

Abstract
Group A Streptococcus (GAS) causes the life-threatening infection in humans known as necrotizing fasciitis (NF). Infected subcutaneous tissues from an NF patient and mice challenged with the same GAS strain possessed high bacterial loads but a striking paucity of infiltrating polymorphonuclear leukocytes (PMNs). Impaired PMN recruitment was attributed to degradation of the chemokine IL-8 by a GAS serine peptidase. Here, we use bioinformatics approach coupled with target mutagenesis to identify this peptidase as ScpC. We show that SilCR pheromone downregulates scpC transcription via the two-component system—SilA/B. In addition, we demonstrate that in vitro, ScpC degrades the CXC chemokines: IL-8 (human), KC, and MIP-2 (both murine). Furthermore, using a murine model of human NF, we demonstrate that ScpC, but not the C5a peptidase ScpA, is an essential virulence factor. An ScpC-deficient mutant is innocuous for untreated mice but lethal for PMN-depleted mice. ScpC degrades KC and MIP-2 locally in the infected skin tissues, inhibiting PMN recruitment. In conclusion, ScpC represents a novel GAS virulence factor functioning to directly inactivate a key element of the host innate immune response.
Keywords: chemokines, group A Streptococcus , peptidase, polymorphonuclear neutrophils, virulence
Introduction
Group A Streptococcus (GAS, Streptococcus pyogenes) is a major Gram-positive bacterial pathogen associated with a wide spectrum of human diseases, ranging from superficial throat and skin infections to life-threatening invasive conditions such as necrotizing fasciitis (NF) (Cunningham, 2000; Bisno et al, 2003). NF is an infection that results in a rapid and progressive destruction of fascia and fat (Kaul et al, 1997) that is frequently complicated by development of toxic shock syndrome (TSS). Even with prompt antibiotic treatment and surgical debridement, reported mortality rates in GAS NF and TSS are high, ranging from 20 to 60% (Davies et al, 1996; Kaul et al, 1997; Hassell et al, 2004). Although NF can be produced by GAS of various serotypes (Johnson et al, 2002; Bisno et al, 2003; Hassell et al, 2004), M1 and M3 strains have been most commonly reported (Cleary et al, 1992; Monnickendam et al, 1997; O'Brien et al, 2002; Sharkawy et al, 2002). Our prospective, population-based study of invasive GAS infections in Israel further identified M14 strains as strongly associated with severe soft tissue infections (Moses et al, 2003).
In vivo screening of a transposon-tagged mutant library from an invasive M14 GAS isolate identified the streptococcal invasion locus (sil) as essential for rapid dissemination of GAS in a murine model of NF (Hidalgo-Grass et al, 2002). GAS sil shares homology with the genetic competence regulon of Streptococcus pneumoniae (Claverys and Havarstein, 2002). It includes the two-component system (TCS) SilA/B and the ABC-type transporter SilD/E. Situated between these entities and preceded by a combox-like promoter (Morrison and Lee, 2000) lies the small open reading frame (ORF) silC. Highly overlapping silC, but transcribed from the reverse strand, is a sixth ORF SilCR, which is a putative competence stimulating peptide (CSP).
In the M14 serotype strains, a start codon mutation eliminates expression of the putative CSP SilCR (Hidalgo-Grass et al, 2002). We have hypothesized that sil, and SilCR in particular, may function to negatively regulate the invasive disease potential of GAS. Support for this hypothesis was provided in experiments in which mice were challenged with M14 GAS JS95 wild-type (WT) strain with or without simultaneous injection of the mature synthetic SilCR peptide. Compared to mice challenged with WT alone, co-administration of SilCR dramatically reduced bacterial proliferation, tissue necrosis, and mortality (Hidalgo-Grass et al, 2004). In mice inoculated with the WT alone, we observed a rapid spread of bacteria into the necrotic soft tissues in conjunction with a striking paucity of polymorphonuclear leukocyte (PMN) infiltration. Administration of SilCR altered the equation, triggering PMN recruitment to the site of infection with restriction of infectious spread (Hidalgo-Grass et al, 2004). The observation that a culture medium of the WT strain degrades in vitro the CXC chemokine IL-8, but when grown in the presence of the SilCR peptide IL-8 degradation is lost (Hidalgo-Grass et al, 2004), offers a potential mechanism for this effect.
