

Abstract
It is becoming apparent that the hormone leptin plays an important role in modulating hippocampal function (Harvey, 2003). Indeed, leptin enhances NMDA receptor activation and promotes hippocampal long-term potentiation (LTP; Shanley et al, 2001). Furthermore obese rodents with dysfunctional leptin receptors display impairments in hippocampal synaptic plasticity (Li et al, 2002). Here we demonstrate that under conditions of enhanced excitability (evoked in Mg2+-free medium or following blockade of GABAA receptors), leptin induces a novel form of long-term depression (LTD) in area CA1 of the hippocampus. Leptin-induced LTD was markedly attenuated in the presence of D-AP5, suggesting that it is dependent on the synaptic activation of NMDA receptors. In addition, low frequency stimulus-evoked LTD occluded the effects of leptin. In contrast, mGluRs did not contribute to leptin-induced LTD as mGluR antagonists failed to either prevent or reverse this process. The signaling mechanisms underlying leptin-induced LTD were independent of the Ras-Raf-MAPK-signaling pathway, but were markedly enhanced following inhibition of either PI 3-kinase or protein phosphatases 1 and 2A. These data indicate that under conditions of enhanced excitability, leptin induces a novel form of homosynaptic LTD, which further underscores the proposed key role for this hormone in modulating NMDA receptor-dependent hippocampal synaptic plasticity.
Keywords: leptin, long-term depression, hippocampus, NMDA receptor-dependent,
Long-term depression (LTD) of excitatory synaptic transmission is a persistent weakening of synaptic strength that is involved in learning and memory processes and neuronal development (Bear & Abraham, 1996). Two main forms exist in the mammalian CNS, that are induced by the synaptic activation of N-methyl-D-aspartate (NMDA; Bear & Abraham, 1996) and metabotropic glutamate receptors (mGluRs; Anwyl, 1999; Kemp & Bashir, 2001), respectively. Homosynaptic NMDA receptor-dependent LTD is expressed postsynaptically and triggered by Ca2+ influx via NMDA channels and subsequent activation of serine/threonine protein phosphatases (Mulkey et al, 1993; 1994). In contrast, mGluR-dependent LTD is independent of Ca2+ (Fitzjohn et al, 2001) and serine/threonine phosphatases (Schnabel et al, 2001), and may have a presynaptic mechanism of expression (Rammes et al, 2003). Insulin can also induce LTD at hippocampal CA1 synapses (Huang et al, 2004), via activation of a phosphoinositide 3-kinase (PI 3-kinase)/PKC-linked pathway.
The hormone leptin is best known for its role in regulating energy homeostasis via actions on specific hypothalamic nuclei (Ahima & Flier, 2000). However, leptin receptors are expressed in numerous brain regions (Elmquist et al, 1998; Shanley et al, 2002), suggesting that leptin has additional functions in the CNS. Indeed, leptin enhances NMDA receptor activity and modulates hippocampal LTP (Shanley et al, 2001). Obese rodents with dysfunctional leptin receptors also display impairments in hippocampal LTP and LTD (Li et al, 2002), and administration of leptin into the dentate gyrus enhances the level of LTP in vivo (Wayner et al, 2004), consistent with the notion that leptin modulates excitatory synaptic strength. However the effects of leptin on hippocampal LTD remain to be established.
Leptin receptors are class I cytokine receptors that signal via association with janus tyrosine kinases (JAKs). Activated JAKs can stimulate various downstream signaling pathways, including PI 3-kinase. Indeed, PI 3-kinase is a key element of leptin receptor signaling in neurones (Shanley et al, 2001, 2002a,Shanley et al, b; Niswender et al, 2001). Another target for activated JAKs is the adaptor protein SHP-2, which initiates the Grb2-Ras-Raf MAPK (mitogen-activated protein kinase) signaling cascade. Stimulation of this pathway by leptin has also been observed in neurones (Shanley et al, 2001). Recent studies suggest that PI 3-kinase and MAPK play a prominent role in hippocampal synaptic plasticity. PI 3-kinase inhibitors block the induction (Opazo et al, 2003; Man et al, 2003) and maintenance of NMDA receptor-dependent LTP (Kelly & Lynch, 2000; Sanna et al, 2002). PI 3-kinase also regulates the synapse-specificity of homosynaptic LTD (Daw et al, 2002), and a PI 3-kinase-linked pathway is required for mGluR- (Hou & Klann, 2004) and insulin-induced LTD (Huang et al, 2004). Additionally, MAPK-dependent signaling pathways regulate the efficacy of excitatory synaptic transmission (Thomas & Huganir, 2004; Thiels et al, 2002), and hippocampal mGluR-dependent LTD (Gallagher et al, 2004).
In this study we examined the effects of leptin on hippocampal excitatory synaptic transmission. We show that under conditions of enhanced excitability, leptin induces a long-lasting depression of excitatory synaptic transmission that is independent of MAPK, but modulated by PI 3-kinase and protein phosphatase activity.
Materials and Methods
Hippocampal slices
Young Sprague Dawley rats (14-18 days old) were killed by cervical dislocation in accordance with Schedule 1 of the U.K. Government Animals (Scientific Procedures) Act, 1986. To avoid variations in the levels of leptin between animals, all animals were maintained under identical conditions and were euthanased at the same time of day. After decapitation the brain was removed and placed in ice-cold artificial cerebrospinal fluid (aCSF) consisting of (mM): NaCl 124; KCl 3; NaHCO3 26; NaH2PO4 1.25; MgSO4 1; CaCl2 2; D-glucose 10 (bubbled with 95% O2/5% CO2; pH 7.4). Transverse hippocampal slices (400 μm) were cut using a Vibratome tissue slicer and were maintained in oxygenated aCSF at room temperature for an hour before use.
