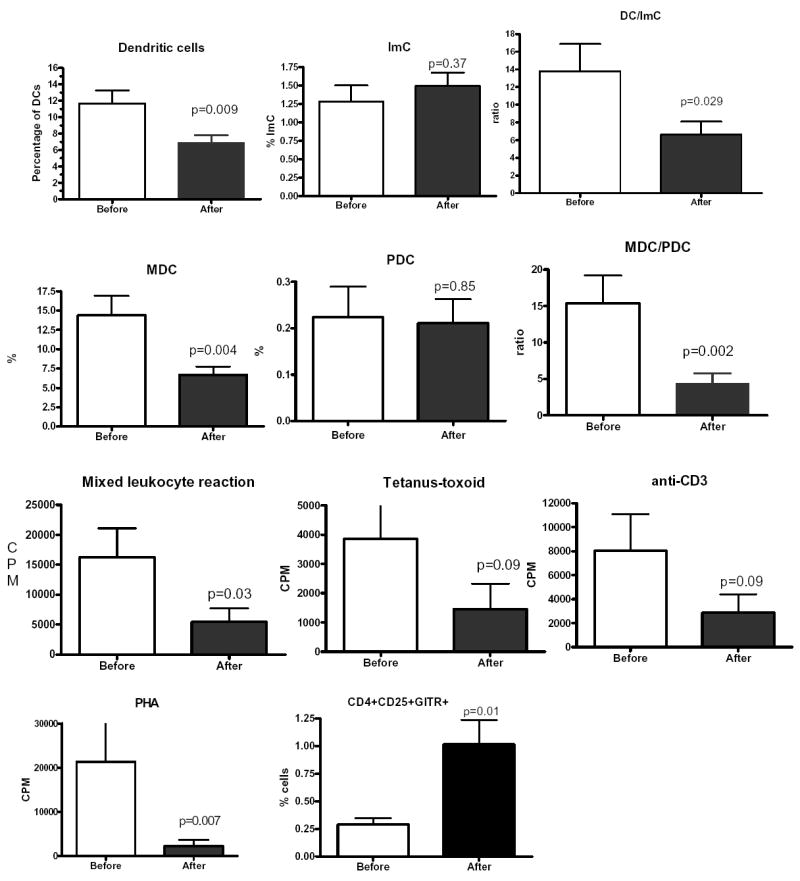
Fifteen patients with RCC were treated with IL-2 alone. Blood samples were collected prior the start of the treatment and after finish of one 4-week cycle of the therapy. Immunological parameters were evaluated as described above. Actual p values in two-sided paired Wilcoxon-Mann-Witney test are shown.
Discussion
It is well established that tumor growth is associated with decreased presence of DCs and accumulation of ImC. Studies from many groups have identified immunosuppressive ImC as one of the major factors contributing to tumor evasion of T cell mediated rejection (2, 34, 35). Elimination of these cells could improve antitumor immune responses and enhance the effect of the vaccines. Previous studies have demonstrated that ATRA could differentiate mouse ImC in vitro (6) and in vivo (26) and patients’ ImC in vitro (8, 36). A naturally-occurring isomer of retinoic acid, ATRA is a molecule capable of induction of differentiation of the human leukemia cell line HL-60 and freshly isolated acute promyelocytic leukemia cells (37, 38). It is successfully used in differentiation induction therapy in patients with acute promyelocytic leukemia (39, 40). ATRA may also affect the growth of normal hematopoietic progenitors and blast progenitors in acute myelogeneous leukemia (41–43). At the molecular level ATRA activates nuclear receptors, which belong to the family of steroid/thyroid/retinoid-activated transcriptional regulators. Two classes of retinoid receptors RARα, RARβ, RARγ and RXRα, RXRβ, RXRγ activate transduction of target genes via complex genetic and epigenetic mechanisms (44). In therapeutic concentrations, ATRA does not directly inhibit the growth of most solid tumors. This could be partly explained by decreased expression of the receptors (45). In this study we addressed, for the first time, the question whether ATRA can affect ImC and DCs in cancer patients. This question is critical not only for understanding the biology of ATRA effects on human myeloid cells but also for the potential use of this treatment in association with cancer vaccines. The trial was performed in patients with metastatic clear cell kidney cancer. The ATRA treatment was followed by an IL-2 schedule for which there is extensive experience.
