

Abstract
12-Oxophytodienoate reductase (OPR) 3, a homologue of old yellow enzyme (OYE), catalyzes the reduction of 9S,13S-12-oxophytodienoate to the corresponding cyclopentanone, which is subsequently converted to the plant hormone jasmonic acid (JA). JA and JA derivatives, as well as 12-oxophytodienoate and related cyclopentenones, are known to regulate gene expression in plant development and defense. Together with other oxygenated fatty acid derivatives, they form the oxylipin signature in plants, which resembles the pool of prostaglandins in animals. Here, we report the crystal structure of OPR3 from tomato and of two OPR3 mutants. Although the catalytic residues of OPR3 and related OYEs are highly conserved, several characteristic differences can be discerned in the substrate-binding regions, explaining the remarkable substrate stereoselectivity of OPR isozymes. Interestingly, OPR3 crystallized as an extraordinary self-inhibited dimer. Mutagenesis studies and biochemical analysis confirmed a weak dimerization of OPR3 in vitro, which correlated with a loss of enzymatic activity. Based on structural data of OPR3, a putative mechanism for a strong and reversible dimerization of OPR3 in vivo that involves phosphorylation of OPR3 is suggested. This mechanism could contribute to the shaping of the oxylipin signature, which is critical for fine-tuning gene expression in plants.
Keywords: flavoprotein, jasmonic acid biosynthesis, plant defense, oxylipin signature, 12-oxophytodienoic acid
Flavoproteins catalyze a wide variety of essential biochemical reactions, including electron transfer, dehydrogenation, and hydroxylation reactions. Old yellow enzyme (OYE), the first flavin-dependent enzyme identified (1), and homologues of OYE from bacteria, yeast, and plants are able to reduce the C C double bond of α,β-unsaturated carbonyl compounds, an activity that is rather uncommon for flavoenzymes (2, 3). This reaction has been shown to proceed by a ping-pong bi-bi mechanism, during which the FMN cofactor is reduced by NAD(P)H before the substrate is bound and reduced by hydride transfer to the substrate's Cβ (4). Despite extensive efforts, the physiological substrate has been revealed only for one member of the OYE family, the plant enzyme 12-oxophytodienoate reductase (OPR) 3, which catalyzes one step in the biosynthesis of the plant hormone jasmonic acid (JA) (5, 6). JA and JA derivatives act as signaling compounds in the defense response against herbivores and pathogens and are involved in the regulation of various developmental processes, such as fruit ripening, pollen maturation, and senescence (7, 8). JA is synthesized from α-linolenic acid, which is oxidized and cyclized, resulting in the cyclopentenone 9S,13S-12-oxophytodienoate (9S,13S-OPDA). OPR3 reduces 9S,13S-OPDA to the corresponding cyclopentanone, which is converted to JA by repeated β-oxidation (Fig. 1) (9). Several OPR isozymes have been identified in plants, including 3 isoforms in Lycopersicon esculentum, 5 in Arabidopsis thaliana, and 13 in Oryza sativa (10, 11). OPR3 from A. thaliana (AtOPR3) and OPR3 from L. esculentum (LeOPR3) reduce all four stereoisomers of OPDA, whereas the large majority of OPR isoforms seem to resemble LeOPR1 and AtOPR1/2 in their substrate specificity (6, 10). These enzymes efficiently reduce 9R,13R-OPDA but fail to react with the enantiomeric JA precursor 9S,13S-OPDA (12, 13).
C double bond of α,β-unsaturated carbonyl compounds, an activity that is rather uncommon for flavoenzymes (2, 3). This reaction has been shown to proceed by a ping-pong bi-bi mechanism, during which the FMN cofactor is reduced by NAD(P)H before the substrate is bound and reduced by hydride transfer to the substrate's Cβ (4). Despite extensive efforts, the physiological substrate has been revealed only for one member of the OYE family, the plant enzyme 12-oxophytodienoate reductase (OPR) 3, which catalyzes one step in the biosynthesis of the plant hormone jasmonic acid (JA) (5, 6). JA and JA derivatives act as signaling compounds in the defense response against herbivores and pathogens and are involved in the regulation of various developmental processes, such as fruit ripening, pollen maturation, and senescence (7, 8). JA is synthesized from α-linolenic acid, which is oxidized and cyclized, resulting in the cyclopentenone 9S,13S-12-oxophytodienoate (9S,13S-OPDA). OPR3 reduces 9S,13S-OPDA to the corresponding cyclopentanone, which is converted to JA by repeated β-oxidation (Fig. 1) (9). Several OPR isozymes have been identified in plants, including 3 isoforms in Lycopersicon esculentum, 5 in Arabidopsis thaliana, and 13 in Oryza sativa (10, 11). OPR3 from A. thaliana (AtOPR3) and OPR3 from L. esculentum (LeOPR3) reduce all four stereoisomers of OPDA, whereas the large majority of OPR isoforms seem to resemble LeOPR1 and AtOPR1/2 in their substrate specificity (6, 10). These enzymes efficiently reduce 9R,13R-OPDA but fail to react with the enantiomeric JA precursor 9S,13S-OPDA (12, 13).
Fig. 1.