Extracellular field recordings
Extracellular recordings of field EPSPs (fEPSPs) were made from the stratum radiatum of area CA1 of slices using glass microelectrodes filled with 4 M NaCl (resistance 1–3 M ). Slices were maintained in a submerged recording chamber and perfused at a rate of 3 ml min−1 with aCSF at 30 °C. Responses were evoked by stimulation of the Schaffer collateral-commissural pathway at a frequency of 0.033 Hz. For paired-pulse studies two identical stimuli separated by a 50ms inter-stimulus interval were utilized. In all experiments the stimulus intensity was subthreshold for generation of population spikes and was set to give a slope value 30–50 % of maximal responses. Recordings were made using an Axopatch 200B amplifier and data were filtered at 5 kHz and digitized at 10 kHz. Electrical signals were recorded and analysed on- and off-line using LTP software (Courtesy of Dr Bill Anderson, University of Bristol, UK). For studies comparing the effects of leptin in Mg2+-free medium and D-AP5, control slices were interleaved. In all other experiments, drug-treated slices were paired with matched controls from the same animal.
Materials
Human recombinant leptin (R & D Systems; 95–98% purity) was prepared as a stock solution in normal aCSF and was diluted in normal aCSF containing 0.2% bovine serum albumin. LY294002, wortmannin, U0126, U0124, okadaic acid, cyclosporine A and PD98059 were obtained from Calbiochem, whereas D-AP5, LY341495, MPEP, cypermethrine and LY367385 were all obtained from Tocris Cookson, UK.
Statistical Analyses
All data are presented as mean ± SEM, and statistical analyses were performed using Student’s t-test for comparison of means or two way ANOVA (analysis of variance) for comparisons between multiple groups (unless otherwise stated). P<0.05 was considered significant.
Results
Leptin induces LTD under conditions of enhanced excitability
In the presence of normal aCSF (1mM Mg2+), application of leptin (50 nM for 15 min) had no effect on the amplitude or slope of the evoked fEPSP (n=7; P>0.05). Similarly following 30 min washout of leptin the amplitude and slope of fEPSPs did not alter significantly (n=7; P>0.05).
Perfusion of slices with Mg2+-free medium or picrotoxin results in an increase in neuronal excitability associated with enhanced NMDA receptor activation following low frequency stimulation (Coan & Collingridge, 1985). In normal conditions, the synaptic activation in the hippocampus is restricted by powerful concurrent inhibition leading to an enhanced voltage-dependent Mg2+ block of NMDA receptor channels (Collingridge et al, 1988). Under Mg2+-free conditions, application of leptin (50 nM; 15min) induced a long-lasting depression of synaptic transmission in 22 out of 30 slices (Fig 1B). During application of leptin, synaptic responses were not significantly affected (98 ± 2.0% of control amplitude). However following washout of leptin (for 30 min) the amplitude of synaptic responses were reduced to 73.2 ± 4.1% of control (n=22; P<0.05); an action that persisted for the duration of recordings (up to 60 min). Similar effects were observed on fEPSP slope (n=22). Likewise, in slices perfused with the GABAA receptor antagonist, picrotoxin (50 μM) in the presence of 1 mM Mg2+, leptin depressed synaptic responses to 84.3 ± 5.1% of control (n=5; Fig 1C) which was not significantly different from the level of depression induced in Mg2+-free conditions (74.7 ± 4.1% of control; n=5; P>0.05; paired t test). These data indicate that under conditions of enhanced excitability leptin induces LTD of synaptic transmission in the hippocampus.
Figure 1. Leptin induces LTD of excitatory synaptic transmission.
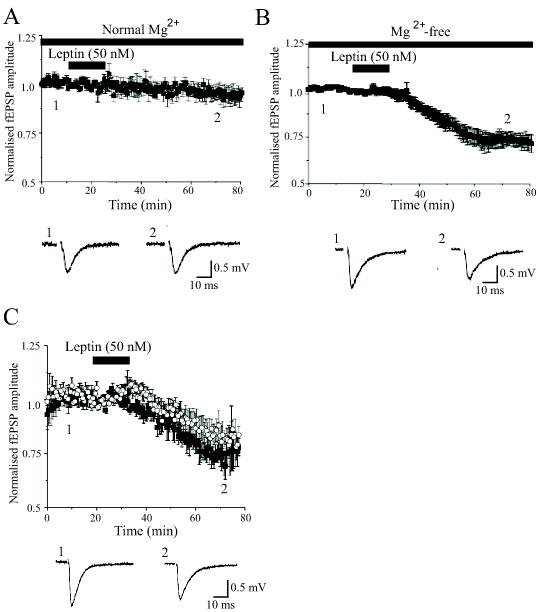
A, Plot of the pooled data illustrating the normalized fEPSP amplitude against time in slices bathed in normal (1 mM Mg2+-containing) aCSF. Application of leptin (50 nM) for the time indicated by the bar failed to evoke any long lasting change in the amplitude of fEPSPs. Below the plot are typical examples of fEPSPs obtained from an individual experiment prior to (1) and following exposure to leptin (50 nM; 15 min). B, In contrast in slices exposed to Mg2+-free aCSF, addition of leptin (50 nM; 15 min) evoked a long lasting depression of synaptic transmission that was sustained for the duration of recordings. Examples of the synaptic records obtained in the absence (1) and following incubation with leptin (2) are depicted below the plot. C, Leptin also induced LTD following blockade of GABAA receptors with picrotoxin (50 μM; open circles) and in paired experiments the level of depression induced was similar to that evoked in Mg2+-free conditions (filled square). D. Examples of the synaptic records obtained in the absence (1) and following exposure to leptin (2) in slices exposed to picrotoxin (50 μM) are depicted below the plot.