Although generally well tolerated, ATRA has pharmacokinetics that are complicated by heterogeneity of absorption and clearance between individuals, and the capacity to induce its own metabolism with chronic dosing. Therefore it was important to establish an optimal dose schedule of the drug. We used a 3-fold range of dose levels, encompassing doses previously identified to be generally well tolerated and safe as well as having a biologic effect (induced maturation and apoptosis of APL cells of promyelocytic leukemia) (32, 39, 46). As anticipated, ATRA pharmacokinetic exhibited wide interpatient variability. Median peak levels declined over the 7-day ATRA treatment period, consistent with previous observations that continuous administration of ATRA resulted in decreased plasma level of this drug (32). Analysis of ATRA levels in patients’ blood revealed the existence of two groups with relatively low and high peak ATRA concentrations. It is important that low concentration group was comprised of all 6 patients that received lowest dose of ATRA (50 mg/m2/day) but also 2 patients who received the 100 mg/m2/day dose and 1 patient with 150 mg/m2/day dose. To evaluate the relationship between ATRA concentration and its effect on ImC and DCs we compared ATRA HC and LC patients. This plasma-level based analysis is more logical than dose-assigned one, because of the heterogeneity issue. Each dose group was relatively small (only 6 patients) and some patients in the groups with higher dose had low ATRA concentration in plasma. The mechanism of this heterogeneity is not entirely clear. ATRA does not require a specific transport mechanism therefore general conditions that influence absorption of the drug during oral administration like quantity and quality of recent food intake, the condition of gastro-intestinal tract, etc may affect the absorption and serum concentration of ATRA. The heterogeneity observed within dose cohorts appears comparable to that seen in previous studies (28, 29, 32, 33). It is likely that in a larger cohort these differences will be less prominent and patients can be separated based only on dose administered. Our data indicated that ATRA concentration below 135 ng/ml did not significantly affect either phenotype or function of DCs and ImC, whereas higher concentration (>150 ng/ml) demonstrated a significant effect. This points to the specific nature of the observed effects of ATRA and suggests that ATRA dose of 150 mg/m2/day could be the best for achieving a biological effect. At the highest dose level, ATRA concentrations in plasma on days 4 and 7 were similar to the ineffective level observed on day 1 of the lower ATRA dose. This provides a basis to conjecture that a shorter duration at the higher dose level could be sufficient to achieve optimal effect.
ATRA treatment did not change the number of white blood cells. Only slight decrease in monocyte count was found immediately after finish of the treatment. It returned to pre-treatment level 7 days later. To reduce inter-experimental variations, MNC collected at different time points from the same patient were thawed and analyzed at the same time. Although no additional cell purification was performed MNC were cultured overnight prior to the analysis. This was important for two reasons. First, the phenotype and especially the function and of the cells analyzed immediately after the thawing can be significantly affected. Second, normal peripheral blood contains small proportion of immature myeloid cells and immature DCs that quickly differentiate during overnight culture. This probably reflects the process that is taking place in tissues. We wanted to distinguish those cells from abnormal ImC accumulated in cancer patients. Overnight incubation would help to achieve this goal. As previously observed in other advanced cancer patients (8, 21), subjects with metastatic kidney cancer have almost three-fold excess of ImC. ATRA significantly reduced that proportion. This was evident when samples from all patients on trial were analyzed together. However, the differences became stronger when we compared patients with low and high ATRA plasma concentration. Slight decrease in ImC was found in former group whereas the latter one showed dramatic reduction of the proportion of these cells; restoring them to the control level. Thus, these data confirmed previous in vitro observations (8, 36). ATRA had little effect on the proportion of total DCs. The ratio between myeloid and lymphoid DCs may play an important role in tumor associated immune defects in light of observations that MDCs primarily responsible for induction of immune response and PDCs under certain circumstances can have immunosuppressive features (2, 47–49). In a number of studies preferential loss of MDCs but not PDCs in cancer patients was reported (21, 50, 51). Similar effect was observed in the patients with metastatic RCC. The proportion of MDCs was significantly reduced, whereas PDCs was not affected. This resulted in dramatic drop in MDC/PDC ratio. At “effective” concentrations ATRA slightly increased the proportion of MDCs and decreased the proportion of PDCs that restored MDC/PDC ratio to the control level. Our previous study in vitro has demonstrated that ImC isolated from cancer patients suppressed allogeneic MLR and antigen-specific T-cell responses (8). Elimination of ImC and increase in MDC/PDC ratio after ATRA treatment apparently was responsible for the significant improvement of allogeneic MLR and antigen-specific T-cell response (T-T), which depends on adequate function of antigen presenting cells. ATRA did not affect T-cell response to immobilized CD3 antibody and PHA-induced T-cell proliferation that requires little or no antigen presenting cell participation. Consistent with preferential impact on myeloid rather than T-cell compartment ATRA did not affect regulatory T cells.
