

Abstract
Herpes simplex virus type 1 ocular infection elicits a potent inflammatory response including the production of the chemokines, CXCL9 and CXCL10, in mice. Since HSV-1 nucleic acid is recognized by pattern receptors including toll-like receptor (TLR) 9, we tested the hypothesis that TLR9 is necessary for the early augmentation of CXCL10 following HSV-1 infection. Similar to wild type controls, TLR9 deficient mice constitutively expressed CXCL10 in the cornea. Following infection or stimulation with the deoxycytidylate-phosphate-deoxyguanylate (CpG) motif, CXCL10 levels were significantly elevated in the cornea of wild type but not TLR9 or type I interferon receptor deficient mice. The reduced CXCL10 response in the cornea of TLR deficient mice was correlative with an increase in virus shedding and a reduction in neutrophil infiltration. This is the first report that shows enhanced CXCL10 expression following neurotropic viral replication requires both intact TLR 9 and type I interferon signaling pathways.
Keywords: Herpes Simplex Virus 1, toll like receptor 9, type I interferon receptor, chemokines, and CXCL10
1. Introduction
Herpes simplex virus 1 (HSV-1) is a highly successful neurotropic virus. Topical corneal inoculation with HSV-1 leads to a period of viral replication in corneal epithelium and stroma. HSV-1 further spreads to the nervous system by entering sensory nerve terminals in the basal aspect of the corneal epithelium. From there, the virus undergoes retrograde transport to the trigeminal ganglia (TG)(Millhouse and Wigdahl, 2000; Rodahl and Haarr, 2000; Valyi-Nagy et al., 1991). Depending upon the effectiveness of innate immunity to control viral replication in the TG, the virus may further spread to the central nervous system. Effective innate immunity requires an inflammatory response that clears virus but does not harm the host by causing permanent damage to terminally differentiated cells of the nervous system.
Innate immune recognition of HSV-1 occurs via the recognition of viral nucleic acids by pattern recognition molecules, such as toll-like receptor 9 (TLR 9). TLR 9 recognizes deoxycytidylate-phosphate-deoxyguanylate (CpG) motifs, which are abundant in viral DNA (Hemmi et al., 2000). In response to recognition of viral infection, TLRs initiate signaling that elicit anti-viral immune responses including: induction of inflammatory cytokines, including interferon (IFN), production of chemokines, and induction of the host adaptive immune response. Upon recognition of CpG motifs, intracellular signal transduction can lead to the activation of NF-κB or ATFs-c-Jun (Kawai and Akira, 2006), which act in the nucleus to control transcription of inflammatory cytokines (Bonizzi and Karin, 2004). Alternatively, intracellular signaling can lead to transcriptional control of IFNs, potent endogenous anti-viral cytokines (Kawai and Akira, 2006). Specifically related to the current report, HSV-1 has been shown to induce IFN-α production via TLR 9 pathways (Hochrein et al., 2004; Krug et al., 2004).
Corneal epithelial cells express TLR2, TLR4, and TLR9 (Johnson et al., 2005). Following HSV-1 infection of mouse cornea, CXCL10 expression is restricted to the epithelial layer co-localizing with HSV-1 antigen expression during the first 72 hr p.i. (Carr, unpublished observation). Of the chemokines surveyed within 36–48 hr following HSV-1 infection including CCL2, CCL3, CCL5, CXCL1, CXCL9, and CXCL10, CCL2 and CXCL10 are the most prominently displayed chemokines expressed within the cornea (Carr and Tomasek, 2006). In the current report our objective was to determine if CXCL10 expression is activated thru TLR 9 and the importance of type I IFN in the induction or augmentation of this process.
2. Materials and Methods
2.1 Virus and cells
African green monkey kidney fibroblasts (Vero cells, ATCC CCL-81, American Type Culture Collection, Manassas, VA) were propagated in RPMI-1640 medium supplemented with 10% FBS, gentamicin (Invitrogen, Calsbad, CA.) and antibiotic-antimycotic solution (Invitrogen) at 37o C, 5% CO2, and 95% humidity. HSV-1 stocks (McKrae strain, originally obtained from Bryan Gebhardt, LSU Health Sciences Center, New Orleans, LA) were propagated in Vero cells. Stocks were stored at –80° C at a concentration of 1x109 plaque forming units (pfu) and diluted in RPMI-1640 immediately before use to infect mice.