NMDA receptor activation is required for leptin-induced LTD
It is well established that two forms of LTD exist in the CNS, which are induced by synaptic activation of NMDA and metabotropic glutamate receptors, respectively (Bear & Abraham, 1996; Kemp & Bashir, 2001). As leptin enhances hippocampal NMDA receptor function (Shanley et al, 2001), and the ability of leptin to evoke LTD is evident under conditions that enhance the synaptic activation of NMDA receptors, we examined whether NMDA receptor activation is required for leptin-induced LTD, using the competitive NMDA receptor antagonist, D-AP5 (50 μM). In control slices perfused with Mg2+-free medium, LTD was induced following exposure to leptin (50 μM; 15 min; n=5; Fig 2A,B). Thus, 30 min following washout of leptin synaptic responses were depressed to 79.1 ± 4.0% of control (n=5; P<0.05). However, the ability of leptin to depress responses was markedly reduced in the presence of D-AP5 (50 μM; Fig 2A,Bs), such that 30 min following washout of leptin synaptic responses were not significantly attenuated by leptin (95.5 ± 3.1% of control; n=5; P>0.05). As low frequency stimulus (LFS)-evoked LTD is also dependent on NMDA receptor activation, we therefore investigated whether saturation of stimulus-evoked LTD, could occlude the effects of leptin under Mg2+-free conditions. Following saturation of LFS-LTD, using two periods of 1Hz stimulation (900 pulses) at a 30 minute interval, application of leptin (50 nM; 15 min) failed to evoke any further reduction in synaptic transmission (n=5; Fig 2E,F). Thus 30 min after LFS, synaptic responses were depressed to 67.5 ± 7.3% of control (n=5) and the level of depression was not significantly altered following subsequent addition of leptin (depressed to 64.1 ± 8.0% of control; n=5; P>0.05). However, following induction of LTD by leptin, subsequent LFS (900 pulses, 1 Hz) was still able to depress synaptic transmission further (n=5; not illustrated). Thus, leptin depressed synaptic responses to 74.6 ± 5.1 % of control (n=5), whereas 30 min after LFS synaptic responses were depressed to 54.2 ± 3.9% of control (pre-LFS baseline) (n=5; P<0.01). Thus, these data suggest that leptin-induced LTD and LFS-induced LTD share at least some similar mechanisms of expression, and that the synaptic activation of NMDA receptors is a prerequisite for leptin-induced LTD.
Figure 2. Leptin-induced LTD is NMDA receptor-dependent and occluded by LFS-induced LTD.
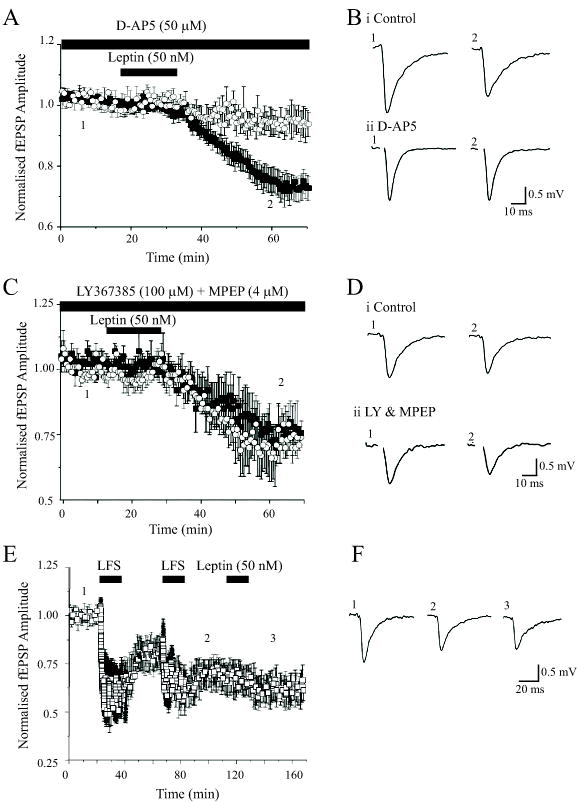
A, Plot of the pooled data illustrating the normalized fEPSP amplitude against time in slices bathed in Mg2+-free aCSF. The leptin-induced depression of synaptic transmission was significantly attenuated in the presence of the competitive NMDA receptor antagonist, D-AP5 (50 μM; filled square) relative to control (open circle). B, Examples of representative synaptic records obtained in the absence (1) and following exposure to leptin (2) in control slices (i) and in slices exposed to D-AP5 (50 μM; ii). C,D, In contrast mGluRs do not contribute to leptin-induced LTD. C. Plot of the pooled data of the mean normalized fEPSP amplitude against time in slices bathed in Mg2+-free medium. The level of depression induced by leptin (50 nM) was not significantly altered in slices exposed to the mGluR antagonists LY367385 (100 μM) and MPEP (4 μM; open circle) compared to control (filled square). D. Representative synaptic records obtained in the absence (1) and following exposure to leptin (2) in control slices (i) and in slices incubated with a combination of LY367385 and MPEP (ii). E. Low frequency stimulation-induced LTD occludes leptin-induced LTD. Plot of the pooled data of the normalized fEPSP amplitude against time in slices bathed in Mg2+-free medium. A low frequency (1Hz for 900 sec) stimulation (LFS) protocol was given at the time indicated by the bar, and this resulted in LTD of excitatory synaptic transmission. LTD evoked by this stimulation protocol was saturated following the second series of LFS. Subsequent addition of leptin (50 nM; 15 min) failed to induce any further inhibition of synaptic transmission. F. Representative synaptic records obtained in control conditions (1), following the second (2) LFS protocol and after exposure to leptin (3).