All patients on this trial were treated with subcutaneous IL-2 therapy following ATRA. Therefore, the duration of ATRA effect could not be clearly established in this study. IL-2 therapy substantially increased the total number of lymphocytes. This effect was reported in previous clinical trials and was attributed to accumulation of NK and LAK cells (rev. in (52, 53)). Seven days after start of IL-2 therapy the proportion of ImC was significantly increased. It was especially dramatic when taking into account the significant increase in the total number of mononuclear cells. Apparently, in addition to stimulation of lymphocyte production, IL-2 also stimulates myeloid lineage that resulted in increased production of immature cells and decreased proportion of DCs. Increased number of neutrophils observed in these patients was consistent with this fact. Importantly, IL-2 also decreased antigen and mitogen driven T-cell proliferation. Although the precise mechanism of this effect is not clear it could be attributed to a significant increase in the proportion of regulatory T cells observed in these patients. Investigation of the effect of IL-2 was not a goal of this study. However, it is consistent with an hypothesis that limited clinical success of IL-2 therapy could be attributable to the accumulation of immunosuppressive ImC, that are able to suppress the very same antigen-specific immune response IL-2 therapy is trying to enhance. It has been previously reported that some patients demonstrated profound activation of NK and LAK without any evidence of tumor regression (53).
Observed IL-2 effects raised important question, whether elimination of the positive effect of ATRA on ImC and DCs was due to IL-2 effect or due to the short duration of ATRA effect. Precise analysis would require new trial in a different group of patients where the effect of ATRA could be monitored for longer period of time. However, in a framework on this study we could partially address this question by evaluating the effect of IL-2 in patients who did not receive ATRA treatment. Our data demonstrated that IL-2 alone exerted exactly the same effect as in combination with ATRA. This strongly suggests that abrogation of ATRA effect was probably due to the effect of IL-2. Our study suggests that for the purpose of improvement of host immune responses and to provide the support for cancer vaccines ATRA probably should not be combined with IL-2. However, IL-2 therapy can have a major therapeutic effect in a subset of patients with renal cell cancer. It is possible that accumulation of ImS and Treg in patients treated with IL-2 could limit the clinical efficacy of this therapy. If this is the case then ATRA could be considered as potentially useful addition to IL-2. Specifically designed clinical trials are needed to address this question.
Thus, at an effective dose, ATRA treatment substantially reduced the presence of ImC and improved myeloid/lymphoid DC ratio. This resulted in improved DC function and antigen-specific immune response. This indicates that impaired dendritic cell phenotype and immune function in patients with advanced cancer can be effectively modulated pharmacologically and provides strong rationale for combining ATRA treatment with different cancer vaccines.
Acknowledgments
The authors appreciate effort by John Seigne, MB BCh, in the development of the clinical trial.