2.2 Mice
Male and female C57BL/6 mice were obtained from The Jackson Labs (Bar Harbor, ME). TLR9 deficient (TLR9 KO) (Hemmi et al., 2000) and type I IFN receptor deficient mice (IFN α/β R KO) (Garvey et al., 2005) mice on a C57BL/6 background were used to establish a colony in the vivarium at the Dean A. McGee Eye Institute (Oklahoma City, OK). The cornea of anesthetized age (5–8 weeks old) and sex-matched (male and female) mice were scarified using a 25-gauge needle, and 1,000 plaque forming units (pfu) of HSV-1 was applied in a volume of 3.3 μl in RPMI-1640. At the indicated time p.i., the mice were euthanized and perfused with PBS (pH 7.4). The corneas, TG, and brain stem (BS) were removed and placed in RPMI 1640 medium for determination of virus quantity by plaque assay. The tissue was homogenized in 500 μl of medium and the supernatant was clarified (10,000xg, 1 min) and used immediately for the detection of virus (by plaque assay). In addition, mouse eyes were swabbed at times p.i. (1–3 days) and assayed for infectious virus by plaque assay. In a second group of mice, the corneas of uninfected or infected TLR9 KO or C57BL/6 mice or mice treated with 20 μg of the CpG (ODN1826, 5’-TCCATCACGTTCCTGACGTT-3’) or CpG control (5’-TCCATCAGCTTCCTGAGCTT-3’) oligonucleotide (InvivoGen, San Diego, CA) in 3 μl medium were removed prior to or 36–42 hr p.i., weighed, and placed in PBS containing a protease inhibitor cocktail set I (Calbiochem, San Diego, CA). Following homogenization on ice, the supernatant was centrifuged (10,000xg, 1 min) and subsequently assessed for chemokine content by ELISA or suspension array. In a third group of mice, the vaginas of uninfected or HSV-2-infected mice were removed 36 hr p.i., weighed and processed in an identical manner as the cornea samples for CXCL9 and CXCL10 content. All procedures were approved by The University of Oklahoma Health Sciences Center and Dean A McGee Eye Institute animal use committees.
2.3 Viral plaque assay
Clarified supernatant from homogenized tissue was serially diluted and placed (100 μl) onto Vero cell monolayers in 96-well cultured plates. After a 1-hr incubation at 37o C in 5% CO2, and 95% humidity, the supernatants were discarded, and 75 μl of an overlay solution (0.5% methylcellulose in RPMI-1640 supplemented with 10% FBS, gentamicin, and antibiotic/anti-mycotic solution) was added on top of the monolayer. The cultures were incubated at 37o C in 5% CO2, and 95% humidity for 28–32 h to observe plaque formation, and the amount of infectious virus was reported as pfu/tissue. Supernatants obtained from uninfected mice rendered no detectable plaques.
2.4 Measurement of Cytokines and Chemokines
The detection of CXCL9 and CXCL10 was performed using commercially available kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. The sensitivity for the detection of the chemokines/cytokines ranged from 3.0–4.0 pg/tissue. Each sample was assayed in duplicate along with a standard provided in the kit to generate a standard curve used to determine the unknown amount of targeted cytokine/chemokine. Standard curves did not fall below a correlation coefficient of .9930. The amount of cytokine/chemokine measured was normalized to the total wet weight of each tissue under study and is expressed as pg/mg ± SEM. The detection of CXCL1, CCL2, CCL3, CCL5, IL-1β, IL-6, IFN-γ, and TNF-α in the corneas or vaginal tissue from C57Bl/6, TLR9 deficient, or IFN αβR KO mice was conducted using a Bio-Plex™ suspension array system (Bio-Rad, Hercules, CA). Each sample was conducted in duplicate following the manufacturer’s suggestions using standards provided in the kit.
2.5 Real time PCR for oligoadenylate synthetases
Total RNA was isolated from uninfected or HSV-1-infected corneas 24 h p.i. in Ultraspect RNA isolation reagent (Biotecx Inc., Houston, TX) according to the manufacturer’s instructions. First-strand cDNA was synthesized using avian myeloblastosis virus reverse transcriptase (Promega, Madison, WI) and an oligo (dT) primer (Promega). Real time PCR and determination of relative levels of oligoadenylate synthetases (OAS) was carried out as described (Härle et al., 2002).
2.6 Whole-mount preparation
Corneas were removed and fixed for 20 minutes in 4% paraformaldehyde at 4° C. Following fixation, the corneas were washed fives times with 1% Triton-X100 in 1X PBS for 15 minutes each and blocked by soaking each cornea in 100ul of PBS-BGEN(3% BSA, .25% Gelatin, 5mM EDTA, .025% Nonidet-P40 in 1X PBS) with 5ug FcBlock (BD Biosciences). After a 2 hour incubation, 5ul of normal rat serum (Jackson ImmunoResearch, West Grove, PA) was added to the blocking buffer and the corneas were incubated overnight at 4° C. The corneas were then stained by adding an additional 2ul of either FITC-conjugated rat anti-mouse neutrophil antibody (clone 7/4, Serotec, Raleigh, NC) or FITC-conjugated rat anti-mouse F4/80 antibody (clone CI:A3-1, BD Biosciences) to the blocking buffer, incubating overnight, then washing five times for a minimum of 15 minutes each in 0.1% Triton-X100 in 1X PBS. Nuclei were stained using Vectashield mounting medium with DAPI (Vector Labs, Burlingame, CA).