Role of mGluRs in leptin-induced LTD
Numerous studies have demonstrated that mGluRs play an important role in synaptic plasticity. In particular group I mGluRs are implicated in a specific type of LTD evoked at hippocampal CA1 synapses (Kemp & Bashir, 2001). Indeed the induction and maintenance phases of DHPG-induced LTD require ongoing activation of mGluRs (Palmer et al, 1997; Rouach & Nicoll, 2003). Thus, in order to assess if mGluR activation was required for the induction phase of leptin-induced LTD, slices were exposed to a combination of LY367385 (100 μM; group Ia mGluR antagonist) and MPEP (4 μM; mGluR5 subtype-selective antagonist) prior to the addition of leptin. In control slices perfused with Mg2+-free medium, application of this combination of mGluR antagonists completely inhibited the ability of (RS)-3,5-dihydroxyphenylglycine (DHPG; 100 μM), a selective group 1 mGluR (mGluR1 and mGluR5) agonist, to induce LTD (n=4; not illustrated). Application of these agents had little or no effect on synaptic transmission per se, and did not affect the ability of leptin to induce LTD (depressed to 68.8 ± 5.1% of control after 30 min washout; n=5; P<0.05; Fig 2C,D). Metabotropic receptors did not contribute to the maintenance phase of leptin-induced LTD as application of either LY341495 (20–100 μM; a selective group II mGluR antagonist; n=6) alone or a combination of LY367385 (100 μM) plus MPEP (4 μM; n=5) failed to reverse leptin-induced LTD (not illustrated). Thus, these data indicate that mGluRs are unlikely to contribute to the LTD evoked by leptin.
Leptin-induced LTD has a postsynaptic locus of expression
It is well established that NMDA receptor-dependent LTD has a postsynaptic locus of expression, whereas mGluR-mediated LTD is likely to have a presynaptic component (Rammes et al, 2003). As leptin receptors are expressed at both pre- and post-synaptic sites at hippocampal CA1 synapses (Shanley et al, 2002), leptin-induced LTD could conceivably be expressed at either locus. In order to examine the locus of leptin-induced LTD, a paired pulse stimulation protocol was utilized such that two stimuli were delivered, with an inter-stimulus interval of 50 ms. Under conditions where leptin depressed synaptic responses (to 72.7 ± 6.1 % of control; n=7), the paired-pulse facilitation (PPF) ratio (1.46 ± 0.07; n=7) was not significantly different to that obtained under control conditions (1.53 ± 0.08; n=7; P>0.05) prior to leptin addition (Figure 3A,B). As a control we compared the effects of adenosine on the PPF ratio, as adenosine inhibits excitatory synaptic transmission in this region of the brain via a presynaptic mechanism (Wu and Saggau, 1994). Application of adenosine (10–20 μM) evoked 40.0 ± 9.0% (n=5) depression of synaptic responses; an action that was accompanied by a change in the PPF ratio from a control value of 1.38 ± 0.09 to 1.75 ± 0.15 (n=5; P<0.05; Figure 3C,D). Together these data suggest that leptin-induced LTD is most likely to be expressed postsynaptically.
Figure 3. Leptin-induced LTD has a postsynaptic locus of expression.
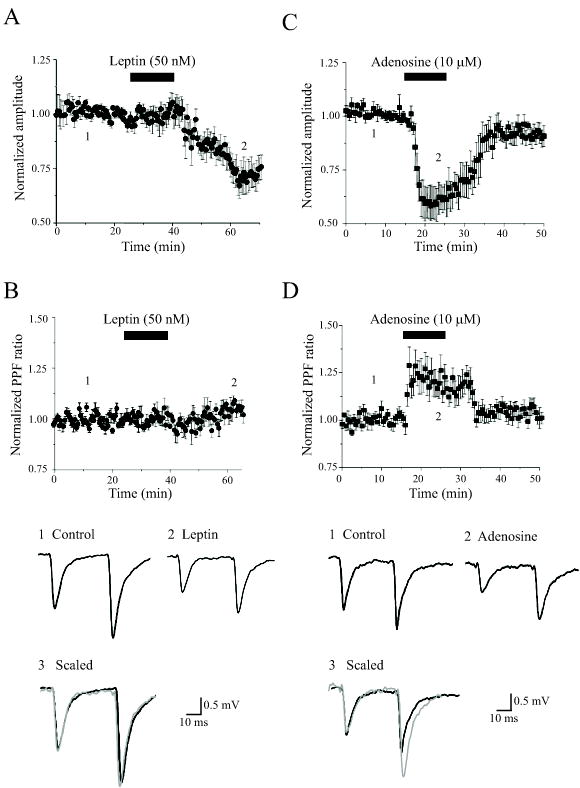
A,B, Leptin-induced LTD was not accompanied by any marked change in the PPF ratio. A, Plot of the pooled data illustrating the normalized fEPSP amplitude against time in slices bathed in Mg2+-free aCSF. Leptin (50 nM) evoked a long lasting depression of synaptic transmission. B, Plot of the mean PPF ratio against time for the experiments depicted in A. Leptin had no significant effect on the PPF ratio. C. In contrast application of adenosine (10 μM) depressed synaptic transmission to a similar extent to leptin. D. The PPF ratio was increased when synaptic transmission was depressed by adenosine. Below the plots are representative pairs of fEPSPs evoked with a 50ms interstimulus interval in control conditions (1) and following exposure to either leptin (2) or adenosine (2). In 3, the first fEPSP in 2 (grey) has been scaled to match the size of the first fEPSP in 1. The depression induced by adenosine, but not leptin, was accompanied by an enhancement in the PPF ratio.