OPR3 catalyzes the reduction of 9S,13S-OPDA to 1S,2S-3-oxo- 2(2′[Z]-pentenyl)-cyclopentane-1-octanoate (OPC-8:0).
In addition to JA, many other biologically active, oxygenated fatty acid derivatives (oxylipins) have been identified in recent years (14, 15). The dynamic mixture of different oxylipins, termed the oxylipin signature, which strikingly resembles the pool of animal prostaglandins in structure and function, is thought to fine-tune gene expression during plant development and defense. One of the oxylipins is OPDA, the substrate of OPR3 that has been shown to be a potent signaling molecule by itself (16, 17). JA deficiency in Arabidopsis caused by a disrupted OPR3 gene results in male sterility due to defective pollen development. Resistance to insects (Bradysia impatients) or fungal pathogens (Alternaria brassicicola) is not impaired, however, suggesting that OPDA or a related cyclopentenone can substitute for JA in plant defense (16). In fact, many defense genes previously known to be regulated by JA were found to be induced by OPDA or related cyclopentenones, whereas other genes are regulated specifically by JA or by OPDA (16, 18).
Here, we present the high-resolution crystal structures of LeOPR3 and of two mutant proteins. The active-site architecture of LeOPR3 provides insight into the remarkable stereoselectivity of the different OPR isoforms and the substrate specificity of the OYE family in general. Unexpectedly, LeOPR3 crystallized as a self-inhibited dimer, making it, to our knowledge, unique among flavoproteins studied so far. Furthermore, the structural data suggest that dimerization, which also occurs in solution, may be regulated by reversible phosphorylation.
Results and Discussion
Overall Structure.
The crystal structures of OPR3 from L. esculentum and of the Glu-291–Lys and the Tyr-364–Phe mutant proteins were determined by molecular replacement and refined at 1.5-, 1.9-, and 1.9-Å resolution, respectively (Table 1). OPR3 exhibits the frequently observed (β/α)8 barrel fold, in which the cylindrical β-sheet composed of eight parallel β-strands is surrounded by eight α-helices (Fig. 2a and b). As in other (β/α)8 structures, all turns at the N terminus of the β-strands are composed of only 3–4 aa, whereas the loops at the C terminus that form the major part of the active site are much more diverse, ranging in length from 3 to 47 aa.
Table 1.
Data collection and refinement statistics
| WT OPR3 | Glu-291-Lys OPR3 | Tyr-364-Phe OPR3 | |
|---|---|---|---|
| Data collection | |||
| Resolution, Å | 1.5 (1.53–1.50) | 1.9 (1.96–1.90) | 1.9 (1.97–1.90) |
| Observed reflections | 364,737 | 107,563 | 161,869 |
| Independent reflections | 119,739 | 57,790 | 59,726 |
| Completeness, % | 95.0 (96.6) | 92.5 (87.3) | 95.2 (91.6) |
| Rmerge, % | 4.6 (40.7) | 6.3 (33.2) | 10.0 (23.2) |
| I/σ(I) | 26.4 (2.0) | 12.3 (2.8) | 11.5 (3.7) |
| Refinement | |||
| Rcryst/Rfree, % | 22.5/25.5 | 20.5/23.5 | 22.6/27.5 |
| No. of atoms | |||
| Protein | 5,790 | 5,620 | 5,649 |
| FMN | 62 | 62 | 62 |
| Ligand | 0 | 24* | 0 |
| No. of water molecules | 493 | 424 | 627 |
| rmsd | |||
| Bonds, Å | 0.009 | 0.006 | 0.012 |
| Angles, ° | 1.40 | 1.31 | 1.55 |
| Bonded Bs, Å2 | 2.03 | 1.65 | 3.55 |
Values in parentheses correspond to the highest-resolution shell.
*A Mes molecule was present in the crystallization buffer.
Fig. 2.

Overall structure of OPR3 from L. esculentum. (a) Stereoview representation of the superimposed Cα traces of OPR3 (green), OPR1 (blue), and OYE (red). (b) Ribbon representation of one OPR3 monomer. The FMN cofactor is depicted as a stick model. (c) Overall structure of the OPR3 dimer. The finger-like loop L6 of protomer A (red) is bound into the substrate-binding pocket of protomer B (yellow) and vice versa. (d) Detailed view of the active site of OPR3 (yellow) blocked by amino acids of loop L6 of the partner protomer (red). (e) Multiple sequence alignment of the insertion (underlined in green) in loop L6 of OPR3-like enzymes. Red circles indicate amino acids that form hydrogen bonds to active-site amino acids.
