

Abstract
Array technologies have made it straightforward to monitor simultaneously the expression pattern of thousands of genes. The challenge now is to interpret such massive data sets. The first step is to extract the fundamental patterns of gene expression inherent in the data. This paper describes the application of self-organizing maps, a type of mathematical cluster analysis that is particularly well suited for recognizing and classifying features in complex, multidimensional data. The method has been implemented in a publicly available computer package, genecluster, that performs the analytical calculations and provides easy data visualization. To illustrate the value of such analysis, the approach is applied to hematopoietic differentiation in four well studied models (HL-60, U937, Jurkat, and NB4 cells). Expression patterns of some 6,000 human genes were assayed, and an online database was created. genecluster was used to organize the genes into biologically relevant clusters that suggest novel hypotheses about hematopoietic differentiation—for example, highlighting certain genes and pathways involved in “differentiation therapy” used in the treatment of acute promyelocytic leukemia.
Array technologies have made it straightforward to monitor simultaneously the expression patterns of thousands of genes during cellular differentiation and response (1–5). The challenge now is to make sense of such massive data sets. For simple experiments comparing just two samples, it is enough to rank the genes by their relative induction. Richer experimental designs, however, could involve hundreds of samples—for example, complete developmental time courses in many cell lines. No two genes are likely to exhibit precisely the same response, and many distinct types of behavior may be present.
A key goal is to extract the fundamental patterns of gene expression inherent in the data. Many mathematical techniques have been developed for identifying underlying patterns in complex data for such diverse applications as object recognition by machine vision systems, phoneme detection in speech processing, bandwidth compression in telecommunications, and signal classification in electrocardiography and sleep research (6–10). The techniques are essentially different ways to cluster points in multidimensional space. They can be directly applied to gene expression by regarding the quantitative expression levels of n genes in k samples as defining n points in k-dimensional space.
Clustering Techniques.
The question is, which clustering techniques are likely to be most useful for interpreting gene expression?
One simple approach is to use direct visual inspection to group together genes with similar expression patterns. This approach was recently used by Cho et al. (4) to cluster genes whose expression correlated with particular phases of the cell cycle. The method is best suited for instances in which the patterns of interest are clear in advance (such as a periodic fluctuation in phase with the cell cycle), but it does not scale well to larger data sets and is less appropriate for discovering unexpected patterns.
A common computational approach is hierarchical clustering (6–8). Data points are forced into a strict hierarchy of nested subsets: the closest pair of points is grouped and replaced by a single point representing their set average, the next closest pair of points is treated similarly, and so on. The data points are thus fashioned into a phylogenetic tree whose branch lengths represent the degree of similarity between the sets. Hierarchical clustering has recently been described for gene expression and has clearly proven valuable (11–13).
Hierarchical clustering, however, has a number of shortcomings for the study of gene expression. Strict phylogenetic trees are best suited to situations of true hierarchical descent (such as in the evolution of species) and are not designed to reflect the multiple distinct ways in which expression patterns can be similar; this problem is exacerbated as the size and complexity of the data set grows. Hierarchical clustering has been noted by statisticians to suffer from lack of robustness, nonuniqueness, and inversion problems that complicate interpretation of the hierarchy (see ref. 14 for a detailed study). Finally, the deterministic nature of hierarchical clustering can cause points to be grouped based on local decisions, with no opportunity to reevaluate the clustering. It is known that the resulting trees can lock in accidental features, reflecting idiosyncrasies of the agglomeration rule.
Various other clustering techniques are used in biological applications but have not yet been applied to the analysis of gene expression. These techniques include Bayesian clustering, k-means clustering, and self-organizing maps (SOMs). Bayesian clustering is a highly structured approach appropriate when a strong prior distribution on the data is available. k-means clustering is a completely unstructured approach, which proceeds in an entirely local fashion and produces an unorganized collection of clusters that is not conducive to interpretation.