In the present study, we apply a bioinformatics analysis and real-time RT–PCR measurement coupled with targeted mutagenesis to identify ScpC as the protease responsible for IL-8 degradation. We show that ScpC degrades the murine CXC chemokines KC and MIP-2 in vitro and in the tissues of experimentally infected mice, thereby critically impairing the PMN innate immune response.
Results
Identification of GAS serine proteases transcriptionally repressed by SilCR
We hypothesized that SilCR may reduce IL-8 degradation by repressing the transcription of the gene encoding the GAS IL-8 peptidase. To identify possible gene candidates for such a peptidase, we used the MEROPS database http://merops.sanger.ac.uk (Rawlings et al, 2004), which classified the GAS serine peptidases into 12 families. Excluding those groups associated with basic cellular maintenance functions, we concentrated on five families S1C, S8A, S9C, S15, and S54 (description of the families of these peptidases and their possible roles in virulence and chemokine degradation is detailed in Supplementary data).
Transcription of each serine peptidase gene in the WT was determined in the presence or absence of SilCR using real-time RT–PCR (Figure 1A), with primers designed to amplify any GAS allele belonging to the indicated families (Supplementary Table I). Of the entire candidate peptidase genes tested, only the transcription of SPy0416 (which we termed ScpC; see below) was significantly affected by SilCR administration, with a 10-fold reduction in mRNA levels. This result suggested that the SPy0416 could represent the serine peptidase activity, under negative regulation of SilCR, which functions to promote GAS chemokine degradation. Indeed, a mutant defective in SilA (silA), the response regulator of the TCS of the sil locus, had a 10-fold lower transcription of the protease than the WT and lost completely the ability to degrade IL-8 in vitro (Supplementary Figure 1S).
Figure 1.
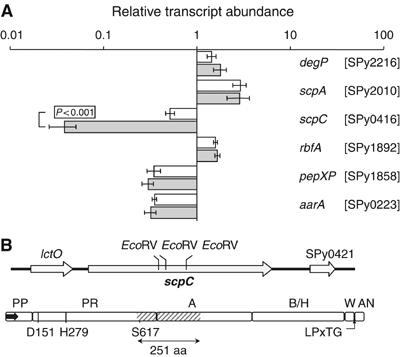
Identification of ScpC as a putative IL-8 protease. (A) The effect of SilCR on the transcription of GAS serine peptidase. The abundance of the indicated serine peptidase transcripts relative to that of gyrA was determined by real-time RT–PCR in RNA derived from WT grown to OD of 0.4 at 600 nm in the absence (clear bars) or presence (shaded bars) of SilCR (10 μg/ml). The values are mean obtained from analysis in duplicate of three independent RNA samples. Error bars represent standard deviation (s.d.). SilCR downregulates the transcription of scpC. P<0.001 (Student's test). (B) Genomic arrangement and domain organization of ScpC. Upper panel: The arrows depict the identified ORFs and their direction of transcription. The 5′ and 3′ EcoRV sites at positions 1801 and 2554 bp from the beginning of scpC were used to replace the internal scpA coding sequence with Ωkm2. spy0421 encodes a product of 236 aa with no homology to characterized proteins. Lower panel: The map of the motifs identified in the predicted sequence of ScpC includes the following: the pre-pro (PP) domain (residues 1–123) containing the signal sequence (residues 1–34) depicted as an arrow, protease domain (PR) (residues 124–688) containing Asp, His, and Ser forming the catalytic triad; the A domain (residues 689–1128) and the B/H domain (residues 1129–1560), the cell wall domain (W) (residues 1561–1613), the cell wall anchor domain (AN) (residues 1613–1647), and the LPxTG motif starting at residue 1613. The DNA region removed by the EcoRV digestion contains a segment of 251 aa (hatched) including Ser617 of the catalytic triad.
Coupled with the knowledge of the recent description of IL-8 degradation by a partially purified cell-envelope peptidase (CEP) preparation SpyCEP (Edwards et al, 2005). We were encouraged to pursue molecular genetic studies to establish the contribution of the protease to disease pathogenesis. There are several different CEPs in GAS, and SpyCEP has an extensive homology to the C5a peptidase ScpA. To conform more closely to standard GAS gene nomenclature, we elected therefore to call this gene and encoded peptidase scpC and ScpC, respectively (for streptococcal chemokine protease).