2.7 Confocal microscopy
Corneas were imaged using an Olympus IX81-FV500 epifluorescence/confocal laser-scanning microscope with a UApo 40X water immersion lens. Samples were excited with 405, 488, and 546 nm wavelength lasers. Scanning images were taken with a step size of 1.2 μm in the z axis and image analysis was performed using Fluoview software (Olympus). For analysis of labeled cells in the cornea, 20X-40X fields were randomly chosen at either the periphery or center of the cornea. MCA771F-stained (neutrophil) and F4/80-stained (macrophage) cells were enumerated by masked observers from 6 corneas/group. Uninfected mice presented with no detectable MCA771F-stained cells but on occasion, an F4/80-stained cell was detectable.
2.8 Statistics
One-way ANOVA and Tukey’s t test were used to determine significance (p<.05) comparing the C57BL/6 wild type (WT) mice to the TLR9 or IFN αβR KO mice using the GBSTAT program (Dynamic Microsystems, Silver Spring, MD).
3. Results
3.1.TLR 9 deficient mice shed more HSV-1 in tears than wild type mice
HSV-1 has been shown to stimulate IFN-α production through both TLR 9 dependent and independent pathways (Hochrein et al., 2004). To determine susceptibility of TLR 9 mice to HSV-1 ocular infection, viral titers were determined in the tear film and infected tissue at times p.i. Whereas there were no differences in viral titers recovered from the infected cornea, TG, or BS of TLR9 KO mice compared to WT controls at day 3 or 7 p.i., TLR9 KO mice shed significantly more HSV-1 in tears (Fig. 1 ).
Figure 1.
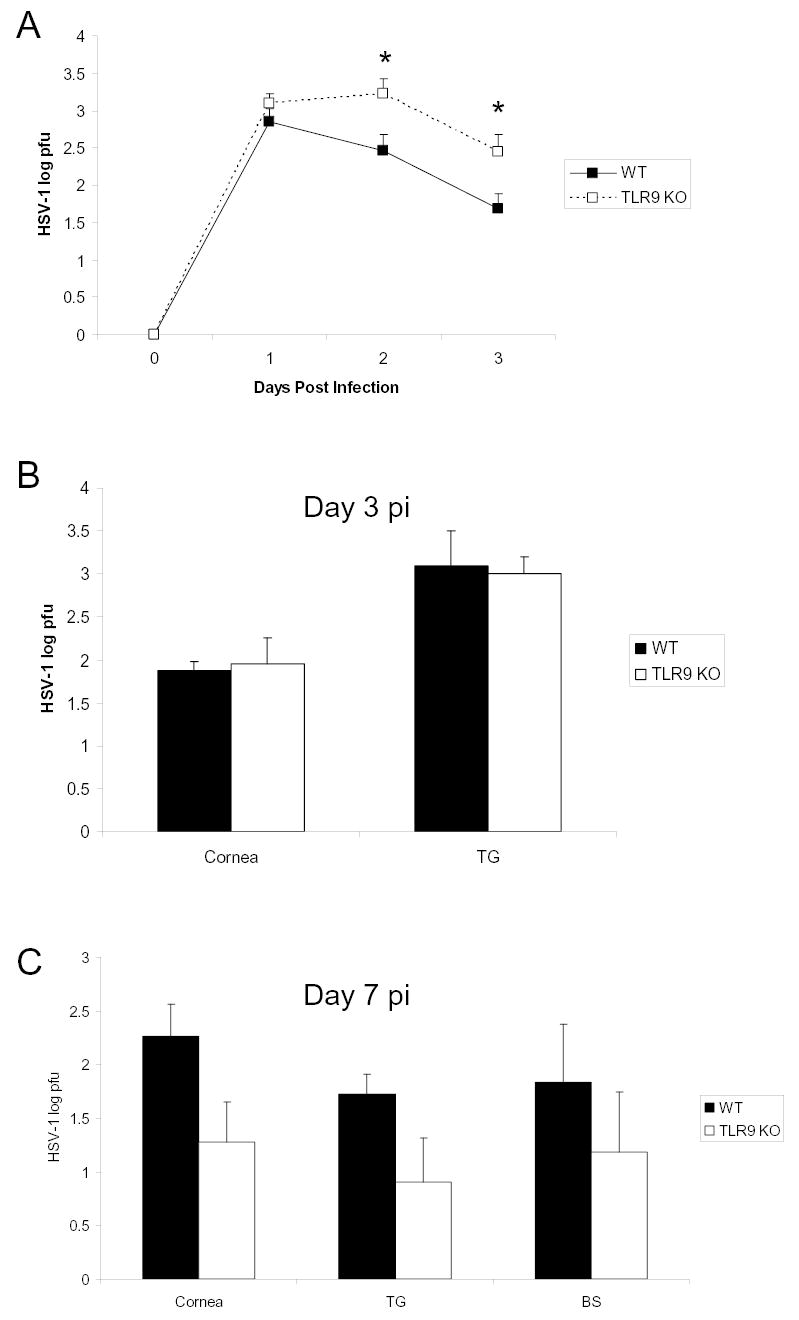
TLR 9 knockout mice shed more HSV-1 in tears than wild type mice. C57BL/6 (WT) and TLR 9 knockout (TLR9 KO) mice were infected with 1,000 pfu HSV-1/scarified cornea. Panel A represents infectious HSV-1 in tears at indicated days post infection. Panels B and C, respectively, depict the levels of infectious HSV-1 recovered in cornea, trigeminal ganglia (TG), and brainstem (BS) at days 3 and 7 post infection. Data in panels A-C represent cumulative means ± SEM from 2–3 separate experiments (n = 12: cornea and TG; n = 6: BS). Statistical significance was determined by Bonferoni’s t-test; *p<0.05, comparing WT to TLR9 KO.