Leptin-induced LTD is enhanced by PI 3-kinase inhibition
Evidence is mounting that PI 3-kinase plays a pivotal role in NMDA receptor dependent LTP (Sanna et al, 2002; Man et al, 2003) and LTD (Daw et al, 2002). Furthermore, PI 3-kinase is a key enzyme activated downstream of neuronal leptin receptors (Shanley et al, 2001; 2002; Harvey, 2003). Thus, we examined the role of PI 3-kinase in leptin-induced LTD, using two structurally unrelated inhibitors, namely wortmannin and LY294002. Application of LY294002 (10–50 μM; 45–60 min) or wortmannin (50 nM; 45–60 min) had little or no effect on synaptic responses per se. However, incubation with either agent did not prevent, but rather facilitated the ability of leptin to induce LTD (Fig 4A,B). Thus in LY294002-treated slices, application of leptin (50 nM; 15 min) depressed synaptic responses to 63.0 ± 5.6% of control (30 min after leptin washout; n=4), which was a significantly greater level of depression induced by leptin in the absence of LY294002 (83.5 ± 5.0% of control; n=4; P<0.05; Fig 4A,B). Similarly exposure to wortmannin enhanced the effects of leptin such that synaptic responses were depressed to 71.7 ± 4.4% (in wortmannin) by leptin compared to 87.1 ± 5.0% in control conditions (n=4; P<0.05; Fig 4B). Thus these data suggest that leptin-induced LTD is negatively regulated by PI 3-kinase.
Figure 4. Inhibition of PI 3-kinase facilitates leptin-induced LTD.
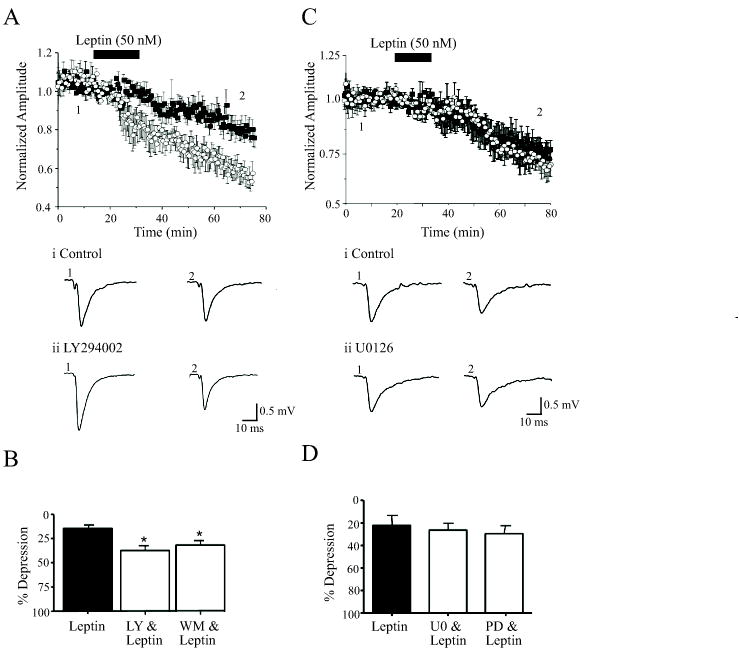
A, Leptin-induced LTD is enhanced in the presence of PI 3-kinase inhibitors. The ability of leptin to depress synaptic transmission is markedly enhanced in slices exposed to LY294002 (10 μM; open circle) relative to control (no LY294002; filled square). Below the plot are representative synaptic records obtained in the absence (1) and following exposure to leptin (2) in control slices (i) and in slices incubated with LY294002 (ii). B. Histogram of the pooled data showing the relative depression induced by leptin in control conditions and in the presence of either LY294002 (10 μM) or wortmannin (50 nM). In this figure, * represents P<0.05. C. In contrast inhibition of MAPK activation with U0126 (open circle) had no effect on leptin-induced LTD (filled square). Representative synaptic records are depicted below the plot as before. D. Histogram showing the relative depressions induced by leptin in control conditions and in the presence of either U0126 (1 μM) or PD98059 (10–50 μM). The ability of leptin to depress synaptic transmission was not altered following inhibition of MAPK activation.
As the Ras-Raf-MAPK signaling pathway also underlies leptin-induced facilitation of NMDA responses (Shanley et al, 2001), and the MAPK pathway plays a key role in hippocampal synaptic plasticity (Thomas & Huganir, 2004), we explored the potential role of this pathway. Application of either PD98059 (10–50 μM) or U0126 (1 μM) had no effect on synaptic transmission per se. Furthermore, the ability of leptin to induce LTD was unaffected by prior exposure to either agent such that leptin depressed responses to 70.5 ± 3.7 % (n=4; P>0.05) and 73.5 ± 5.8% (n=4; P>0.05) in the presence of either PD98059 or U0126, respectively (Fig 4C,D). Thus a MAPK-driven signaling cascade is unlikely to underlie leptin-induced LTD.