SOMs (9, 10) have a number of features that make them particularly well suited to clustering and analysis of gene expression patterns. They are ideally suited to exploratory data analysis, allowing one to impose partial structure on the clusters (in contrast to the rigid structure of hierarchical clustering, the strong prior hypotheses used in Bayesian clustering, and the nonstructure of k-means clustering) and facilitating easy visualization and interpretation. SOMs have good computational properties and are easy to implement, reasonably fast, and scalable to large data sets.
SOMs have been well studied and empirically tested on a wide variety of problems (15, 16). For example, Mangiameli et al. (17) applied SOMs and seven hierarchical methods to 252 “messy” data sets with real-world data imperfections such as dispersion, irrelevant variables, outliers, and nonuniform densities. SOMs were found to be significantly superior in both robustness and accuracy.
SOMs.
SOMs are constructed as follows (see Materials and Methods for details). One chooses a geometry of “nodes”—for example, a 3 × 2 grid. The nodes are mapped into k-dimensional space, initially at random, and then iteratively adjusted (Fig. 1). Each iteration involves randomly selecting a data point P and moving the nodes in the direction of P. The closest node NP is moved the most, whereas other nodes are moved by smaller amounts depending on their distance from NP in the initial geometry. In this fashion, neighboring points in the initial geometry tend to be mapped to nearby points in k-dimensional space. The process continues for 20,000–50,000 iterations.
Figure 1.

Principle of SOMs. Initial geometry of nodes in 3 × 2 rectangular grid is indicated by solid lines connecting the nodes. Hypothetical trajectories of nodes as they migrate to fit data during successive iterations of SOM algorithm are shown. Data points are represented by black dots, six nodes of SOM by large circles, and trajectories by arrows.
SOMs impose structure on the data, with neighboring nodes tending to define related clusters. An SOM based on a rectangular grid is analogous to an entomologist’s specimen drawer, with adjacent compartments holding similar insects. Alternative structures can be imposed on the data through different initial geometries, such as grids, rings, and lines, with different numbers of nodes.
We developed a computer package, genecluster, to produce and display SOMs of gene expression data. The program was then applied to various data sets involving the yeast cell cycle and hematopoietic differentiation to evaluate its ability to assist in interpretation of gene expression.
MATERIALS AND METHODS
SOMs.
An SOM has a set of nodes with a simple topology (e.g., two-dimensional grid) and a distance function d(N1,N2) on the nodes. Nodes are iteratively mapped into k-dimensional “gene expression” space (in which the ith coordinate represents the expression level in the ith sample). The position of node N at iteration i is denoted fi(N). The initial mapping f0 is random. On subsequent iterations, a data point P is selected and the node NP that maps nearest to P is identified. The mapping of nodes is then adjusted by moving points toward P by the formula:
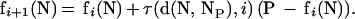 |
The learning rate τ decreases with distance of node N from NP and with iteration number i. The point P used at each iteration is determined by random ordering of the n data points generated once and recycled as needed. The function τ is defined by τ(x, i) = 0.02T/(T + 100 i) for x = ρ(i) and τ(x, i) = 0 otherwise, where radius ρ(i) decreases linearly with i (ρ(0) =3) and eventually becomes zero and T is the maximum number of iterations. genecluster is written in C, runs under UNIX, and requires a Web browser. genecluster is available from the authors.
Data Preprocessing.
A variation filter was used to eliminate genes that did not change significantly across samples. Genes were eliminated if they did not show a relative change of xand an absolute change of y units, with (x, y) = (2, 35) for yeast data and (x, y) = (3,100) for human data. Expression levels were then normalized to have mean = 0 and variance = 1. For yeast data, expression levels were normalized within each of the two cell cycles. For the human data, expression levels were normalized within the time points for each cell line.
Cell Culture.