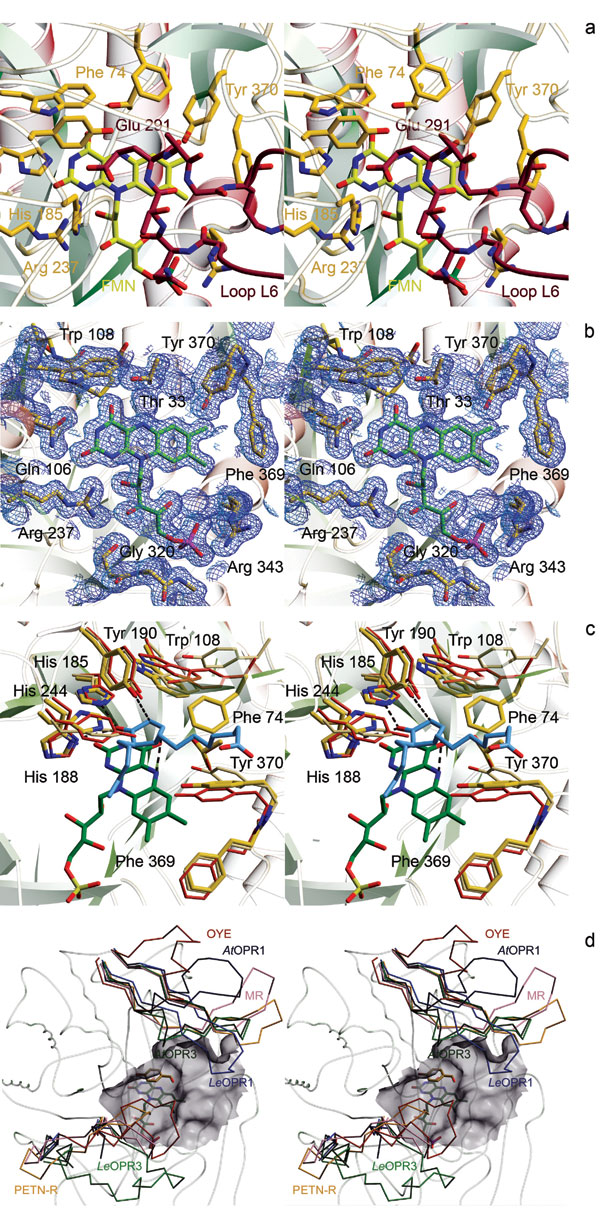
The overall structure of OPR3 resembles the structure of OPR1 and the structures of other members of the OYE flavoprotein family (Fig. 2a) (19, 20). A short hairpin loop at the N terminus and helix αB, which is part of loop L8,‡‡ on the opposite side of the barrel are conserved among proteins of the OYE family. Amino acids of helix αB contribute to a common phosphate-binding motif (21) that anchors the phosphate group of the FMN in the OYE family. Helix αA, found in OPR3 as well as OPR1 and OYE, is located in loop L4 near the active site and helps to polarize the carbonyl group of the substrate. In the OPR subfamily, the fourth extra secondary element consists of two short β-sheets in loop L3 that are arranged in a cross-shaped conformation and form the ceiling of the substrate-binding pocket above the FMN. The corresponding loop of OYE adopts an αβαβ structure and is positioned far above the active site (Fig. 2a). In contrast, loop L6 borders the active site in OYE, whereas it adopts an extended conformation in OPR3 and protrudes like a finger away from the protomer and into the active-site cavity of the molecular neighbor (Fig. 2 c and d). Vice versa, loop L6 of that molecular neighbor is hooked into the protomer's active site, which results in a mutually self-inhibited OPR3 dimer with almost perfect local C2 symmetry in the crystal. The interaction surface encompasses 1,642 Å2 of solvent-accessible surface area per monomer, corresponding to a large interface compared with other protein dimers (22). At the tip of loop L6, Glu-291 binds directly above the FMN cofactor into the substrate-binding pocket, resulting in a complete block of the active site (Fig. 2d).
To study the functional significance of the unexpected OPR3 dimer observed in the crystal, we mutated Glu-291 to lysine, which should prevent binding of loop L6 into the adjacent active site. The Glu-291–Lys mutant crystallized in the same space group as WT OPR3 but with strikingly different cell constants (see Materials and Methods). Intriguingly, the structure solution revealed that the Glu-291–Lys mutant is present as a monomer in the crystal. Its L6 loop is disordered, and none of the specific dimer contacts of WT OPR3 is retained. These data show that dimerization of WT OPR3 in the crystal is actively driven by the mutual binding of the two L6 loops into the two active sites and is not caused by the combined action of dominant crystal contacts distant from the dimeric interface. Interestingly, loop L6 of the OPR3 subgroup (including LeOPR3, AtOPR3, and a few other plant OPRs) contains an insertion of 7 aa compared with the monomeric OPR1 and OPR1-like isozymes (Fig. 2e and Fig. 6a, which is published as supporting information on the PNAS web site). In addition, the amino acids at the tip of loop L6 are highly conserved among OPR3-like enzymes, supporting the assumption that the observed OPR3 dimer forms on purpose.
Active-Site Architecture.