Analysis of ScpC domains
The GAS gene encoding ScpC resides between a gene encoding lactate oxidase (lctO) and an ORF of unknown function (Spy0421) (Figure 1B). By homology inference to other subtilisin-like serine CEPs (Siezen, 1999) (detailed in Supplementary data), we found that ScpC is composed of six putative domains: (PP) the pre-pro domain, which begins with a signal sequence; (PR) the peptidase domain, which contains the candidate catalytic triad of Asp+His+Ser; (A) and (B/H) domains, potentially serving as spacers for proper positioning of the active site; (W) and (AN), cell wall-anchoring domains that are separated by the LPxTG motif (Ton-That et al, 2004) (Figure 1B). The scpC gene is present in all 12 available GAS genomes encoding proteins that are highly homologous (87.8% amino-acid (aa) identity, 99.6% aa similarity).
Construction of ScpA- and ScpC-deficient mutants and assessment of their role in virulence
ScpA and ScpC prevent adequate recruitment and activation of PMN in vitro (DeMaster et al, 2002; Edwards et al, 2005); thus, the two peptidases might have overlapping roles in GAS pathogenesis. To test the contribution to virulence of ScpC in the absence of ScpA, we first constructed a ΔscpA mutant in the WT M14 GAS strain. The ΔscpA mutant was obtained by replacing 2752 bp from the coding region of the gene with aad9 (Supplementary Tables I and II). Loss of ScpA expression in the mutant was confirmed by dot blot analysis (Supplementary Figure 2S). Secondly, we deleted scpC in the scpA-deficient background. The ΔscpA/ΔscpC double mutant was constructed by replacing a 753 bp internal EcoRV fragment of scpC coding region (Figure 1B), including the critical serine residue of the catalytic triad with Ω-Km2 element. The fidelity of scpC replacement was confirmed as detailed in Materials and methods.
The WT GAS parental strain and its isogenic ΔscpA and ΔscpA/ΔscpC mutants demonstrated equivalent logarithmic growth in Todd–Hewitt Yeast (THY) media (Supplementary Figure 3Sa) and similar proliferation rates in nonimmune whole human blood (Supplementary Figure 3Sb). This result indicates that neither of the peptidases contributes significantly to GAS whole blood survival. As surface-expressed M and M-like proteins are absolutely required for GAS resistance to phagocytic killing by complement and non-stimulated PMNs (Bisno et al, 2003), these results also imply that both ΔscpA and ΔscpA/ΔscpC mutants maintain adequate levels of M protein expression. Despite different strategies used and several attempts, we were unable to inactivate scpC without first inactivating scpA (for details see Supplementary Figure 4S). This may imply that there could be a functional linkage between ScpA and ScpC in the M14 strain containing the sil locus.
Next, we tested the relative contribution of ScpA and ScpC to virulence in a murine model of GAS NF. All mice challenged with either the WT or the ΔscpA mutant experienced a rapidly progressing infection and death between 2 and 7 days after challenge (Figure 2A). The Kaplan–Meier analysis showed that the rates of death of mice inoculated with the WT and the ΔscpA mutant were similar. In marked contrast, only three mice out of the 28 challenged with the ΔscpA/ΔscpC mutant succumbed to the infection (Figure 2A), proving it to be significantly less virulent than either the WT or the ΔscpA mutant. As shown, weight changes paralleled the data shown in Figure 2A. Mice challenged with WT and the ΔscpA mutant showed progressive weight loss before death, whereas mice challenged with the ΔscpA/ΔscpC mutant regained weight by day 2 with progressive weight gain thereafter (Figure 2B). Mice challenged with either the WT or the ΔscpA mutant developed visible necrotic skin lesions of similar size 1 day after the inoculation (Figure 2C). At day 2 after inoculation, the lesion size of the WT and the ΔscpA mutant expanded and continued to increase in size until mice died by day 4 after the infection. In contrast, mice challenged with the ΔscpA/ΔscpC double mutant developed small lesions peaking in size on day 2 (Figure 2C), followed by slow resolution and complete healing by day 14 (not shown). Grossly, the small lesions caused by ΔscpA/ΔscpC mutant were superficial in nature, whereas the lesions produced by either the WT GAS or the ΔscpA mutant were deeper and more necrotic.
Figure 2.