HL-60 and U937 cells were provided by American Type Culture Collection, Jurkat cells by S. Burakoff (Dana–Farber Cancer Institute, Boston, MA), and NB4 cells line by M. Lanotte (Hôpital St. Louis, Paris, France). All trans-retinoic acid (ATRA)-resistant lines have been described (18). Cells were grown in RPMI medium 1640 with 10% fetal bovine serum. HL-60, U937, and Jurkat cells were stimulated with 10 nM phorbol 12-myristate 13-acetate (PMA) (Sigma) for 0, 0.5, 6, or 24 hours; NB4 cells were stimulated with 1 μM ATRA (Sigma) for 0, 6, 24, 48, or 72 hours. Final concentration for dimethyl sulfoxide stimulations was 1.25%.
Yeast Experiments.
Yeast data was downloaded from http://genomics.stanford.edu. The 90-minute time point was excluded because of difficulties with scaling.
Expression Analysis.
A detailed protocol is at http://www.genome.wi.mit.edu/MPR. Briefly, 1 μg of mRNA was used to generate first-strand cDNA by using a T7-linked oligo(dT) primer. After second-strand synthesis, in vitro transcription (Ambion) was performed with biotinylated UTP and CTP (Enzo Diagnostics), resulting in 40- to 80-fold linear amplification of RNA. Forty micrograms of biotinylated RNA was fragmented to 50- to 150-nt size before overnight hybridization to Affymetrix (Santa Clara, CA) HU6000 arrays. Arrays contain probe sets for 6,416 human genes (5,223 known genes and 1,193 expressed sequence tags). Because probe sets for some genes are present more than once on the array, the total number on the array is 7,227. After washing, arrays were stained with streptavidin–phycoerythrin (Molecular Probes) and scanned on a HewlettPackard scanner. Intensity values were scaled such that overall intensity for each chip of the same type was equivalent. Intensity for each feature of the array was captured by using genechip software (Affymetrix, Santa Clara, CA), and a single raw expression level for each gene was derived from the 20 probe pairs representing each gene by using a trimmed mean algorithm. A threshold of 20 units was assigned to any gene with a calculated expression level below 20, because discrimination of expression below this level could not be performed with confidence.
Northern Blotting.
Ten to twenty micrograms of total RNA was electrophoresed through denaturing agarose gels and transferred to Hybond-N nylon membranes (Amersham Pharmacia). Hybridization was performed by using Rapid-Hyb buffer (Amersham Pharmacia). A 476-bp G0S2 probe was generated corresponding to nucleotides 41–516 of the published sequence (GenBank accession no. M69199). Probes were 32P-labeled by random hexamer priming (Stratagene).
RESULTS
genecluster accepts an input file of expression levels from any gene-profiling method (e.g., oligonucleotide arrays or spotted cDNA arrays), together with a geometry for the nodes.
The program begins with two preprocessing steps that greatly improve the ability to detect meaningful patterns. First, the data are passed through a variation filter to eliminate those genes with no significant change across the samples. This prevents nodes from being attracted to large sets of invariant genes. Second, the expression level of each gene is normalized across experiments. This focuses attention on the “shape” of expression patterns rather than on absolute levels of expression.
An SOM is then computed, typically in about 1 min for large data sets such as below. genecluster uses a Web-based interface to visualize the clusters. Each cluster is represented by its average expression pattern, making it easy to discern similarities and differences among the patterns (Fig. 2a). The variation around the pattern can be visualized by means of error bars or by overlaying the patterns of all members of the cluster (Fig. 2b).
Figure 2.

Yeast Cell Cycle SOM. (a) 6 × 5 SOM. The 828 genes that passed the variation filter were grouped into 30 clusters. Each cluster is represented by the centroid (average pattern) for genes in the cluster. Expression level of each gene was normalized to have mean = 0 and SD = 1 across time points. Expression levels are shown on y-axis and time points on x-axis. Error bars indicate the SD of average expression. n indicates the number of genes within each cluster. Note that multiple clusters exhibit periodic behavior and that adjacent clusters have similar behavior. (b) Cluster 29 detail. Cluster 29 contains 76 genes exhibiting periodic behavior with peak expression in late G1. Normalized expression pattern of 30 genes nearest the centroid are shown. (c) Centroids for SOM-derived clusters 29, 14, 1, and 5, corresponding to G1, S, G2 and M phases of the cell cycle, are shown. (d) Centroids for groups of genes identified by visual inspection by Cho et al. (4) as having peak expression in G1, S, G2, or M phase of the cell cycle are shown.