OPR3 binds the FMN cofactor noncovalently at the C terminus of the β-strands that form the barrel. The amino acids that perform side-chain interactions with FMN are 100% conserved within the OYE family, indicating similar chemophysical properties. Key interactions involve hydrogen bonds between the hydroxyl and amide group of Thr33§§ to FMN O (4) and N (5), respectively, and hydrogen bonds between Gln-106 and Arg-237 to N (3) and O (2) of the isoalloxazine ring (Figs. 3a and 6). Above the FMN, the active-site amino acids His-185 and His-190 activate the Cα-Cβ double bond of the substrate by forming strong hydrogen bonds with the carbonyl oxygen of the substrate (20, 23). As shown for OYE, the activated substrate is reduced by hydride transfer from FMNH− N (5) to Cβ and protonated at Cα by Tyr-190 (19, 24). Superposition of the structures of OPR3, OYE, and OPR1 in complex with 9R,13R-OPDA shows that the position and conformation of His-185, His-188, and Tyr-190 are virtually identical (Fig. 3b). This structural correspondence is also found for nearly all structurally characterized members of the OYE family (data not shown) with the exception of His-188, which is replaced by asparagine in several enzymes (19, 20, 25–28). The corresponding atoms Asn Nδ2 and His Nδ1 that bind the carbonyl oxygen overlay perfectly, however, indicating a similar effect on substrate activation and positioning. On the other hand, pronounced differences are found at the opening of the substrate-binding cavity (Fig. 3b). Tyr-246 and Tyr-78 of OPR1 form a narrow entrance of the cavity above the FMN, and the corresponding amino acids of OPR3, whereas the smaller His-244, and Phe-74, which turns away from the active site, give rise to a widening of the cavity in OPR3. These differing geometries most likely account for the high degree of substrate stereoselectivity of OPR1 and the more “relaxed” specificity of OPR3. In sharp contrast to the highly conserved catalytic amino acids, the two gating amino acids at the opening of the cavity vary widely within the OYE family. These differences recur in the loops L3 and L6, which show diverse lengths and conformations (Fig. 3c). Both loops have been shown to contribute to ligand binding in enzymes of the OYE family (19, 20, 25). In conclusion, the structural comparison reveals a pronounced structural conservation of catalytically important amino acids and a high variability of loops that interact with the nonreactive part of the substrates. It seems likely that the as yet unknown physiological substrates of OYE-like enzymes are α,β-unsaturated carbonyl compounds like the OPR3 substrate 9S,13S-OPDA, with their particular chemical composition and stereochemistry depending on the specific enzyme.
Fig. 3.

Active site of OPR3. (a) FMN-binding site. FMN (green) and amino acids that contact the FMN (yellow) are shown as stick models. The 2Fo − Fc electron density map is calculated at 1.5 Å and contoured at 1.0σ. (b) Superposition of the substrate-binding pockets of OPR3 (yellow), OPR1 (red), and OYE (beige). In addition, the FMN of OPR3 (green) and the OPR1 substrate 9R,13R-OPDA (blue) are shown to visualize interactions that are essential for catalysis. (c) Cα backbone of OPR3 and superimposed L3 and L6 loops of members of the OYE family. LeOPR3, light green; AtOPR3, dark green, LeOPR1, blue; AtOPR1, dark blue; OYE, red; morphinone reductase (MR), magenta; pentaerythritol tetranitrate reductase (PETN-R), yellow. In addition, the surface of the substrate-binding pocket of OPR3 and p-hydroxy benzaldehyde complexed to OYE are shown.
Homodimerization in Solution.
To investigate whether the OPR3 dimer observed in the crystal is also present in solution, we biophysically characterized OPR3 in solution. Analytical size-exclusion chromatography of OPR3 showed a shift to shorter elution times with increasing protein concentration, indicating an oligomerization process with rapid conversion of the involved protomer species. In contrast, elution times of the monomeric OPR1 did not depend on the protein concentration. Similarly, dynamic light-scattering experiments revealed a concentration-dependent shift of the hydrodynamic radius of OPR3 corresponding to Mapp ≈ 40 kDa at a concentration of 22 μM and Mapp ≈ 80 kDa at 358 μM. To determine the dissociation constant of the OPR3 dimer, we performed analytical ultracentrifugation experiments. At a protein concentration of 2 μM, OPR3 exhibited a molecular mass of 46.9 ± 2.3 kDa, close to the real molecular mass of 44.6 kDa (Fig. 4a and data not shown). The apparent molecular mass of OPR3 was concentration-dependent. Quantitative analysis of the data yielded a Kd of 30 μM for the OPR3 dimer (Fig. 4a). In contrast, the Glu-291–Lys mutant did not show any concentration-dependent behavior and was present as a monomer at all concentrations (Fig. 4a), which is in agreement with the structural data. In summary, the results from size-exclusion chromatography, dynamic light scattering, and analytical ultracentrifugation point to a rapid monomer-dimer equilibrium with a high dissociation constant in vitro.
Fig. 4.

Biochemical analysis of the OPR3 dimer. (a) Sedimentation equilibrium of WT OPR3 (filled circles) and the Glu-291–Lys mutant (open circles) was analyzed at a protein concentration of 1–34 μM in the presence of 50 μM FMN. For WT OPR3, the apparent molecular masses were fitted to a monomer dimer equilibrium, yielding a dissociation constant (Kd) of 30 μM. (b) Enzyme-monitored turnover. WT OPR3 (solid line) and Glu-291–Lys OPR3 (dashed line) at 86 μM were mixed in the stopped-flow instrument with 1.4 mM NADPH and 1.5 mM trans-hex-2-enal (100 mM potassium phosphate, pH 7; 25°C). In both cases, rapid reduction by NADPH was observed (time = 0 s), followed by a steady-state phase. After exhaustion of NADPH, the enzyme returned to its oxidized state, as indicated by the recovery of absorbance at 450 nm.
Enzymatic Activity of OPR3.