3.2. Increased CXCL9 and CXCL10 expression following HSV-1 infection in mouse cornea requires intact TLR 9 signaling
As a means to address whether CXCL10 is a downstream effector molecule of HSV-1 induced TLR 9 signaling, CXCL10 protein levels were quantified following HSV-1 infection in WT and TLR 9 KO mice. CXCL10 levels rose 19-fold in corneas from infected versus uninfected WT mice (Fig. 2A). Another related chemokine, CXCL9, was also found to rise significantly (9-fold) in corneas following HSV-1 infection of WT mice (Fig. 2A). By comparison, the cornea levels for CXCL9 and CXCL10 protein did not significantly change following HSV-1 infection in TLR9 KO mice compared to uninfected samples (Fig. 2A). In the absence of infection, both TLR9 KO and WT mice expressed CXCL9 and CXCL10 in the cornea (Fig. 2A). Consistent with this observation CpG containing oligodeoxynucleotide enhanced the expression of CXCL10 in the cornea of WT but not TLR9 KO mice or WT mice treated with a control oligonucleotide (Fig. 2B). In order to determine the tissue specificity of TLR9 control of CXCL9 and CXCL10 expression, uninfected and HSV-2-infected vaginal tissue were surveyed for chemokine expression comparing WT to TLR9 KO mice. Unlike cornea tissue, CXCL9 and CXCL10 expression were not hampered in the vaginal tissue in the absence of TLR9 following HSV-2 infection (Fig. 2C).
Figure 2.
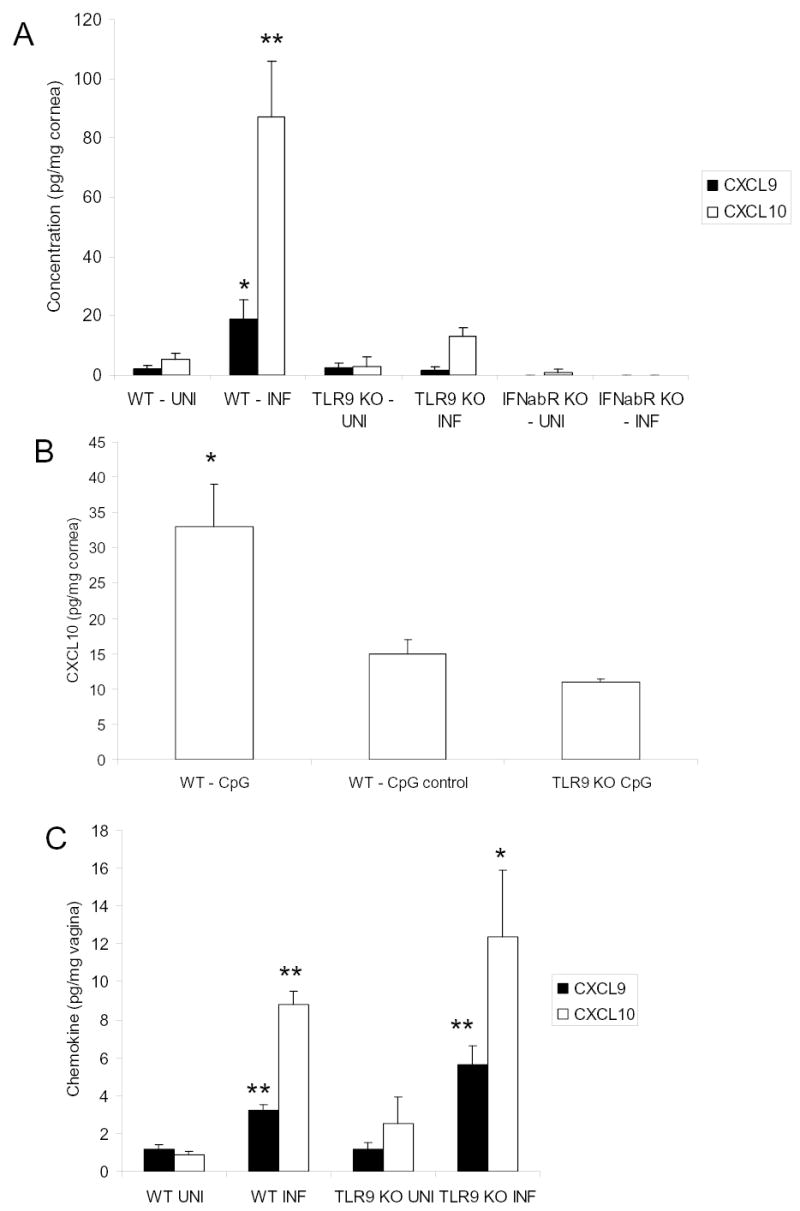
Intact TLR 9 and type I interferon signaling pathways are required for HSV-1 stimulated CXCL9 and CXCL10 protein expression in the corneas of infected mice. A. Wild type (WT), TLR 9 knockout (TLR9 KO), and interferon α/β receptor knockout (IFNabR KO) mouse corneas were scarified and vehicle (UNI) or 1,000 pfu HSV-1/cornea (INF) was topically administered. Thirty-six to 42 hr post infection, mice were euthanized and corneas were dissected, homogenized, and the indicated chemokines were assayed by ELISA. B. The corneas of wild type (WT) and TLR9 knockout (TLR9 KO) mice were scarified and 20 μg of CpG oligodeoxynucleotide (CpG) or scrambled