SOMs are particularly well suited for exploratory data analysis, to expose the fundamental patterns in the data. The underlying structure can be readily explored by varying the geometry of the SOM. With only a few nodes, one tends not to see distinct patterns and there is large within-cluster scatter. As nodes are added, distinctive and tight clusters emerge. Beyond this point, the addition of further nodes tends to produce no fundamentally new patterns. Although there is no strict rule governing such exploratory data analysis, straightforward inspection quickly identified an appropriate SOM geometry in each of the examples below.
Yeast Cell Cycle.
We first tested genecluster on a previously published data set to determine whether it could automatically expose known patterns without using prior knowledge. For this purpose, we used data from a recent study of Cho et al. (4). These authors synchronized Sacharomyces cerevisiae in G1, released the cells, and collected RNA at 10-min intervals over two cell cycles (160 min). Expression levels of 6,218 yeast ORFs were measured by using oligonucleotide arrays. From the set of genes passing a variation filter, the authors used visual inspection to identify 416 genes showing peaks of expression in early G1, late G1, S, G2, or M phase.
We used genecluster to reanalyze the data, rapidly settling on a 6 × 5 SOM. As shown in Fig. 2a, the SOM automatically and quickly (computation time 82 sec) extracted the cell-cycle periodicity as among the most prominent features in the data. The neighboring clusters (24, 28 and 29, for example) contain genes with peak expression in late G1 phase (25–45 min and 85–105 min; Fig. 2 a and b). The genes agree well with those identified by visual inspection. Of the 105 late G1-peaking genes reported in ref. 4 that passed our variation filter, 91 (87%) were contained in the three G1-associated clusters identified by the SOM. Of the 14 remaining genes, 7 were located in neighboring clusters. More broadly, the SOM-derived clusters corresponding to the G1, S, G2, and M phases of the cell cycle (Fig. 2c) closely match those identified visually (Fig. 2d).
Macrophage Differentiation in HL-60 cells.
We next applied SOMs to models of human hematopoietic differentiation. This process is largely controlled at the transcriptional level, and blocks in the developmental program likely underlie the pathogenesis of leukemia. Cell lines modeling the differentiation process have been extensively used over the past decade to study expression of dozens of individual genes. Our goal was to take a more global approach by creating a reference database describing the behavior of some 6,000 genes.
We began by studying the myeloid leukemia cell line HL-60, which undergoes macrophage differentiation on treatment with the phorbol ester PMA. Nearly 100% of HL-60 cells become adherent and exit the cell cycle within 24 hours of PMA treatment. To monitor this process at the transcriptional level, antisense cRNA was prepared from cells harvested at 0, 0.5, 4, and 24 hrs after PMA stimulation (see Materials and Methods). Samples then were hybridized to expression-monitoring arrays from Affymetrix (Santa Clara, CA) containing oligonucleotide probes for 5,223 known human genes and 1,193 expressed sequence tags, and hybridization intensities were determined for each gene. The list of genes on the arrays and all expression data are available at http://www.genome.wi.mit.edu/MPR.
Expression levels were normalized for the 567 genes (9%) that passed the variation filter. A 4 × 3 SOM was used to organize the genes into 12 clusters (Fig. 3). Although generated without preconceptions, the clusters correspond to patterns of clear biological relevance. Most of the known genes found to be regulated have, in fact, been previously identified in the extensive literature on macrophage differentiation. Our study, however, identified the vast majority of these genes in a single experiment and also uncovered additional ones not previously known to be regulated.
Figure 3.