A functional role of the dimerization of OPR3 observed in the crystal structure is also supported by kinetic analysis of OPR3 activity. In steady-state experiments using trans-hex-2-enal as a model substrate, a kcat of 14 s−1 was determined at saturating NADPH concentrations. Typically, these experiments were performed at a nanomolar range of OPR3 (10–150 nM). However, when the oxidative half-reaction was analyzed in a stopped-flow spectrophotometer, a much lower rate of reoxidation by trans-hex-2-enal (kox = 0.3 s−1) was found and was independent of substrate concentration. Because the concentration of OPR3 used in the stopped-flow experiments was in the micromolar range (15–30 μM OPR3), the apparent inconsistency of the rate of oxidation and the turnover rate can be rationalized by partial dimerization of OPR3 at the higher concentrations used in the stopped-flow experiments. This interpretation is also consistent with the observed independence of the reoxidation rate from substrate concentration, which suggests that a step other than substrate binding limits reoxidation. To gain further insight into the influence of dimerization on catalysis, WT OPR3 and the Glu-291–Lys mutant were compared in a steady-state experiment at protein concentrations near the estimated dissociation constant of the OPR3 dimer (see above). In these experiments, turnover is monitored by observing the redox state of the FMN cofactor at 450 nm (“enzyme-monitored turnover”). Under these conditions, the Glu-291–Lys mutant exhibited a 6-fold faster turnover than WT OPR3, clearly indicating that dimerization reduces the activity of OPR3 (Fig. 4b).
Regulation of OPR3 Activity by Dimerization.
The structural and biochemical data presented in this study show that LeOPR3 from tomato can exist as a self-inhibited dimer. In this context, it seems surprising that the recently reported structure of AtOPR3 does not show an obvious dimer (28). Yet, the two “monomers” observed in the asymmetric unit of AtOPR3 crystals can be superimposed with the corresponding protomers of the LeOPR3 dimer, suggesting a similar dimeric structure for AtOPR3. Although loop L6 was too flexible to be traced completely in the electron density of AtOPR3, it clearly protrudes toward the active site of the molecular neighbor in a similar fashion as in LeOPR3. This structural analogy and the observation that the inhibitory extension of loop L6 is present and highly conserved only in the OPR3 subfamily (Fig. 2e) point to a general mechanism of self-inhibition for OPR3.
Self-inhibition is a common regulatory mechanism used by various proteins, such as proteinases, kinases, and receptors. Many cases of intramolecular self-inhibition (autoinhibition) are known (29, 30), but examples of intermolecular inhibition by homodimerization have been reported as well (31–36). Proteolytic cleavage, phosphorylation, ligand binding, and even light-induced conformational changes represent different possibilities to initiate or abrogate self-inhibition. We have identified OPR3 as a self-inhibited dimer that is weakly associated in vitro, a feature that is, to the best of our knowledge, so far unique among flavoproteins. Intriguingly, the structure of OPR3 also suggests a hypothetical mechanism by which a strong dimerization may be achieved in vivo and by which this dimerization can be regulated. In the dimer interface of OPR3, a sulfate ion is bound, which forms hydrogen bonds with two arginines of protomer A and one arginine of loop L6 of the partner protomer B (Fig. 5). Apart from directly cross-linking the two OPR3 protomers, the sulfate ion reorients two of the arginine side chains [Arg-366(A) and Arg-294(B)], which enables them to further stabilize the dimer by the formation of an extensive hydrogen-bond network. Interestingly, the sulfate ion is located in close proximity to the hydroxyl group of Tyr-364, positioned perfectly to mimic the phosphate group of a Tyr-364 phosphorylated in vivo (Fig. 5). To elucidate the relevance of sulfate binding, and possibly phosphorylation, for dimerization, we determined the crystal structure of the Tyr-364–Phe mutant, in which the strong hydrogen bond between Tyr-364 and the sulfate ion cannot be formed. Like the Glu-291–Lys mutant, Tyr-364–Phe OPR3 crystallizes as a monomer with none of the dimer interactions retained, confirming the suspected importance of the sulfate-mediated interactions for dimerization.
Fig. 5.

Central section of the dimer interface of protomer A (red; amino acids are marked with an asterisk) and protomer B (yellow) of OPR3. In the crystal structure, a sulfate ion mediates various intermolecular contacts. The position of the sulfate ion would fit very well with that of the phosphate group of a phosphorylated Tyr-364 (blue).
Reversible protein phosphorylation is known to be involved in the regulation of wound signaling in plants and has even been reported to influence JA levels, as well as JA-responsive gene expression (37–41). A role of posttranslational modifications in the regulation of the JA biosynthesis itself is supported by the observation that JA begins to accumulate almost instantly after wounding, whereas maximum transcript levels of induced genes coding for enzymes of JA biosynthesis are reached only after 2 h (10, 42, 43). These findings indicate that JA is initially produced by JA pathway enzymes that are constitutively expressed at low levels, with the formation of JA being limited by the availability of intermediates and/or by the posttranslationally regulated activity of the enzymes involved (42, 44). Our data support the latter assumption by demonstrating that OPR3 activity is regulated by self-inhibitory dimerization, which might be controlled by reversible phosphorylation of Tyr-364 in vivo.
Materials and Methods
Expression and Purification of OPR3 and Mutant Proteins.