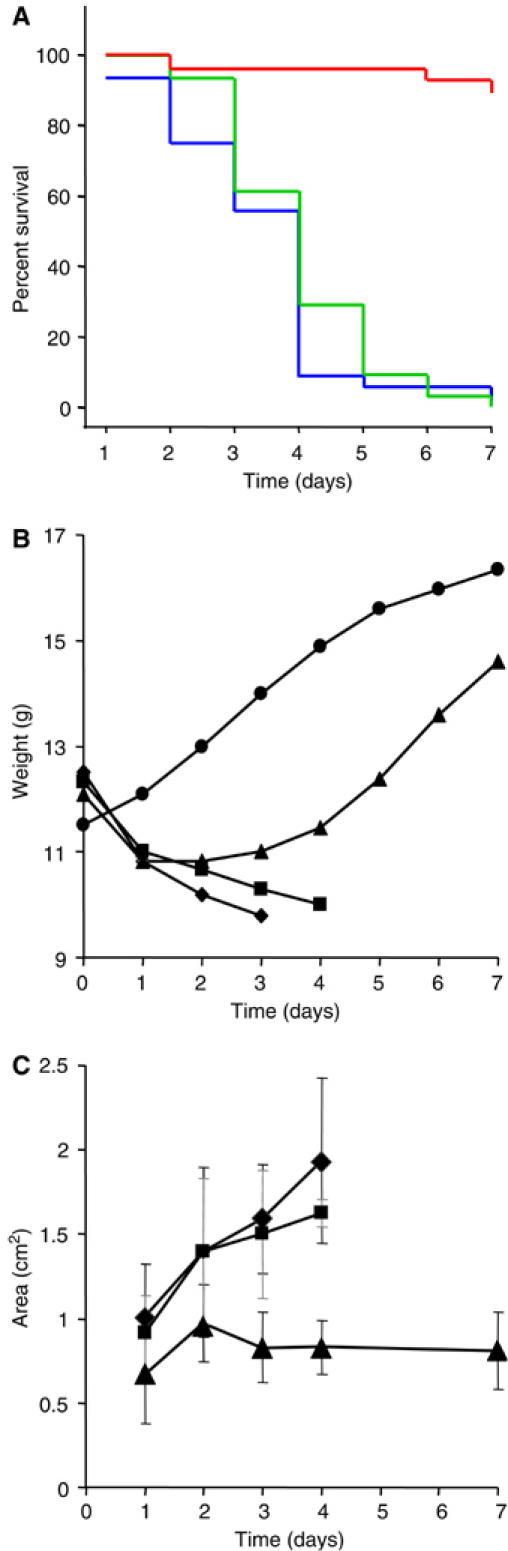
ScpC significantly contributes to virulence in the murine model of GAS necrotizing soft tissue infections. (A) Inactivation of scpC but not of scpA abolished lethality of GAS. Mice were injected subcutaneously with 1 × 108 CFU of WT ( n=32), ΔscpA (
n=32), ΔscpA ( n=31), and ΔscpA/ΔscpC (
n=31), and ΔscpA/ΔscpC ( n=28) and survival was monitored daily. The Kaplan–Meier analysis performed on five different experiments shows P<0.001 for ΔscpA/ΔscpC versus WT or versus ΔscpA and P>0.05 for WT versus ΔscpA (log rank (Mantel–Cox) test). (B) Weight change in control mice (•) and mice challenged with 1 × 108 CFU of WT (♦), ΔscpA (▪), and ΔscpA/ΔscpC (▴). Experiments were repeated five times with similar results. (C) Mean total lesion size (cm2). Mice were injected with 1 × 108 CFU of WT (♦ n=9), ΔscpA (▪ n=15), and ΔscpA/ΔscpC (▴ n=9), photographed daily, and the area was calculated using ImageJ software. Error bars represent s.d. WT and ΔscpA versus ΔscpA/ΔscpC P<0.05 (Student's test) for time points 1 and 2 and P<0.001 (Student's test) for time points 3 and 4. WT versus ΔscpA P>0.05 (Student's test) at all time points.
n=28) and survival was monitored daily. The Kaplan–Meier analysis performed on five different experiments shows P<0.001 for ΔscpA/ΔscpC versus WT or versus ΔscpA and P>0.05 for WT versus ΔscpA (log rank (Mantel–Cox) test). (B) Weight change in control mice (•) and mice challenged with 1 × 108 CFU of WT (♦), ΔscpA (▪), and ΔscpA/ΔscpC (▴). Experiments were repeated five times with similar results. (C) Mean total lesion size (cm2). Mice were injected with 1 × 108 CFU of WT (♦ n=9), ΔscpA (▪ n=15), and ΔscpA/ΔscpC (▴ n=9), photographed daily, and the area was calculated using ImageJ software. Error bars represent s.d. WT and ΔscpA versus ΔscpA/ΔscpC P<0.05 (Student's test) for time points 1 and 2 and P<0.001 (Student's test) for time points 3 and 4. WT versus ΔscpA P>0.05 (Student's test) at all time points.