oligodeoxynucleotide (CpG control) were placed onto the surface of the cornea in 3 μl of medium. Thirty six hr post application, the mice were anesthetized, perfused, and the corneas were processed for detection of CXCL10 by ELISA. C. Wild type (WT) and TLR 9 knockout (TLR9 KO) mice were treated with 2 mg DepoProvera and five days later infected with 2,000 pfu/vagina of HSV-2. Thirty six hr post infection, the mice were anesthetized, perfused, and vaginal tissue removed and processed for chemokine levels by ELISA. Uninfected (UNI) mice served as controls to determine induction of the chemokines. Bars represent the mean ± SEM from 2–3 separate experiments, n=4–12/group). For CpG experiment, the results are from a single experiment, n=2–3 mice/group. Statistical significance was determined by Bonferoni’s t-test, **p<.01, *p<.05 comparing INF to UNI group for each genotype of mice.
3.3. HSV-1 induced increase in CXCL9 and CXCL10 expression is mediated through a functional type I IFN receptor
TLR9 can induce CXCL10 expression in the cornea following HSV-1 infection by at least two pathways including IFN-α or NFκB pathways (Bhattacharjee and Akira, 2005). Therefore, we examined the expression of these mediators as a means to determine the principal signaling cascade. Corneas removed from HSV-1-infected WT mice showed no measurable NFκB by ELISA 24 p.i. (data not shown). In order to evaluate the presence of IFN following HSV-1 infection, the IFN-inducible gene family oligoadenylate synthetases (OAS) were measured by real time PCR. Both WT and TLR 9 knockout mice had similar levels of increased OAS expression (2.3 – 2.8 fold increase) in infected versus uninfected corneas. These results inferred a local induction of type I IFN in the presence or absence of TLR9 in response to HSV-1 infection. Consequently, we investigated the role of type I IFN induction of CXCL9 and CXCL10 expression using IFN α/βR KO mice. IFN α/βR KO mice prior to or after HSV-1 infection showed little to no CXCL9 or CXCL10 expression implicating type I IFN as a major stimulus for CXCL9 and CXCL10 expression in the cornea in response to HSV-1 (Fig. 2A).