Footnotes
This work was supported by NIH grants CA101324, CA84488 to DIG and in part by grant from Chiron to MF
References
- 1.Gabrilovich D, Pisarev V. Tumor escape from immune response: mechanisms and targets of activity. Curr Drug Targets. 2003;4:525–36. doi: 10.2174/1389450033490849. [DOI] [PubMed] [Google Scholar]
- 2.Gabrilovich D. The mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–52. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 3.Subiza J, Vinuela J, Rodriguez R, De la Concha E. Development of splenic natural suppressor (NS) cells in Ehrlich tumor-bearing mice. Int J Cancer. 1989;44:307–14. doi: 10.1002/ijc.2910440220. [DOI] [PubMed] [Google Scholar]
- 4.Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP. Unopposed Production of Granulocyte-Macrophage Colony-Stimulating Factor by Tumors Inhibits CD8+ T Cell Responses by Dysregulating Antigen-Presenting Cell Maturation. J Immunol. 1999;162:5728–37. [PMC free article] [PubMed] [Google Scholar]
- 5.Kusmartsev S, Li Y, Chen S-H. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. 2000;165:779–85. doi: 10.4049/jimmunol.165.2.779. [DOI] [PubMed] [Google Scholar]
- 6.Gabrilovich DI, Velders M, Sotomayor E, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 7.Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–45. doi: 10.1182/blood-2003-01-0190. [DOI] [PubMed] [Google Scholar]
- 8.Almand B, Clark JI, Nikitina E, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients. A mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–89. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 9.Pandit R, Lathers D, Beal N, Garrity T, Young M. CD34+ immune suppressive cells in the peripheral blood of patients with head and neck cancer. Ann Otol Rhinol Laryngol. 2000;109:749–54. doi: 10.1177/000348940010900809. [DOI] [PubMed] [Google Scholar]
- 10.Bronte V, Serafini P, Appoloni E, Zanovello P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunoth. 2001;24:431–46. doi: 10.1097/00002371-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Seung L, Rowley D, Dubeym P, Schreiber H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Natl Acad Sci U S A. 1995;92:6254–8. doi: 10.1073/pnas.92.14.6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, Letterio JJ, Paul WE, Berzofsky JA. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–52. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci USA. 1996;93:13119–24. doi: 10.1073/pnas.93.23.13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, Zanovello P. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270–8. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Van Ginderachter J, Brys L, De Baetselier P, Raes G, Geldhof A. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J Immunol. 2003;170:5064–74. doi: 10.4049/jimmunol.170.10.5064. [DOI] [PubMed] [Google Scholar]
- 16.Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet. 1997;350(9092):1647–54. [PubMed] [Google Scholar]
- 17.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo N, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838. [PMC free article] [PubMed] [Google Scholar]
- 18.Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J Leukoc Biol. 2003;74:186–96. doi: 10.1189/jlb.0103010. [DOI] [PubMed] [Google Scholar]
- 19.Li Q, Pan PY, Gu P, Xu D, Chen SH. Role of immature myeloid Gr-1+ cells in the development of antitumor immunity. Cancer Res. 2004;64:1130–9. doi: 10.1158/0008-5472.can-03-1715. [DOI] [PubMed] [Google Scholar]
- 20.Danna EA, Sinha P, Gilbert M, Clements VK, Pulaski BA, Ostrand-Rosenberg S. Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease. Cancer Res. 2004;64:2205–11. doi: 10.1158/0008-5472.can-03-2646. [DOI] [PubMed] [Google Scholar]
- 21.Almand B, Resser J, Lindman B, Nadaf S, Clark J, Kwon E, Carbone D, Gabrilovich D. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–66. [PubMed] [Google Scholar]