A construct of OPR3 from L. esculentum carrying an N-terminal His tag cloned into pET21-d (10) was expressed recombinantly in Escherichia coli strain BL21-CP(DE3)-RIL. OPR3 was purified by Ni-nitrilotriacetic acid affinity chromatography (Qiagen, Hilden, Germany) followed by size-exclusion chromatography using a Superdex 75 column (GE Healthcare, Freiburg, Germany) equilibrated with 25 mM Tris·HCl (pH 7.5) and 150 mM NaCl. All mutants of OPR3 were constructed by using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, Heidelberg, Germany) and verified by sequencing. They were purified and assayed under the same conditions as the WT protein.
Crystallization and Structure Solution.
Crystallization was carried out at 20°C by using the sitting drop vapor-diffusion method. Crystals of OPR3 were grown by mixing 1 μl of protein (15 mg/ml) with 1 μl of 100 mM Mes-Tris (pH 6.5), 16% PEG 8000, and 50 mM ammonium sulfate. Monoclinic crystals with dimensions of 100 × 100 × 350 μm3 appeared after 2 days. For cryogenic measurements, crystals were transferred to the mother liquor supplied with 20% 2-methyl-2,4-pentanediol and rapidly frozen in a 100-K stream of nitrogen gas. PEG-ion screening resulted in the optimized crystallization condition for Glu-291–Lys OPR3 (100 mM Tris·HCl, pH 7.5/14% PEG 8000/50 mM ammonium tartrate) and for Tyr-364–Phe OPR3 (100 mM Hepes-NaOH, pH 7.5/10% PEG 8000/200 mM calcium acetate).
High-resolution data sets of OPR3 and the mutant proteins were collected at the European Synchrotron Radiation Facility (Grenoble, France) (beamline ID14-4, λ = 0.9393 Å) by using a Q4R CCD detector (ADSC, San Diego, CA). Data were integrated by using DENZO and scaled with SCALEPACK (45). All crystals exhibited space group P21 with two molecules in the asymmetric unit (OPR3: a = 57.9 Å, b = 89.7 Å, c = 81.4 Å, β = 109.5°; Glu-291–Lys OPR3: a = 49.2 Å, b = 92.2 Å, c = 90.1 Å, β = 99.4°; Tyr-364–Phe OPR3: a = 49.2 Å, b = 93.4 Å, c = 89.5 Å, β = 97.7°). The structure of OPR3 was determined by molecular replacement by using the program MOLREP (46) and OPR1 from L. esculentum (20) as a search model. Electron density maps based on the coefficients 2Fo − Fc and 3Fo − 2Fc were used to build atomic models in O (47). Refinement and model rebuilding proceeded smoothly by means of rigid-body, positional, and B factor optimization in CNS (48) and converged at an R/Rfree of 22.5%/25.5%. The structure was checked by using simulated annealing composite omit maps. Twenty amino acids at the N terminus and 12 aa at the C terminus were not visible in the electron density. The refined OPR3 structure was used as a search model to elucidate the structures of the two mutants. Data collection and refinement parameters are summarized in Table 1. Figures were prepared by using the programs MOLSCRIPT (49), BOBSCRIPT (50), GRASP (51), and Raster3D (52).
Biophysical Analysis.
Analytical size-exclusion chromatography.
Analytical runs were performed with a SMART system by using a Superdex S200 PC3.2 column (GE Healthcare) equilibrated with 25 mM Tris·HCl (pH 7.5)/0.15 M NaCl.
Dynamic light scattering.
Protein samples were filtered and centrifuged at 16,000 × g for 10 min. Dynamic light scattering was analyzed in 25 mM Tris·HCl (pH 7.5)/0.15 M NaCl by using a DynaPro MS DLS instrument (Protein Solutions, High Wycombe, U.K.). Hydrodynamic radii were calculated with the software DYNALS (Protein Solutions), and molecular weights were approximated by using the protein standard curve of the DYNAMICS software (Protein Solutions).
Analytical ultracentrifugation.
OPR3 was analyzed at initial protein concentrations of 0.1–1.5 mg/ml in 50 mM phosphate (pH 8.0)/0.15 M ammonium sulfate/1 mM EDTA/50 μM FMN by using an Optima XL-A centrifuge (Beckman, Fullerton, CA) and an An50Ti rotor. Sedimentation equilibrium measurements (absorption at 280 and 450 nm) were carried out in six channel cells at 10,000 rpm and 20°C. The apparent molecular masses were determined by using the software provided by Beckman Instruments (Palo Alto, CA). From these data, the monomer dimer equilibrium was calculated as described in ref. 53.
Kinetic Measurements.
Steady-state kinetic assays were carried out as described in ref. 54. Rapid reaction kinetics were performed by using a stopped-flow spectrophotometer equipped with a thermostated 1-cm path length cell and a diode array detector (SI Spectroscopy Instruments, Gutach, Germany). Analyses of the data were performed by using the program Specfit/32 (Spectrum Software Associates, Marlborough, MA). Enzyme-monitored turnover experiments were carried out at a 43 μM final concentration of OPR3 and the Glu-291–Lys mutant. The enzyme solution was mixed in the stopped-flow instrument with 0.7 mM NADPH and 0.75 mM trans-hex-2-enal, monitoring turnover at 450 nm.