HL-60 SOM. HL-60 cells were treated with PMA for 0, 0.5, 4, or 24 hours, and expression levels of more than 6,000 genes were measured at each time point. The 567 genes passing the variation filter were grouped by a 4 × 3 SOM.
Cluster 11, for example, contains 32 genes with gradual induction over the time course, during which time cells gradually lose proliferative capacity and acquire hallmarks of the macrophage lineage. Four of the genes are duplicates on the array, reducing the cluster to 28 distinct genes (Table 1). Two are expresses sequence tags for which no coding sequence is available. The remaining 26 can be divided into 18 that would be expected based on current knowledge of hematopoietic differentiation [such as the antiapoptosis genes Bfl-1 and A20 and Macrophage Inflammatory Protein 1α (MIP1α)] and 8 that would be unexpected.
Table 1.
Genes in cluster 11 (PMA-induced genes in HL-60 cells)
| Gene | Description |
|---|---|
| Expected | |
| MIP1α | Macrophage Inflammatory Protein 1 α |
| BFL-1 (Bcl-2 related) | |
| PEA-15 | Major astrocytic phosphoprotein |
| CD83 antigen | |
| DTR | Diphteria toxin receptor (heparin-binding EGF-like growth factor) |
| JUNB | Protooncogene |
| P4HA | Procollagen-proline, 2-oxoglutarate 4-dioxygenase (proline 4-hydroxylase), polypeptide |
| DAF | Decay accelerating factor for complement (CD55) |
| EGR2 | Early growth response 2 |
| SLP-76 | 76-kDa tyrosine phosphoprotein |
| TNFAIP1 | Tumor necrosis factor α-inducible protein A20 |
| KNG | Kininogen |
| Fc-receptor γ-chain | |
| Tryptophanyl-tRNA synthetase | |
| BTG1 | B cell translocation gene 1 |
| RASA1 | GTPase-activating protein ras p21 (RASA) |
| CRFB4 | Cytokine receptor family II, member 4 |
| Homeobox c1 protein | |
| Unexpected | |
| GLVR1 | Leukemia virus receptor 1 |
| PTPN12 | Protein tyrosine phosphatase, non-receptor type 12 |
| FKBP25 | FK506-binding protein |
| CSNK1A1 | Casein kinase 1, alpha 1 |
| CSNK2A2 | Casein kinase 2, alpha prime polypeptide |
| RPL3 | Ribosomal protein L3 |
| RPL4 | Ribosomal protein L4 |
| HIP | Putative tumor suppressor (HNC6) |
| EST | GenBank accession no. H80240 |
| EST | GenBank accession no. T53118 |
Four of the unexpected genes (FKBP25, caseine kinases I and II, and HIP) suggest that an immunophilin-mediated pathway may play a role in macrophage differentiation. FKBP25 is a member of the immunophilin family of FK506-binding proteins, which are thought to play important roles in protein folding and trafficking (19). Caseine kinase II has been shown to play a role in activation of another immunophilin, FKBP52 (20). The HIP protein interacts with the molecular chaperone protein hsc70, which in turn acts in concert with immunophilins and antiapoptotic proteins (21).
Cluster 10 has 142 genes showing late induction. These include many genes known to be involved in macrophage differentiation (e.g., CSF1 receptor, IL1β, and cathepsin B). Cluster 2 contains 64 genes showing down-regulation on terminal differentiation induced by PMA. These include cell cycle-related genes, such as those encoding cyclin D2, cyclin D3, CDK2, and PCNA. Cluster 4 has 71 genes whose expression peaks within 30 min of PMA treatment, suggesting an immediate early response. These include serum response factor (SRF) and the early growth response gene EGR1.
These results suggest that the SOM captured the predominant patterns of gene regulation in this simple model of macrophage differentiation.
Hematopoietic Differentiation Across Four Cell Lines.