Leptin-induced LTD is regulated by serine/threonine protein phosphatases
It is well documented that serine/threonine protein phosphatases play a role in both NMDA receptor-dependent and mGluR-dependent LTD in the hippocampus. Thus activation of protein phosphatase 2B (PP2B) is required for NMDA receptor-dependent LTD (Mulkey et al, 1993), whereas protein phosphatase 1/2A (PP1/2A) inhibitors facilitate mGluR-induced LTD (Schnabel et al, 2001). Thus the role of PP1/2A and PP2B in leptin-induced LTD were examined. Application of either cypermethrin (1 μM; 45 min) or cyclosporine A (2 μM; 45 min), inhibitors of PP2B, had little effect on synaptic transmission per se. Moreover, incubation with either agent failed to affect the ability of leptin to induce LTD (Fig 5A,B), such that leptin (50 nM; 15 min) depressed synaptic responses to 74.9 ± 3.0% of control (n=4) in slices incubated with cypermethrine, and 70.4 ± 6.2% of control (n=5) in cyclosporine A-treated slices. These values did not differ significantly from those obtained in paired control slices in the absence of either cypermethrine (72.14 ± 4.5% of control; n=4; P>0.05) or cyclosporine A (74.2 ± 6.3% of control; n=5; P>0.05). In contrast, in slices incubated with the PP1/2A inhibitor okadaic acid (1 μM; 45 min; Fig 5C), the level of depression induced by leptin (to 61.7 ± 5.5% of control; n=4) was significantly greater than in control conditions (79.0 ± 5.2%; n=4; P<0.05). Thus these data suggest that leptin-induced LTD is also regulated by PP1/2A.
Figure 5. Role of serine/threonine phosphatases in leptin-induced LTD.
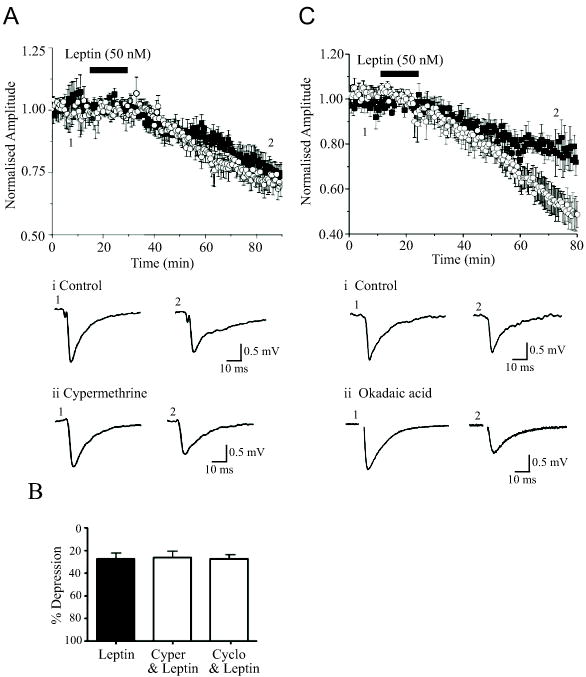
A. The ability of leptin to induce LTD is unaffected by inhibitors of PP2B. Leptin depressed synaptic transmission to a similar degree in the absence (open circle) and presence of cypermethrine (1 μM; filled square). Examples of the synaptic records obtained in the absence (1) and following exposure to (2) leptin obtained, in control conditions (i) and in the presence of cypermethrine (ii) are depicted below the plot. B. Histogram showing the relative depressions induced by leptin in control conditions and in the presence of either cypermethrine (1 μM) or cyclosporine A (2 μM). C. Leptin-induced LTD is enhanced in the presence of a PP1/2A inhibitor. The ability of leptin to depress synaptic transmission is markedly enhanced in slices exposed to okadaic acid (250 nM; open circle) relative to control (no okadaic acid; filled square). Examples of the synaptic records obtained in the absence (1) and following exposure to (2) leptin obtained, in control conditions (i) and in the presence of okadaic acid (ii) are depicted below the plot.
Discussion
The present study demonstrates that under conditions that enhance the synaptic activation of NMDA receptors, leptin induced a long lasting depression of excitatory synaptic transmission in the hippocampal CA1 region. In contrast, leptin failed to evoke any long lasting changes in the efficacy of excitatory synaptic transmission in control (1 mM Mg2+) conditions. It is well established that there at least two distinct forms of LTD, that are dependent on the synaptic activation of NMDA receptors and mGluRs, respectively (Nicoll et al, 1998; Palmer et al, 1997). In this study the synaptic activation of NMDA receptors was required for the depressant effects of leptin, as the competitive NMDA receptor antagonist D-AP5 prevented leptin-induced LTD. In contrast, mGluR activation was not a pre-requisite for leptin-induced LTD as prior exposure to broad spectrum or group-specific mGluR antagonists did not prevent the actions of leptin. Furthermore the mGluR antagonists did not reverse the LTD induced by leptin, indicating that mGluR activation is unlikely to contribute to the maintenance of leptin-induced LTD. We have shown previously that leptin receptors are expressed at both pre- and post-synaptic sites at hippocampal CA1 synapses (Shanley et al, 2002). Moreover NMDA receptor-dependent LTD is thought to have a postsynaptic locus, whereas mGluR-mediated LTD is likely to have a presynaptic component (Rammes et al, 2003). In this study leptin-induced LTD was not accompanied by any significant change in the PPF ratio. In contrast, adenosine, which depresses hippocampal excitatory synaptic transmission via a presynaptic mechanism, evoked a marked change in the PPF ratio. Given that both leptin and low frequency stimulus evoked-LTD both involve NMDA receptor activation it was perhaps not surprising that saturation of stimulus-evoked LTD occluded the effects of leptin. However, the converse was not the case, suggesting that the present leptin exposure protocol did not fully saturate LTD.