In vitro degradation of chemokines by ScpC
To provide direct evidence that ScpC was responsible for chemokine degradation, we tested the set of the isogenic mutants using an in vitro IL-8 degradation assay. The ΔscpA mutant degraded IL-8 as efficiently as the parental WT strain (Figure 3A). In contrast, the isogenic ΔscpA/ΔscpC double mutant showed minimal IL-8-degrading ability (Figure 3A). To further demonstrate that ScpC was specifically responsible for IL-8 degradation, the ΔscpA/ΔscpC mutant was transformed with the expression vector pLZ (Husmann et al, 1995) harboring a full copy of the scpC gene (pLscpC; Supplementary Table II). Whereas the ΔscpA/ΔscpC mutant complemented with the vector alone (pLZ) did not degrade IL-8, the ΔscpA/ΔscpC mutant complemented with pLscpC demonstrated full restoration of IL-8 proteolysis (Figure 3A). These results indicate that ScpC but not ScpA is required for IL-8 degradation.
Figure 3.
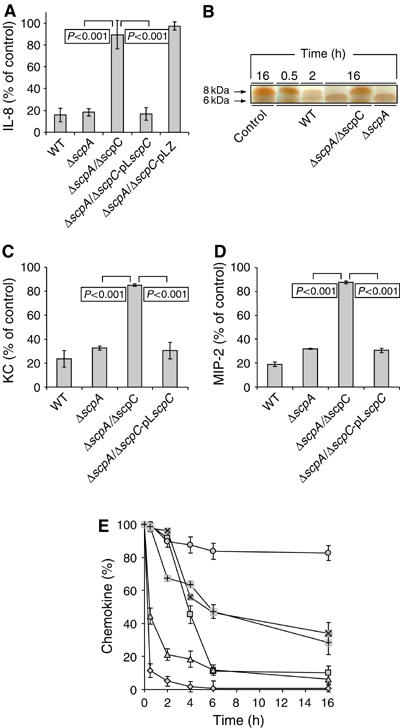
ScpC is absolutely required for chemokine degradation. (A) ScpC is responsible for IL-8 degradation. The determination of IL-8 after 2 h of proteolysis in control (in the absence of bacterial supernatant, 100%) and in supernatants of WT, ΔscpA, ΔscpA/ΔscpC, ΔscpA/ΔscpC-pLscpC, and ΔscpA/ΔscpC-pLZ was conducted by ELISA. The values are the mean obtained from analysis in duplicate (n=3). Error bars represent s.d. (B) IL-8 is cleaved by WT supernatant. Control represents IL-8 incubated for 16 h in the absence of bacterial supernatant. Samples containing IL-8 and supernatants from WT, ΔscpA/ΔscpC, and ΔscpA were incubated for the indicated time points and subjected to 17.5% SDS–PAGE, which was then silver stained. The arrows represent the molecular weights of IL-8 and of the generated 6 kDa form. (C) ScpC is responsible for KC degradation. The determination of KC after 16 h of proteolysis in control (in the absence of bacterial supernatant, 100%) and supernatants of WT, ΔscpA, ΔscpA/ΔscpC, and ΔscpA/ΔscpC-pLScpC was conducted by ELISA. The values are the mean obtained from analysis in duplicate (n=3). Error bars represent s.d. (D) ScpC is responsible for MIP-2 degradation. The determination of MIP-2 after 16 h of proteolysis in control (in the absence of bacterial supernatant, 100%) and supernatants of WT, ΔscpA, ΔscpA/ΔscpC, and ΔscpA/ΔscpC-pLScpC was conducted by ELISA. The values are mean obtained from analysis in duplicate (n=3). Error bars represent s.d. (E) Kinetics of chemokine degradation. WT supernatant was incubated with IL-8 ( ), KC (
), KC ( ), MIP-2 (
), MIP-2 ( ), RANTES (
), RANTES ( ), LIX74 (
), LIX74 ( ), or LIX93 (
), or LIX93 ( ) and the concentration of the chemokines at the indicated time points was determined by ELISA. 100% represents the concentration of chemokines at time zero. Error bars represent s.d.