3.4 The absence of TLR9 reduces the infiltration of neutrophils but not macrophages into the cornea of HSV-1-infected mice
The transient neutralization of CXCL10 has previously been reported to significantly dampen the inflammatory response as measured by infiltration of leukocytes and expression of selective cytokines/chemokines in the cornea following HSV-1 infection (Carr et al., 2003). Since CXCL10 levels were significantly reduced in the TLR9 KO mice compared to the WT controls initially following HSV-1 infection, we investigated the level of inflammation by measuring neutrophil and macrophage recruitment into the cornea and cytokine/chemokine levels in the cornea following infection. Since neutrophils and cytokines/chemokines are readily detectable in the cornea of mice by 72 hr p.i. (Carr et al., 2006), we used this time point to assess the level of inflammation. In comparing the infiltration of neutrophils into the corneal stroma of WT to TLR9 KO mice, we found a significant reduction in neutrophil infiltration into the peripheral cornea in TLR9 KO mice in comparison to WT animals (Fig. 3). In addition, there was a trend in the infiltration of neutrophils into the center of the cornea comparing the TLR9 KO (3.3 ± 1.7 cells/section) to WT (23.7 ±14.9 cells/section) mice but the difference did not reach significance due to the high variability in the WT mice. In contrast, there was no difference in macrophage recruitment into the center or peripheral cornea of WT and TLR9 KO mice with a range of between 5–10 cells/section in each group (data not shown). Even though neutrophil recruitment into the cornea was limited in the TLR9 KO mice, there were no significant differences in cytokine/chemokine (IL-1β, IFN-γ, TNF-α, CXCL1, CCL2, CCL3, and CCL5) levels at 36 or 72 hrs post infection (Table 1). Although the list of soluble mediators expressed in the cornea is not exhaustive, the specific change in CXCL9 and CXCL10 within the cornea following HSV-1 infection does implicate these chemokines in the early recruitment of leukocytes into the tissue within the early phase of acute virus infection.
Figure 3.
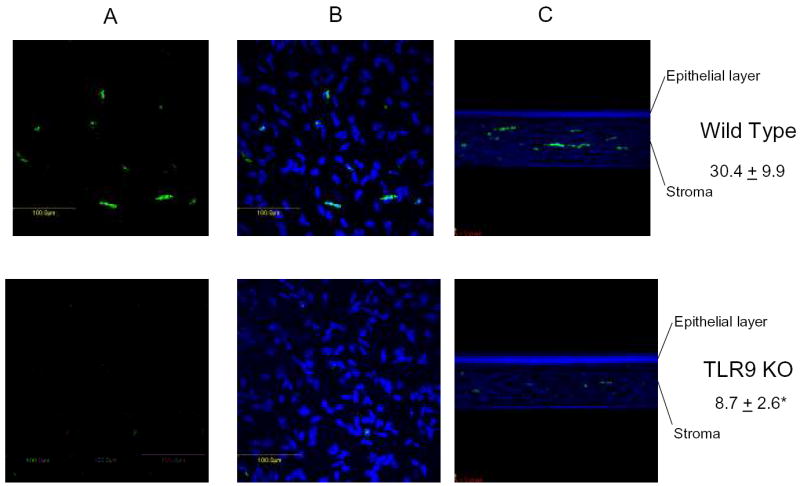
Neutrophil infiltration is reduced in HSV-1-infected TLR9 deficient mice. C57BL/6 wild type and TLR9 knockout (TLR9 KO) mice were infected with 1,000 pfu/eye HSV-1. Seventy-two hr post infection, the mice were anesthetized, perfused, and the corneas removed and processed for detection of neutrophils and macrophages by confocal microscopy. Panel A represents FITC-labeled neutrophils within a 16 μm composition section from wild type and TLR9 KO peripheral cornea. Panel B includes DAPI staining of the section shown in panel A. Panel C represents a cross section composite image of a peripheral aspect of the cornea showing full thickness. Panel A and B are 400x and panel C 200x magnification. Numbers represent mean neutrophil count ± SEM, n=12 samples/group. *p<.05 comparing the wild type to TLR9 KO mice.
Table 1.
Cytokine/Chemokine Expression in the Cornea of HSV-1-infected mice
| Group | CXCL1 | CCL2 | CCL3 | CCL5 | IL-1β | IL-6 | TNF-α | IFN-γ |
|---|---|---|---|---|---|---|---|---|
| WT – 36 | 1.1 ± 0.2 | 114 ± 43 | 0.6 ± 0.4 | 12.9 ± 0.6 | 0.5 ± 0.3 | 0.2 ± 0.1 | 0 | 0 |
| TLR9 KO – 36 | 0.7 ± 0.3 | 39 ± 11 | 1.2 ± 0.8 | 8.9 ± 2.5 | 0.5 ± 0.4 | 0.3 ± 0.2 | 0 | 0 |
| WT – 72 | 17 ± 17 | 28 ±13 | 5.4 ± 4.2 | 25 ± 9.0 | 1.4 ± 1.4 | 0.8 ± 0.6 | 0.1 ± 0.1 | 0 |
| TLR9 KO - 72 | 14 ± 11 | 85 ± 42 | 4.7 ± 2.4 | 50 ± 20 | 2.4 ± 2.4 | 1.0 ± 0.9 | 0 | 0 |
C57BL/6 wild type (WT) and TLR9 knockout (TLR9 KO) mice (n=6–9/group/timepoint) were infected with HSV-1 (1,000 pfu/eye).