We next investigated whether the SOM approach could be applied to more complex data sets involving multiple cell lines: HL-60 and the similar myeloid cell line U937, which also undergoes macrophage differentiation in response to PMA; Jurkat, a T cell line that acquires many hallmarks of T cell activation in response to PMA; and NB4, an acute promyelocytic leukemia cell line that undergoes neutrophilic differentiation in response to ATRA. A total of 17 RNA samples were generated, yielding 6,416 data points in 17-dimensional space. Of these, 1,036 genes passed the variation filter. The genes were classified with a 6 × 4 SOM (Fig. 4), thereby grouping the 1,036 genes into 24 categories.
Figure 4.

Hematopoietic-Differentiation SOM. The 1,036 genes varying in at least one of four cell lines were used to generate a 6 × 4 SOM. Time courses for four cell lines are shown (Left to Right): HL-60 + PMA, U937 + PMA, NB4 + ATRA, Jurkat + PMA.
Cluster 21 contains 21 genes induced in the closely related cell lines HL-60 and U937, whereas the adjacent clusters 17 and 20 contain genes induced in one of the two lines. This indicates that whereas HL-60 and U937 have similar macrophage maturation responses to PMA stimulation, there are transcriptional responses that distinguish the two cell lines. Cluster 22 contains genes up-regulated in the three myeloid lines, but not the lymphoid cell line Jurkat.
We focused on Cluster 15, which contains 154 genes induced by ATRA in NB4 cells but not regulated in the other three cell lines. NB4 cells harbor a t(15;17) translocation that fuses the PML and RARα genes, resulting in a fusion protein that blocks normal neutrophil differentiation (22, 23). ATRA stimulation restores neutrophil differentiation. This response is the presumed basis of “differentiation therapy,” which is part of standard treatment for patients with acute promyelocytic leukemia, but the precise mechanism of differentiation remains uncertain.
Most of the genes in Cluster 15 encode markers of neutrophil differentiation (such as granulocyte colony stimulating factor receptor, CD59, and defensin α4) or proteins known to be induced by retinoic acid in various systems (such as the RIG-E gene and the interferon-inducible genes IFI56, INP10, and IRF1). Some unexpected genes, however, provide interesting insights into NB4 differentiation.
Of the genes showing unexpected ATRA regulation, the most strongly induced was the G0S2 gene, which encodes a protein of unknown function reported as a cyclohexamide-inducible protein in T cells (24). Northern analysis confirmed G0S2 induction as early as 6 hours after ATRA treatment of NB4 cells (Fig. 5). Interestingly, we also found that G0S2 is not up-regulated in ATRA-induced neutrophil differentiation of HL-60 cells (which lack PML/RARα); in dimethyl sulfoxide-induced neutrophil differentiation of NB4 cells; or in ATRA stimulation of ATRA-resistant NB4 cells (carrying an inactivating point mutation in the PML/RARα fusion) (Fig. 5). Whether G0S2 induction is seen in patients treated with ATRA in vivo remains to be determined, but its early induction in NB4 cells is consistent with the hypothesis that G0S2 is a candidate PML/RARα-specific, ATRA-mediated regulator of neutrophil differentiation.
Figure 5.

G0S2 Regulation. Cells were treated with the neutrophil-differentiating agents ATRA or dimethyl sulfoxide for the times (in hours) indicated. RNA was subjected to Northern analysis with a G0S2 probe (Upper). The blots were then reprobed for glyceraldehyde-3-phosphate dehydrogenase as a loading control (Lower). NB4-S1 is an ATRA-sensitive subclone of NB4. NB4-R1 and NB4-R2 are subclones that fail to differentiate after ATRA treatment. NB4-R2 has a point mutation in PML/RARα; the mechanism of ATRA resistance in NB4-R1 is unknown.