The present data indicate that leptin-induced LTD does not involve activation of the Ras-Raf-MAPK-driven signaling cascade as inhibition of this pathway failed to attenuate the level of depression evoked by leptin. However, leptin depressed synaptic responses to a significantly greater extent in slices exposed to PI 3-kinase inhibitors, suggesting that leptin-induced LTD is negatively regulated by PI 3-kinase. This contrasts with a previous study investigating insulin-induced LTD, which was NMDA-independent, but was dependent on PI 3-kinase activity (Huang et al, 2004). Thus, although biochemical studies suggest that leptin and insulin activate similar signaling cascades, they appear to modulate synaptic plasticity via distinct mechanisms. As leptin-induced LTD requires NMDA receptor activation, it is unlikely that the leptin-induced enhancement of NMDA responses reported previously (Shanley et al, 2001) underlies this effect, as PI3K inhibition blocks the direct effects of leptin on NMDA responses (Shanley et al, 2001), but enhances leptin-induced LTD. Moreover, it is likely that leptin-induced LTD requires coincident activation of NMDA receptors and a leptin-driven intracellular signaling element, the precise identity of which remains to be determined. The finding that okadaic acid, but not cyclosporin A or cypermethrin, potentiates leptin-induced LTD suggests that this leptin receptor-driven signalling process involves phosphorylation of one or more serine/threonine kinases, and that it is regulated by PP1 and/or PP2A, but not PP2B. Interestingly, a similar mechanism underlies hippocampal LTD induced by mGluRs (Schnable et al, 2001), whereas NMDA receptor-dependent LTD is prevented by inhibitors of PP1/2A as well as PP2B (Mulkey et al, 1993; 1994). In conclusion, the intracellular signaling mechanisms underlying leptin-induced LTD appear to be unique and do not fit with classical LFS-induced LTD or mGluR-evoked LTD, however leptin-induced LTD shares some similarities with both forms.
It is now clear that leptin exerts a number of different actions on hippocampal function, including enhancement of NMDA receptor function (Shanley et al, 2001) and facilitation of LTP (Shanley et al, 2001; Wayner et al, 2004), as well as stimulation of BK channel activity (Shanley et al, 2002a) and inhibition of hyperexcitability (Shanley et al, 2002b). Although leptin-induced LTD in the present study was dependent on NMDA receptor activation, leptin facilitation of NMDA responses and enhancement of LTP is likely to be mediated via a distinct PI 3-kinase-dependent mechanism. In some experiments a small, transient enhancement of synaptic transmission was observed in the presence of leptin, which may reflect the facilitation of NMDA receptor-mediated currents (Shanley et al, 2001). Thus it is likely that the effects we observe in the present study reflect a balance between leptin-induced enhancement of NMDA responses and leptin-induced LTD, which would tend to counteract one another during initial exposure to leptin. Indeed, in the presence of PI 3-kinase inhibitors, that block the enhancement of NMDA responses by leptin (Shanley et al, 2001), the onset of leptin-induced LTD was observed much more rapidly.
Previous studies have also shown that leptin can reduce neuronal hyperexcitability and the mechanism underlying this process is likely to involve, at least in part, activation of BK channels (Shanley et al, 2002b). Thus spontaneous Ca2+ oscillations in cultured neurons exposed to Mg2+-free medium and interictal activity observed in hippocampal slices are inhibited by leptin. The effects of leptin on BK channels are also blocked by inhibitors of PI 3-kinase, suggesting that a different signaling mechanism to leptin-induced LTD is involved. However the combination of channel activation coupled with a reduction in excitatory synaptic strength suggests that leptin could act as a powerful endogenous anticonvulsant. Indeed the frequency of spontaneous interictal activity, evoked following exposure to Mg2+-free medium for prolonged periods (2–3 hours) coupled with strong synaptic activation, is enhanced in leptin receptor-deficient rats (Zucker fa/fa) and suggests a tonic influence of leptin in regulating hippocampal hyperexcitability (Shanley et al, 2002b).
It is known that leptin circulates in the plasma in amounts proportional to body weight, crosses the blood brain barrier and reaches the brain via the cerebrospinal fluid at low nanomolar concentrations (10–20 nM; Caro et al, 1996). There is also evidence that leptin mRNA and protein are found in specific neuronal populations including the hippocampus (Ur et al, 2002), suggesting that leptin may also be released locally. Thus the actual concentration of leptin that reaches hippocampal synapses is likely to be derived from both the cerebrospinal fluid and locally released leptin. As leptin is a large peptide, and it is known that the access of large peptide molecules in acute slices may be restricted by layers of tissue damaged in the slicing process, the effective concentration of leptin used in this study is likely to lie well within the physiological range. However it is apparent that leptin is capable of reaching hippocampal synapses at sufficiently high concentrations to influence synaptic function in vivo as rodents that are either unable to produce leptin or are insensitive to leptin display deficits in hippocampal synaptic function. Indeed, ob/ob mice show reduced levels of a number of synaptic proteins in the hippocampus compared to wild type mice and these deficits can be reversed by leptin addition (Ahima et al, 1999). Moreover, rodents that are insensitive to leptin (db/db mice or fa/fa rats) display impairments in hippocampal LTP and LTD (Li et al, 2002).