- 22.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–60. [PubMed] [Google Scholar]
- 23.Hengesbach L, Hoag K. Physiological concentrations of retinoic acid favor myeloid dendritic cell development over granulocyte development in cultures of bone marrow cells from mice. J Nutr. 2004;134:2653–9. doi: 10.1093/jn/134.10.2653. [DOI] [PubMed] [Google Scholar]
- 24.Kuwata T, Wang I, Tamura T, Ponnamperuma R, Levine R, Holmes K, Morse H, De Luca L, Ozato K. Vitamin A deficiency in mice causes a systemic expansion of myeloid cells. Blood. 2000;95:3349–56. [PubMed] [Google Scholar]
- 25.Walkley C, Yuan Y, Chandraratna R, McArthur G. Retinoic acid receptor antagonism in vivo expands the numbers of precursor cells during granulopoiesis. Leukemia. 2002;16:1763–72. doi: 10.1038/sj.leu.2402625. [DOI] [PubMed] [Google Scholar]
- 26.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich DI. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–9. [PubMed] [Google Scholar]
- 27.Yang JC, Sherry RM, Steinberg SM, Topalian SL, Schwartzentruber DJ, Hwu P, Seipp CA, Rogers-Freezer L, Morton KE, White DE, Liewehr DJ, Merino MJ, Rosenberg SA. Randomized study of high-dose and low-dose interleukin-2 in patients with metastatic renal cancer. J Clin Oncol. 2003;21:3127–33. doi: 10.1200/JCO.2003.02.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanvers C, Reinhardt D, Dubbers A, Wagner-Bohn A, Creutzig U, Ritter J, Boos J. Pharmacology of all-trans-retinoic acid in children with acute promyelocytic leukemia. Med Pediatr Oncol. 2003;40:293–301. doi: 10.1002/mpo.10257. [DOI] [PubMed] [Google Scholar]
- 29.Muindi JR, Frankel SR, Huselton C, DeGrazia F, Garland WA, Young CW, Warrell RP., Jr Clinical pharmacology of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res. 1992;52:2138–42. [PubMed] [Google Scholar]
- 30.Wu C, Njar V, Brodie A, Borenstein M, Nnane I. Quantification of a novel retinoic acid metabolism inhibitor, 4-(1H-imidazol-1-yl)retinoic acid (VN/14-1RA) and other retinoids in rat plasma by liquid chromatography with diode-array detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;810:203–8. doi: 10.1016/j.jchromb.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Chang WY, Prins GS, van Breemen RB. Simultaneous determination of all-trans, 9-cis, 13-cis retinoic acid and retinol in rat prostate using liquid chromatography-mass spectrometry. J Mass Spectrom. 2001;36:882–8. doi: 10.1002/jms.189. [DOI] [PubMed] [Google Scholar]
- 32.Adamson PC. All-trans-retinoic acid pharmacology and its impact on the treatment of acute promyelocytic leukemia. The Oncologist. 1996;1:305–14. [PubMed] [Google Scholar]
- 33.Conley BA, Egorin MJ, Sridhara R, Finley R, Hemady R, Wu S, Tait NS, Van Echo DA. Phase I clinical trial of all-trans-retinoic acid with correlation of its pharmacokinetics and pharmacodynamics. Cancer Chemother Pharmacol. 1997;39:291–9. doi: 10.1007/s002800050575. [DOI] [PubMed] [Google Scholar]
- 34.Kusmartsev S, Gabrilovich DI. Role Of Immature Myeloid Cells in Mechanisms of Immune Evasion In Cancer. Cancer Immunol Immunother. 2006;55:237–45. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. doi: 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 36.Dannull J, Kuebler HR, Tseng TT, Zhang A, Su Z, Dahm P, Vieweg J. 2006. Immature myeloid cell (ImC)- mediated immunosuppression in advanced renal cell cancer (RCC) Abstract # 3866. [Google Scholar]
- 37.Breitman TR, Collins SJ, Keene BR. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood. 1981;57:1000–4. [PubMed] [Google Scholar]
- 38.Hittelman WN, Agbor P, Petkovic I, Anderson B, Kantarjian H, Walters R, Koller C, Beran M. Detection of lekemic clone maturation in vivo by premature chromosome condensation. Blood. 1988;72:1950–60. [PubMed] [Google Scholar]