) and the concentration of the chemokines at the indicated time points was determined by ELISA. 100% represents the concentration of chemokines at time zero. Error bars represent s.d.
In experiments reported by Edwards et al (2005) GAS cleave IL-8 at the C-terminus between Glu59 and Arg60 to generate peptides of ∼6 and ∼2 kDa, the former of which could be clearly identified on 17.5% SDS–PAGE (Edwards et al, 2005). Elimination of the last 14 aa at the C-terminus of IL-8 is known to markedly impair the chemokines' ability to stimulate PMN chemotaxis (25-fold reduction) and elastase release (50-fold reduction) (Clark-Lewis et al, 1991; Edwards et al, 2005).
IL-8 proteolysis by WT culture supernatant was followed by SDS–PAGE using a starting concentration of 3.0 μg/ml of the chemokine peptide (Figure 3B). The 6 kDa proteolytic fragment of IL-8 was detected clearly after 2 h and by 16 h all the IL-8 had been cleaved to the 6 kDa form. IL-8 was not degraded by culture supernatant from the ΔscpA/ΔscpC mutant, yet it was completely processed into the 6 kDa fragment by culture supernatant of the ΔscpA mutant (Figure 3B). These data show that ScpC is solely responsible for the secreted IL-8 proteolytic activity of GAS.
KC and MIP-2 are functional murine homologs of IL-8, playing significant roles in both wound healing (Gillitzer and Goebeler, 2001) and the host innate immune responses to Gram-positive bacterial skin infection (Miller et al, 2006). In addition, the CXC chemokine murine granulocyte chemotactic protein 2, also termed as LPS-induced CXC chemokine (LIX), was shown to induce PMN infiltration in mice, particularly after N- and C-terminal truncation (Wuyts et al, 1999).
Using our isogenic mutants, we found that the WT GAS parental strain and the ΔscpA mutant each efficiently degraded KC and MIP-2 (Figure 3C and D). In contrast, the isogenic ΔscpA/ΔscpC mutant demonstrated a complete loss of KC and MIP-2 degradation (Figure 3C and D), showing that ScpC is solely responsible for the degradation of these murine chemokines. MIP-2 is cleaved at the C-terminus by a partially purified ScpC preparation but less effectively than IL-8 (Edwards et al, 2005). To compare the relative rate of CXC chemokine degradation by ScpC, we subjected IL-8, KC, MIP-2, LIX (two forms of the mature peptide stretching from aa 5 to 78 (LIX74) and from aa 1 to 93 (LIX93)) and the CC mouse chemokine RANTES to proteolysis by WT supernatant. Comparative analysis of chemokine degradation by WT GAS supernatant revealed that 90% of IL-8 and 60% of KC were degraded within 30 min, whereas MIP-2 degradation was slower but completed after 6 h of incubation. In contrast, the two forms of LIX were only partially degraded, even after prolonged incubation of 16 h. RANTES was not affected at all (Figure 3E). These results demonstrate that the cleavage of CXC chemokines by ScpC is specific and the preferable substrates in a descending order are IL-8, KC, MIP-2, and LIX.
In vivo degradation of chemokines by ScpC and its impact on PMN recruitment
We sought to establish a connection between the ability of ScpC to degrade CXC chemokines in vitro (Figure 3), and the clear contribution of ScpC to GAS virulence in the murine model of NF (Figure 2). KC, MIP-2, and LIX protein levels in extracts from skin biopsies taken at 0, 6, 12, 24, and 48 h after inoculation of mice were measured by ELISA. KC levels in the skin of mice infected with ΔscpA/ΔscpC mutant rose rapidly within 6 h of bacterial challenge, peaking at 15 ng/mg extracted protein at 12 h. KC levels in the skin of mice challenged with either WT or the ΔscpA mutant did not increase significantly and remained 6- to 13-fold lower than the corresponding levels measured for the double mutant (Figure 4A). Skin levels of MIP-2 exhibited a similar behavior, peaking at 9 ng/mg of extracted protein for ΔscpA/ΔscpC-infected mice, and showing 2- to 4.8-fold lower quantities for animals challenged with either the WT or the ΔscpA mutant (Figure 4B). In contrast, mice challenged with either the WT or the mutants had similar skin levels of LIX, peaking at 3.5 ng/mg of extracted protein (Figure 4C). The deferential ability of ScpC to degrade KC, MIP-2, and LIX in vivo, as reflected in the results of Figure 4A–C, respectively, is in agreement with the cleavage specificity of ScpC as determined in vitro (Figure 3E). In contrast to the deferential degradation of KC and MIP-2 in the skin, the levels of these chemokines in spleens (Figure 4D and E) and sera (Figure 4F and G) from all three groups of mice did not differ significantly. These findings demonstrate that GAS ScpC acts locally in the infected skin.