There is accumulating evidence that leptin plays an important role in synaptic plastic mechanisms involved in neuronal development and learning and memory. Indeed, leptin deficiency in rodents results in abnormal brain development (Ahima et al, 1999; Bouret et al, 2004) and alterations in the efficacy of hypothalamic synaptic transmission (Pinto et al, 2004). Moreover rodents with dysfunctional leptin receptors (db/db mice; Zucker fa/fa rats) display deficits in hippocampal synaptic plasticity and in spatial memory tasks (Li et al, 2002). It is known that both pre and postsynaptic forms of LTD coexist at hippocampal CA1 synapses (Oliet et al, 1997), which enables greater flexibility in the long term modulation of synaptic efficacy. Thus the ability of leptin to induce a novel form of NMDA receptor-dependent LTD should not only facilitate the understanding of the molecular basis of LTD but it also gives a valuable insight into how this process is influenced by hormonal systems.
Evidence is accumulating that the development of obesity is attributable to leptin resistance, as obese individuals are unresponsive to leptin, even under conditions of hyperleptinaemia. Moreover, as leptin has the ability to modulate the cellular processes that are thought to underlie learning and memory formation it is feasible that leptin resistance may have a significant impact on cognitive function in obese individuals. As our studies were performed on slices obtained from relatively young animals, which may correlate developmentally to early childhood, this may in turn have important implications for development of the hippocampus in obese children, as the incidence of childhood obesity, associated with resistance to leptin, is steadily increasing.
Acknowledgments
This work was supported by The Wellcome Trust (grant no 075821), The Anonymous Trust and The Cunningham Trust.
References
- Ahima RS, Flier JS. Leptin. Ann Rev Physiol. 2000;62:413–37. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in hippocampus. Ann Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–10. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, Lynn RB, Zhang PL, Sinha MK, Considine RV. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 1996;348:159–61. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- Coan EJ, Collingridge GL. Magnesium ions block an N-methyl-D-aspartate receptor-mediated component of synaptic transmission in rat hippocampus. Neurosci Lett. 1985;53:21–6. doi: 10.1016/0304-3940(85)90091-6. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Herron CE, Lester RA. Synaptic activation of N-methyl-D-aspartate receptors in the Schaffer collateral-commissural pathway of rat hippocampus. J Physiol. 1988;399:283–300. doi: 10.1113/jphysiol.1988.sp017080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Bortolotto ZA, Saulle E, Zaman S, Collingridge GL, Isaac JT. Phosphatidylinositol 3 kinase regulates synapse specificity of hippocampal long-term depression. Nat Neurosci. 2002;5:835–6. doi: 10.1038/nn903. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain J. Comp. Neurol. 1998;395:535–47. [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol. 2001;537:421–30. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24:4859–64. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey J. Leptin: a multifaceted hormone in the central nervous system. Mol Neurobiol. 2003;28:245–58. doi: 10.1385/MN:28:3:245. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–61. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lee CC, Hsu KS. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J Neurochem. 2004;89:217–31. doi: 10.1111/j.1471-4159.2003.02307.x. [DOI] [PubMed] [Google Scholar]
- Kelly A, Lynch MA. Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacol. 2000;39:643–51. doi: 10.1016/s0028-3908(99)00169-0. [DOI] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI. Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol. 2001;265:339–65. doi: 10.1016/s0301-0082(01)00013-2. [DOI] [PubMed] [Google Scholar]
- Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neurosci. 2002;113:607–15. doi: 10.1016/s0306-4522(02)00162-8. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, et al. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–24. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–8. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–5. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–5. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. euron. 1997;18:969–82. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O’Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–88. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacol. 1997;36:1517–32. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–5. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Rouach N, Nicoll RA. Endocannabinoids contribute to short-term but not long-term mGluR-induced depression in the hippocampus. Eur J Neurosci. 2003;18:1017–20. doi: 10.1046/j.1460-9568.2003.02823.x. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–65. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammes G, Palmer M, Eder M, Dodt HU, Zieglgansberger W, Collingridge GL. Activation of mGlu receptors induces LTD without affecting postsynaptic sensitivity of CA1 neurons in rat hippocampal slices. J Physiol. 2003;546:455–60. doi: 10.1113/jphysiol.2002.033514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schabel R, Kilpatrick IC, Collingridge GL. Protein phosphatase inhibitors facilitate DHPG-induced LTD in the CA1 region of the hippocampus. Br J Pharmacol. 2001;132:1095–101. doi: 10.1038/sj.bjp.0703905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci. 2001;21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Rae MG, Ashford ML, Harvey J. Leptin inhibits rat hippocampal neurons via activation of large conductance calcium-activated K+ channels. Nat Neurosci. 2002a;5:299–300. doi: 10.1038/nn824. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, O’Malley D, Irving AJ, Ashford ML, Harvey J. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J Physiol. 2002b;545:933–44. doi: 10.1113/jphysiol.2002.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiels E, Kanterewicz BI, Norman ED, Trzaskos JM, Klann E. Long-term depression in the adult hippocampus in vivo involves activation of extracellular signal-regulated kinase and phosphorylation of Elk-1. J Neurosci. 2002;22:2054–62. doi: 10.1523/JNEUROSCI.22-06-02054.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–83. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Ur E, Wilkinson DA, Morash BA, Wilkinson M. Leptin immunoreactivity is localized to neurons in rat brain. Neuroendocrinol. 2002;75:264–72. doi: 10.1159/000054718. [DOI] [PubMed] [Google Scholar]
- Wayner MJ, Armstrong DL, Phelix CF, Oomura Y. Orexin-A (Hypocretin-1) and leptin enhance LTP in the dentate gyrus of rats in vivo. Peptides. 2004;25:991–6. doi: 10.1016/j.peptides.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–48. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]