Supplementary Material
Acknowledgments
P.M. thanks Eva Pointner for skillful technical assistance and Dr. Mario Klimacek for help with the stopped-flow experiments. Work at the Research Institute of Molecular Pathology was supported by Boehringer Ingelheim.
Abbreviations
- OPDA
12-oxophytodienoate
- OPR
OPDA reductase
- OYE
old yellow enzyme
- JA
jasmonic acid
- AtOPR
OPR from Arabidopsis thaliana
- LeOPR
OPR from Lycopersicon esculentum.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2HSA, 2HS6, and 2HS8).
Loops are numbered according to the preceding β-strand (e.g., L8 follows strand β8).
Numbering refers to OPR3 from L. esculentum.
References
- 1.Warburg O, Christian W. Biochem Z. 1935;266:377–411. [Google Scholar]
- 2.Massey V. Biochem Soc Trans. 2000;28:283–296. [PubMed] [Google Scholar]
- 3.Williams RE, Bruce NC. Microbiology. 2002;148:1607–1614. doi: 10.1099/00221287-148-6-1607. [DOI] [PubMed] [Google Scholar]
- 4.Karplus PA, Fox KM, Massey V. FASEB J. 1995;9:1518–1526. doi: 10.1096/fasebj.9.15.8529830. [DOI] [PubMed] [Google Scholar]
- 5.Stintzi A, Browse J. Proc Natl Acad Sci USA. 2000;97:10625–10630. doi: 10.1073/pnas.190264497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaller F, Biesgen C, Müssig C, Altmann T, Weiler EW. Planta. 2000;210:979–984. doi: 10.1007/s004250050706. [DOI] [PubMed] [Google Scholar]
- 7.Schaller F, Schaller A, Stintzi A. J Plant Growth Regul. 2004;23:179–199. [Google Scholar]
- 8.Wasternack C, Stenzel I, Hause B, Hause G, Kutter C, Maucher H, Neumerkel J, Feussner I, Miersch O. J Plant Physiol. 2006;163:297–306. doi: 10.1016/j.jplph.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Vick BA, Zimmerman DC. Plant Physiol. 1984;75:458–461. doi: 10.1104/pp.75.2.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strassner J, Schaller F, Frick UB, Howe GA, Weiler EW, Amrhein N, Macheroux P, Schaller A. Plant J. 2002;32:585–601. doi: 10.1046/j.1365-313x.2002.01449.x. [DOI] [PubMed] [Google Scholar]
- 11.Sobajima H, Takeda M, Sugimori M, Kobashi N, Kiribuchi K, Cho EM, Akimoto C, Yamaguchi T, Minami E, Shibuya N, et al. Planta. 2003;216:692–698. doi: 10.1007/s00425-002-0909-z. [DOI] [PubMed] [Google Scholar]
- 12.Schaller F, Hennig P, Weiler EW. Plant Physiol. 1998;118:1345–1351. doi: 10.1104/pp.118.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biesgen C, Weiler EW. Planta. 1999;208:155–165. doi: 10.1007/s004250050545. [DOI] [PubMed] [Google Scholar]
- 14.Farmer EE, Almeras E, Krishnamurthy V. Curr Opin Plant Biol. 2003;6:372–378. doi: 10.1016/s1369-5266(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 15.Howe GA, Schilmiller AL. Curr Opin Plant Biol. 2002;5:230–236. doi: 10.1016/s1369-5266(02)00250-9. [DOI] [PubMed] [Google Scholar]
- 16.Stintzi A, Weber H, Reymond P, Browse J, Farmer EE. Proc Natl Acad Sci USA. 2001;98:12837–12842. doi: 10.1073/pnas.211311098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mithofer A, Maitrejean M, Boland W. J Plant Growth Regul. 2004;23:170–178. [Google Scholar]
- 18.Taki N, Sasaki-Sekimoto Y, Obayashi T, Kikuta A, Kobayashi K, Ainai T, Yagi K, Sakurai N, Suzuki H, Masuda T, et al. Plant Physiol. 2005;139:1268–1283. doi: 10.1104/pp.105.067058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fox KM, Karplus PA. Structure (London) 1994;2:1089–1105. [PubMed] [Google Scholar]
- 20.Breithaupt C, Strassner J, Breitinger U, Huber R, Macheroux P, Schaller A, Clausen T. Structure (London) 2001;9:419–429. doi: 10.1016/s0969-2126(01)00602-5. [DOI] [PubMed] [Google Scholar]
- 21.Wilmanns M, Hyde CC, Davies DR, Kirschner K, Jansonius JN. Biochemistry. 1991;30:9161–9169. doi: 10.1021/bi00102a006. [DOI] [PubMed] [Google Scholar]
- 22.Wodak SJ, Janin J. Adv Protein Chem. 2002;61:9–73. doi: 10.1016/s0065-3233(02)61001-0. [DOI] [PubMed] [Google Scholar]
- 23.Brown BJ, Deng Z, Karplus PA, Massey V. J Biol Chem. 1998;273:32753–32762. doi: 10.1074/jbc.273.49.32753. [DOI] [PubMed] [Google Scholar]
- 24.Kohli RM, Massey V. J Biol Chem. 1998;273:32763–32770. doi: 10.1074/jbc.273.49.32763. [DOI] [PubMed] [Google Scholar]