Figure 4.
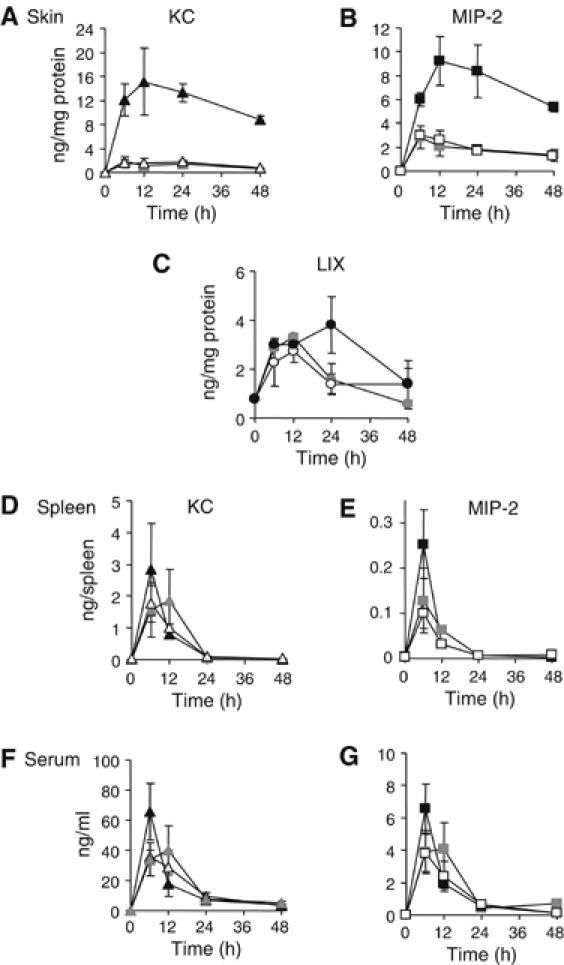
ScpC is responsible for degradation of KC and MIP-2 in infected skin. (A) KC levels (ng/mg protein) extracted from skin lesions of mice inoculated with WT ( ), ΔscpA (▵), and ΔscpA/ΔscpC (▴). Each point represents mean of the determinations performed on three mice killed at the indicated time points after infection. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P<0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point. P>0.05 (Student's test) for WT versus ΔscpA. (B) MIP-2 levels (ng/mg protein) extracted from skin lesions of mice inoculated with WT (
), ΔscpA (▵), and ΔscpA/ΔscpC (▴). Each point represents mean of the determinations performed on three mice killed at the indicated time points after infection. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P<0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point. P>0.05 (Student's test) for WT versus ΔscpA. (B) MIP-2 levels (ng/mg protein) extracted from skin lesions of mice inoculated with WT ( ), ΔscpA (□), and ΔscpA/ΔscpC (▪). Each point represents mean of the determinations performed on three mice killed at the indicated time points after infection. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P<0.01 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA. P>0.05 (Student's test) for WT versus ΔscpA for each time point. (C) LIX levels (ng/mg protein) extracted from skin lesions of mice inoculated with WT (
), ΔscpA (□), and ΔscpA/ΔscpC (▪). Each point represents mean of the determinations performed on three mice killed at the indicated time points after infection. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P<0.01 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA. P>0.05 (Student's test) for WT versus ΔscpA for each time point. (C) LIX levels (ng/mg protein) extracted from skin lesions of mice inoculated with WT ( ), ΔscpA (○), and ΔscpA/ΔscpC (•). Each point represents mean of the determinations performed on three mice killed at the indicated time points after infection. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA. P>0.05 (Student's test) for WT versus ΔscpA for each time point. (D) KC levels (ng/spleen) in spleens of three mice from the same groups of mice described in panel A. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point. (E) MIP-2 levels (ng/spleen) in spleens of three mice from the same groups of mice described in panel B. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point. (F) KC levels (ng/ml) in sera of three mice from the same groups of mice described in panel A. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point. (G) MIP-2 levels (ng/ml) in sera of three mice from the same groups of mice described in panel B. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point.