At 36 or 72 hr post infection, the mice were anesthetized, perfused, and the corneas were removed and assayed for cytokine/chemokine content by suspension array. Numbers are expressed as mean pg/mg cornea ± SEM.
4. Discussion
Pathogen associated molecular patterns are structural motifs found in a variety of microbial pathogens that are recognized by pattern recognition receptors including TLRs. The present study was undertaken to identify the principal pathway that elicits CXCL9 and CXCL10 expression in the cornea following infection with the neurotropic virus, HSV-1. In the absence of TLR9, virus-induced up-regulation of CXCL9 and CXCL10 was no longer found. However, baseline expression of these chemokines was still evident suggesting another pathway(s) is involved in the regulation of constitutive expression. In fact, in the absence of TLR9 expression, the type I IFN response was not attenuated as measured by IFN-responsive gene expression. The involvement of type I IFNs in CXCL9 and CXCL10 expression in the cornea in response to HSV-1 infection was reinforced by showing little to no chemokine expression prior to or following infection in IFN α/βR KO mouse corneas. TLR3 is another TLR that can elicit type I IFN production following virus infection (Kawai and Akira, 2006) and is found in cornea epithelial cells (Kumar et al., 2005). Although not formally proven, it appears likely TLR3 is responsible for constitutive expression of CXCL9 and CXCL10 in the cornea of C57BL/6 mice, and TLR9 is involved in the up-regulation of these chemokines following HSV-1 infection. Therefore, we propose both TLRs operate through the central pathway, type I IFNs, in driving CXCL9 and CXCL10 expression as the absence of this pathway negates chemokine expression in the cornea (constitutive or induced).
Whereas the presence of TLR9 was required for HSV-1-induced up-regulation of CXCL9 and CXCL10 expression in the cornea, it was not required for chemokine expression or up-regulation in the vaginal tissue of mice following HSV-2 infection. The absence of chemokine induction by TLR9 in the genitalia is not due to a lack of local expression (Ashkar et al., 2003; McCLuskie et al., 2006). It is likely that other pattern recognition receptors including TLR3 are responsible for control of CXCL9 and CXCL10 in the vaginal tissue (Ashkar et al., 2004).
Since the absence of TLR9 significantly reduced the expression of CXCL9 and CXCL10 in the cornea following HSV-1 infection and neutralization of CXCL10 has previously been reported to reduce the inflammatory response to ocular HSV-1 infection (Carr et al., 2003), we characterized the inflammatory response in HSV-1-infected TLR9 KO mice. By confocal microscopy, we found a significant reduction in neutrophil but not macrophage infiltration into the stroma but not changes in the expression of other chemokines (CXCL1, CCL2, CCL3, or CCL5) or pro-inflammatory cytokines (IL-1β, IL-6, IFN-γ or TNF-α) of TLR9 KO compared to WT mice suggesting either CXCL9, CXCL10, or other soluble mediators including chemokines are also involved in the recruitment of neutrophils into the infected tissue.
In the present report, virus titers were similar between WT and TLR9 KO mouse cornea and TG during acute infection (i.e., day 3 and day 7 p.i.). However, virus shedding in the tear film was significantly elevated in TLR9 KO mice 48–72 hr p.i. These results are in contrast to a previous report showing no difference in virus shedding (Krug et al., 2004). Differences in the strain of HSV-1 employed as well as mouse strain used in the former study may explain the differences. As a means to determine why TLR9 KO mice shed more virus compared to WT controls, we compared virus shedding in TLR9 KO mice compared to mice deficient in CXCL10 expression. The lack of CXCL10 did not affect the levels of virus in the tear film of infected mice (data not shown) indicating the lack or muted CXCL10 response does not impact on the host in the control of virus shedding. Whether or not CXCL10 deficient mice show a reduction in neutrophil influx into the stroma following HSV-1 infection has yet to be determined. Collectively, TLR9 is one of many elements within the host’s armamentarium to control virus replication and the inflammatory response to infection in the eye. These control elements underscore the breadth of regulatory mechanisms required to clear an infectious pathogen and simultaneously preserve the visual axis.
Acknowledgments
This work was supported by USPHS grants EY015566 and EY12190 as well as a Jules and Doris Stein Research to Prevent Blindness research professorship.