- 25.Barna TM, Khan H, Bruce NC, Barsukov I, Scrutton NS, Moody PCE. J Mol Biol. 2001;310:433–447. doi: 10.1006/jmbi.2001.4779. [DOI] [PubMed] [Google Scholar]
- 26.Barna T, Messiha HL, Petosa C, Bruce NC, Scrutton NS, Moody PC. J Biol Chem. 2002;277:30976–30983. doi: 10.1074/jbc.M202846200. [DOI] [PubMed] [Google Scholar]
- 27.Fox BG, Malone TE, Johnson KA, Madson SE, Aceti D, Bingman CA, Blommel PG, Buchan B, Burns B, Cao J, et al. Proteins. 2005;61:206–208. doi: 10.1002/prot.20533. [DOI] [PubMed] [Google Scholar]
- 28.Malone TE, Madson SE, Wrobel RL, Jeon WB, Rosenberg NS, Johnson KA, Bingman CA, Smith DW, Phillips GN, Markley JL, et al. Proteins Struct Funct Bioinf. 2005;58:243–245. doi: 10.1002/prot.20162. [DOI] [PubMed] [Google Scholar]
- 29.Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann V, Clarkson B, Superti-Furga G, Kuriyan J. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 30.Pufall MA, Graves BJ. Annu Rev Cell Dev Biol. 2002;18:421–462. doi: 10.1146/annurev.cellbio.18.031502.133614. [DOI] [PubMed] [Google Scholar]
- 31.Stoker AW. J Endocrinol. 2005;185:19–33. doi: 10.1677/joe.1.06069. [DOI] [PubMed] [Google Scholar]
- 32.Liu GZ, Bafico A, Harris VK, Aaronson SA. Mol Cell Biol. 2003;23:5825–5835. doi: 10.1128/MCB.23.16.5825-5835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majeti R, Bilwes AM, Noel JP, Hunter T, Weiss A. Science. 1998;279:88–91. doi: 10.1126/science.279.5347.88. [DOI] [PubMed] [Google Scholar]
- 34.Merz GS, Benedikz E, Schwenk V, Johansen TE, Vogel LK, Rushbrook JI, Wisniewski HM. J Cell Physiol. 1997;173:423–432. doi: 10.1002/(SICI)1097-4652(199712)173:3<423::AID-JCP15>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 35.Bilwes AM, denHertog J, Hunter T, Noel JP. Nature. 1996;382:555–559. doi: 10.1038/382555a0. [DOI] [PubMed] [Google Scholar]
- 36.Wickner S, Hoskins J, McKenney K. Proc Natl Acad Sci USA. 1991;88:7903–7907. doi: 10.1073/pnas.88.18.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stratmann JW, Ryan CA. Proc Natl Acad Sci USA. 1997;94:11085–11089. doi: 10.1073/pnas.94.20.11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rojo E, Titarenko E, Leon J, Berger S, Vancanneyt G, Sanchez-Serrano JJ. Plant J. 1998;13:153–165. doi: 10.1046/j.1365-313x.1998.00020.x. [DOI] [PubMed] [Google Scholar]
- 39.Seo S, Okamoto M, Seto H, Ishizuka K, Sano H, Ohashi Y. Science. 1995;270:1988–1992. doi: 10.1126/science.270.5244.1988. [DOI] [PubMed] [Google Scholar]
- 40.Seo S, Sano H, Ohashi Y. Plant Cell. 1999;11:289–298. doi: 10.1105/tpc.11.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaller A, Oecking C. Plant Cell. 1999;11:263–272. doi: 10.1105/tpc.11.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ziegler J, Keinanen M, Baldwin IT. Phytochemistry. 2001;58:729–738. doi: 10.1016/s0031-9422(01)00284-9. [DOI] [PubMed] [Google Scholar]
- 43.Ryan CA. Biochim Biophys Acta. 2000;1477:112–121. doi: 10.1016/s0167-4838(99)00269-1. [DOI] [PubMed] [Google Scholar]
- 44.Stenzel I, Hause B, Maucher H, Pitzschke A, Miersch O, Ziegler J, Ryan CA, Wasternack C. Plant J. 2003;33:577–589. doi: 10.1046/j.1365-313x.2003.01647.x. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Macromolecular Crystallography. Vol 276. San Diego: Elsevier Academic; 1997. pp. 307–326. Part A. [Google Scholar]
- 46.Vagin A, Teplyakov A. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 47.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 48.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 49.Kraulis PJ. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 50.Esnouf RM. J Mol Graphics Model. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 51.Nicholls A, Sharp KA, Honig B. Proteins Struct Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 52.Merritt EA, Murphy MEP. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 53.Bar D, Golbik R, Hubner G, Lilie H, Muller EC, Naumann M, Otto A, Reuter R, Breunig KD, Kriegel TM. J Biol Chem. 2003;278:39280–39286. doi: 10.1074/jbc.M305706200. [DOI] [PubMed] [Google Scholar]
- 54.Strassner J, Fürholz A, Macheroux P, Amrhein N, Schaller A. J Biol Chem. 1999;274:35067–35073. doi: 10.1074/jbc.274.49.35067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}