- 39.Castaigne S, Chomienne C, Daniel MT, Ballerini P, Berger R, Fenaux P, Degos L. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I Clinical results Blood. 1990;76:1704–9. [PubMed] [Google Scholar]
- 40.Warrell RP, Frankel SR, Miller WH, Scheinberg DA, Itri LM, Hittelman WN, Vyas R, Andreeff M, Tafuri A, Jakubowski Aea. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid) New Engl J Med. 1991;324:1385–93. doi: 10.1056/NEJM199105163242002. [DOI] [PubMed] [Google Scholar]
- 41.Van Bockstaele DR, Lenjou M, Snoeck H-W, Lardon F, Stryckmans P, Peetermans ME. Direct effects of 13-cis and all-trans retinoic acid on normal bone marrow (BM) progenitors: Comparative study on BM mononuclear cells and on isolated CD34+ BM cells. Annals of Hematology. 1993;66:61–6. doi: 10.1007/BF01695885. [DOI] [PubMed] [Google Scholar]
- 42.Gratas C, Menot ML, Dresch C, Chomienne C. Retinoic acid supports granulocytic but not erythroid differentiation of myeloid progenitors in normal bone marrow cells. Leukemia. 1993;7:1156–62. [PubMed] [Google Scholar]
- 43.Tohda S, Minden MD. Modulation of growth factor receptors on acute myeloblastic leukemia cells by retinoic acid. Jap J Cancer Res. 1994;85:378–83. doi: 10.1111/j.1349-7006.1994.tb02370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. doi: 10.1016/j.gene.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Xu X, Wong W, Goldberg L, Baer S, Wolf J, Ramsdell W, Alberts D, Lippman S, Lotan R. Progressive decreases in nuclear retinoid receptors during skin squamous carcinogenesis. Cancer Res. 2001;61:4306–10. [PubMed] [Google Scholar]
- 46.Chen GQ, Shen ZX, Wu F, Han JY, Miao JM, Zhong HJ, Li XS, Zhao JQ, Zhu J, Fang ZW, Chen SJ, Chen Z, Wang ZY. Pharmacokinetics and efficacy of low-dose all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Leukemia. 1996;10:825–8. [PubMed] [Google Scholar]
- 47.Arpinati M, Chirumbolo G, Urbini B, Perrone G, Rondelli D, Anasetti C. Role of plasmacytoid dendritic cells in immunity and tolerance after allogeneic hematopoietic stem cell transplantation. Transpl Immunol. 2003;11:345–56. doi: 10.1016/S0966-3274(03)00055-8. [DOI] [PubMed] [Google Scholar]
- 48.Zou W, Machelon V, Coulomb-L’Hermin A, Borvak J, Nome F, Isaeva T, Wei S, Krzysiek R, Durand-Gasselin I, Gordon A, Pustilnik T, Curiel DT, Galanaud P, Capron F, Emilie D, Curiel TJ. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat Med. 2001;7:1339–46. doi: 10.1038/nm1201-1339. [DOI] [PubMed] [Google Scholar]
- 49.Treilleux I, Blay JY, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla JP, Bremond A, Goddard S, Pin JJ, Barthelemy-Dubois C, Lebecque S. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res. 2004;10:7466–74. doi: 10.1158/1078-0432.CCR-04-0684. [DOI] [PubMed] [Google Scholar]
- 50.Bella SD, Gennaro M, Vaccari M, Ferraris C, Nicola S, Riva A, Clerici M, Greco M, Villa ML. Altered maturation of peripheral blood dendritic cells in patients with breast cancer. Br J Cancer. 2003;89:1463–72. doi: 10.1038/sj.bjc.6601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffmann TK, Muller-Berghaus J, Ferris RL, Johnson JT, Storkus WJ, Whiteside TL. Alterations in the frequency of dendritic cell subsets in the peripheral circulation of patients with squamous cell carcinomas of the head and neck. Clin Cancer Res. 2002;8:1787–93. [PubMed] [Google Scholar]
- 52.Khatri VP, Baiocchi RA, Bernstein ZP, Caligiuri MA. Immunotherapy with low-dose interleukin-2: rationale for prevention of immune-deficiency-associated cancer. Cancer J Sci Am. 1997;3(Suppl 1):S129–36. [PubMed] [Google Scholar]
- 53.Sosman JA, Hank JA, Moore KH, Borchert A, Schell K, Kohler PC, Goldstein D, Bechhofer R, Storer B, Albertini MR, et al. Prolonged interleukin-2 (IL-2) treatment can augment immune activation without enhancing antitumor activity in renal cell carcinoma. Cancer Invest. 1991;9:35–48. doi: 10.3109/07357909109032798. [DOI] [PubMed] [Google Scholar]