Footnotes
Todd Wuest, Ph: 405-271-7194; Fax: 405-271-8781; E-mail: todd-wuest@ouhsc.edu
Bobbie Ann Austin, Ph.D., Ph: 405-271-7194; Fax: 405-271-8781; E-mail: bobbie-austin@ouhsc.edu
Satoshi Uematsu, M.D., Ph.D., Ph: 81-6-6879-8303; Fax: +81-6-6879-8305; E-mail: uemattsu@biken.osaka-u.ac.jp
Manoj Thapa, Ph: 405-271-7194; Fax: 405-271-8781; E-mail: manoj-thapa@ouhsc.edu
Shizuo Akira, Ph.D., Ph: +81-66-879-8303; Fax: +81-66-879-8305; E-mail: sakira@biken.osaka-u.ac.jp
References
- Ashkar AA, Bauer S, Mitchell WJ, Vieira J, Rosenthal KL. Local delivery of CpG oligodeoxynucleotides induces rapid changes in the genital mucosa and inhibits replication, but not entry, of herpes simplex virus type 2. J Virol. 2003;77:8948–8956. doi: 10.1128/JVI.77.16.8948-8956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkar AA, Yao XD, Gill N, Sajic D, Patrick AJ, Rosenthal KL. Toll-like receptor (TLR)-3, but not TLR4, agonist protects against genital herpes infection in the absence of inflammation seen with CpG DNA. J Infect Dis. 2004;190:1841–1849. doi: 10.1086/425079. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee RN, Akira S. Toll-like receptor signaling: Emerging opportunities in human diseases and medicine. Curr Immunol Rev. 2005;1:81–90. [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Carr DJ, Chodosh J, Ash J, Lane TE. Effect of anti-CXCL10 monoclonal antibody on herpes simplex virus type 1 keratitis and retinal infection. J Virol. 2003;77:10037–10046. doi: 10.1128/JVI.77.18.10037-10046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DJ, Tomanek L. Herpes simplex virus and the chemokines that mediate the inflammation. Current Topics in Microbiology & Immunology. 2006;303:47–65. doi: 10.1007/978-3-540-33397-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DJ, Ash J, Lane TE, Kuziel WA. Abnormal immune response of CCR5-deficient mice to ocular infection with herpes simplex virus type 1. J Gen Virol. 2006;87:489–499. doi: 10.1099/vir.0.81339-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey TL, Dyer KD, Ellis JA, Bonville CA, Foster B, Prussin C, Easton AJ, Domachowske JB, Rosenberg HF. Inflammatory responses to pneumovirus infection in IFN-αβR gene-deleted mice. J Immunol. 2005;175:4735–4744. doi: 10.4049/jimmunol.175.7.4735. [DOI] [PubMed] [Google Scholar]
- Härle P, Cull V, Agbaga MP, Silverman R, Williams BRG, James C, Carr DJJ. Differential effect of murine alpha/beta interferon transgenes on antagonization of herpes simplex virus type 1 replication. J Virol. 2002;76:6558–6567. doi: 10.1128/JVI.76.13.6558-6567.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Hochrein H, Schlatter B, O'Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2004;101:11416–11421. doi: 10.1073/pnas.0403555101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AC, Heinzel FP, Diaconu E, Sun Y, Hise AG, Golenbock D, Lass JH, Pearlman E. Activation of toll-like receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Invest Ophthalmol Vis Sci. 2005;46:589–595. doi: 10.1167/iovs.04-1077. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- Kumar A, Zhang J, Yu FSX. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunol. 2005;117:11–21. doi: 10.1111/j.1365-2567.2005.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskie MJ, Cartier JL, Patrick AJ, Sajic D, Weeratna RD, Rosenthal KL, Davis HL. Treatment of intravaginal HSV-2 infection in mice: a comparison of CpG oligodoxynucleotides and resiquimod (R-848) Antiviral Res. 2006;69:77–85. doi: 10.1016/j.antiviral.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Millhouse S, Wigdahl B. Molecular circuitry regulating herpes simplex virus type 1 latency in neurons. J Neurovirol. 2000;6:6–24. doi: 10.3109/13550280009006378. [DOI] [PubMed] [Google Scholar]
- Rodahl E, Haarr L. A herpes simplex virus type 1 vector as marker for retrograde neuronal tracing: characterization of lacZ transcription and localization of labelled neuronal cells in sensory and autonomic ganglia after inoculation of the anterior segment of the eye. Exp Eye Res. 2000;71:495–501. doi: 10.1006/exer.2000.0905. [DOI] [PubMed] [Google Scholar]
- Valyi-Nagy T, Deshmane S, Dillner A, Fraser NW. Induction of cellular transcription factors in trigeminal ganglia of mice by corneal scarification, herpes simplex virus type 1 infection, and explantation of trigeminal ganglia. J Virol. 1991;65:4142–4152. doi: 10.1128/jvi.65.8.4142-4152.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]