Another interesting observation is the specific induction in NB4 cells of two genes, LMP7 and UBE1L, related to ubiquitin-mediated proteolysis. Proteasome-dependent degradation of the leukemogenic PML/RARα fusion protein has been shown to occur after ATRA stimulation (25) and is thought to be a critical step in differentiation therapy, but the mechanism is unknown. Induction of LMP7, encoding a chain of the multisubunit proteasome (26), is consistent with regulation of proteolysis though induction of specific proteasome subunits. In addition, LMP7 has been shown recently to be regulated by the wild-type PML protein (27). UBE1L encodes a protein highly similar to the ubiquitin-activating enzyme E1, involved in ubiquitination of proteins targeted for degradation (28). The fact that UBE1L is specifically induced, whereas E1 itself is constitutively expressed in NB4 cells, raises the possibility that degradation of the PML/RARα protein in response to ATRA is achieved through transcriptional induction of specific components of the proteolytic apparatus. Additional experiments are required to fully test this hypothesis.
DISCUSSION
Comparative expression studies have long been known to provide important insight into biological processes. Such studies have historically proceeded one gene at a time, but the advent of array technologies has now made it possible to collect data on thousands of genes simultaneously. Such global views of gene expression are likely to reveal previously unrecognized patterns of gene regulation.
Extracting the maximum information from global expression analysis will likely require a wide range of mathematical tools, each providing different types of insight. Several recent papers, such as the study by Chu et al. (5), have employed hierarchical clustering algorithms to organize genes into a phylogenetic tree, reflecting similarity in expression patterns. Hierarchical clustering of 6,000 genes results in 5,999 nested clusters. The interpretation of these clusters—that is, the recognition of the fundamental patterns—is left to the observer.
SOMs take a fundamentally different approach. They attempt to provide an “executive summary” of a massive data set by extracting the n most prominent patterns (where n is the number of nodes in the geometry) and arranging them so that similar patterns occur as neighbors in the SOM. As with all exploratory data analysis tools, the use of SOMs involves inspection of the data to extract insights.
SOMs are widely used in data mining because they have many desirable mathematical properties, including scaling well to large data sets. In our own hands, we have indeed found them valuable in analyses involving hundreds of experiments.
The examples presented above involve relatively small data sets but illustrate the value of SOMs. Cell cycle periodicity was automatically recovered as among the most prominent patterns during yeast growth. Analysis of more complex data sets of hematopoietic differentiation identified the genes and pathways previously known to be important in this process and generated new hypotheses warranting further study. The success of the SOM methodology in identifying the predominant gene expression patterns in these well characterized model systems suggests that genome-wide expression profiling, together with appropriate computational tools, is likely to provide valuable insights into biological processes that are not yet understood at the molecular level.
Acknowledgments
We thank D. Lockhart, T. Vasicek, and N. Siemers for advice on gene expression analysis; S. Rozen and J. Theilhaber for developing computational tools; J. Park and K. Nowillo for technical assistance; J. Tang and S. Burakoff for the Jurkat stimulations; and M. Lanotte for NB4 cells. This work was supported in part by grants from Bristol-Myers Squibb, Millennium Pharmaceuticals, Affymetrix, the National Institutes of Health, and Whitehead Institute to E.S.L., and by National Institutes of Health support to E.D.
ABBREVIATIONS
- SOM
self-organizing maps
- ATRA
all trans-retinoic acid
- PMA
phorbol 12-myristate 13-acetate
References
- 1.Lockhart D J, Dong H, Byrne M C, Follettie M T, Gallo M V, Chee M, Mittmann M, Wang C, Kobayashi M, Horton, et al. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 2.De Risi J, Iyer V, Brown P. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 3.Wodicka L, Dong H, Mittmann M, Ho M, Lockhart D. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 4.Cho R J, Campbell J J, Winzeler E A, Steinmetz L, Conway A, Wodicka L, Wolfsberg T G, Gabrielian A E, Landsman D, Lockart D J, et al. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 5.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 6.Jobson J. Applied Multivariate Data Analysis: Categorical and Multivariate Methods. NewYork: Springer; 1992. [Google Scholar]
- 7.Hartigan J. Clustering Algorithms. New York: Wiley; 1975. [Google Scholar]
- 8.Gordon A E. Classification: Methods for the Exploratory Analysis of Multivariate Data. New York: Chapman & Hall; 1981. [Google Scholar]
- 9.Kohonen T. Proc IEEE. 1991;78:1464–1480. [Google Scholar]
- 10.Kohonen T. Self-Organizing Maps. Berlin: Springer; 1997. [Google Scholar]
- 11.Spellman P T, Sherlock G, Zhang M Q, Iyer V R, Anders K, Eisen M B, Brown P O, Botstein D, Futcher B. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen X, Fuhrman S, Michaels G S, Carr D B, Smith S, Barker J L, Somogyi R. Proc Natl Acad Sci USA. 1998;95:334–339. doi: 10.1073/pnas.95.1.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan B J T, Ray A P G. Appl Stat. 1995;44:117–134. [Google Scholar]
- 15.Kaski S, Kangas J, Kohonen T. Neural Comp Surv. 1997;1:102–350. [Google Scholar]
- 16.van Osdol W W, Myers T G, Paull K D, Kohn K W, Weinstein J N. J Natl Cancer Inst. 1994;86:1853–1859. doi: 10.1093/jnci/86.24.1853. [DOI] [PubMed] [Google Scholar]
- 17.Mangiameli P, Chen S K, West D. Eur J Oper Res. 1996;93:402–417. [Google Scholar]
- 18.Nason-Burchenal K, Maerz W, Albanell J, Allopenna J, Martin P, Moore M A, Dmitrovsky E. Differentiation. 1997;61:321–331. doi: 10.1046/j.1432-0436.1997.6150321.x. [DOI] [PubMed] [Google Scholar]
- 19.Jin Y, Burakoff S, Bierer B. J Biol Chem. 1992;267:10942–10945. [PubMed] [Google Scholar]
- 20.Miyata Y, Chambraud B, Radanyi C, Leclerc J, Lebeau M C, Renoir J M, Shirai R, Catelli M G, Yahara I, Baulieu E E. Proc Natl Acad Sci USA. 1997;94:14500–14505. doi: 10.1073/pnas.94.26.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hohfeld J, Jentsch S. EMBO J. 1997;16:6209–6216. doi: 10.1093/emboj/16.20.6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de The H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. Cell. 1991;66:675–684. doi: 10.1016/0092-8674(91)90113-d. [DOI] [PubMed] [Google Scholar]
- 23.Kakizuka A, Miller W H, Jr, Umesono K, Warrell R P, Jr, Frankel S R, Murty V V, Dmitrovsky E, Evans R M. Cell. 1991;66:663–674. doi: 10.1016/0092-8674(91)90112-c. [DOI] [PubMed] [Google Scholar]
- 24.Russell L, Forsdyke D. DNA Cell Biol. 1991;10:581–591. doi: 10.1089/dna.1991.10.581. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida H, Kitamura K, Tanaka K, Omura S, Miyazaki T, Hachiya T, Ohno R, Naoe T. Cancer Res. 1996;56:2945–2948. [PubMed] [Google Scholar]
- 26.Beck S, Kelly A, Radley E, Khurshid F, Alderton R P, Trowsdale J. J Mol Biol. 1992;228:433–441. doi: 10.1016/0022-2836(92)90832-5. [DOI] [PubMed] [Google Scholar]
- 27.Zheng P, Guo Y, Niu Q, Levy D E, Dyck J A, Lu S, Sheiman L A, Liu Y. Nature (London) 1998;396:373–376. doi: 10.1038/24628. [DOI] [PubMed] [Google Scholar]
- 28.Kok K, Hofstra R, Pilz A, van den Bergh A, Terpstra P, Buys C H, Carritt B. Proc Natl Acad Sci USA. 1993;90:6071–6075. doi: 10.1073/pnas.90.13.6071. [DOI] [PMC free article] [PubMed] [Google Scholar]