), ΔscpA (○), and ΔscpA/ΔscpC (•). Each point represents mean of the determinations performed on three mice killed at the indicated time points after infection. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA. P>0.05 (Student's test) for WT versus ΔscpA for each time point. (D) KC levels (ng/spleen) in spleens of three mice from the same groups of mice described in panel A. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point. (E) MIP-2 levels (ng/spleen) in spleens of three mice from the same groups of mice described in panel B. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point. (F) KC levels (ng/ml) in sera of three mice from the same groups of mice described in panel A. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point. (G) MIP-2 levels (ng/ml) in sera of three mice from the same groups of mice described in panel B. Assays by ELISA were conducted in duplicate. Error bars represent s.d. P>0.05 (Student's test) of ΔscpA/ΔscpC versus WT or versus ΔscpA for each time point.
To ascertain that the WT, the ΔscpA, and the ΔscpA/ΔscpC mutants triggered similar immune responses in the murine infected skin, we quantified the mRNA levels of KC, IL-6, and IL-1β by real-time RT–PCR. The corresponding mRNA levels (normalized to that of β-actin) were considerably induced in challenged animals compared to animals injected with PBS alone, reaching similar levels in the three groups of mice (Supplementary Figure 5S).
MIP-2 and KC, which were degraded both in vitro (Figure 3) and in vivo (Figure 4), were the most effective CXC chemokines in mobilizing murine PMNs, as determined in migration assays of isolated PMNs from mouse bone marrow (Supplementary Figure 6S). Neither LIX93 nor LIX74 induced a significant chemotactic activity at a concentration of 100 ng/ml, under which both MIP-2 and KC fully stimulated PMN migration (Supplementary Figure 6S).
We assessed myeloperoxidase (MPO) activity, which is a well-established marker for PMN activity (Hansson et al, 2006), and wound histopathology in mice infected with the isogenic GAS strains. Homogenates of skin lesions produced by either the WT GAS or the ΔscpA mutant strains possessed significantly less (at least 3.5-fold) MPO activity than homogenates from skin lesions of mice challenged with the ΔscpA/ΔscpC mutant (Figure 5A). To demonstrate that the PMN response is local, we challenged mice with the WT and the ΔscpA/ΔscpC mutant by injecting the same mouse at opposite flanks with each of the bacterial strains. After 48 h, we found that the MPO activity in the lesions created by the WT was significantly lower (by 2.3-fold) than that in the lesions created by the ΔscpA/ΔscpC mutant (Supplementary Figure 7Sa). An opposite relationship was observed for the lesion size and its macroscopic appearance (Supplementary Figure 7Sb).
Figure 5.
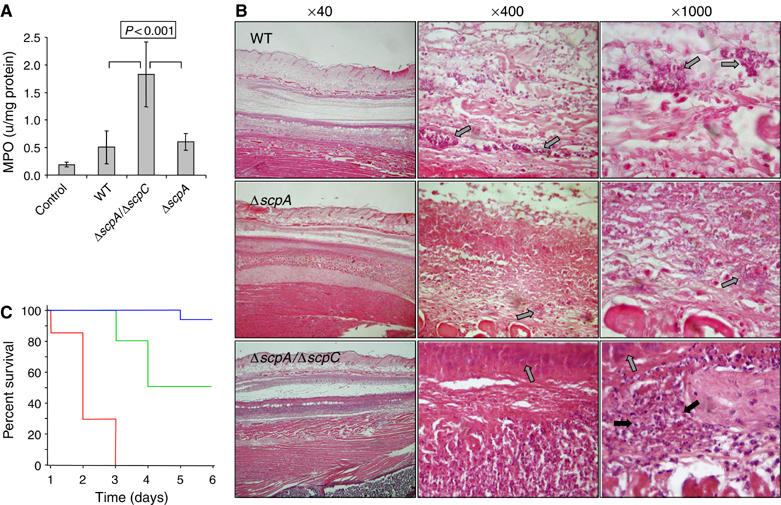
ScpC impairs PMN recruitment. (A) Forty-eight hours after inoculation, lesional (GAS) and control (PBS) 6 mm punch biopsy specimens were taken and the amount of MPO activity (units/mg protein) was determined. Each bar represents the mean±s.d. of two determinations conducted on four specimens. P<0.001 (Student's test) of ΔscpA/ΔscpC versus either WT or ΔscpA. (B) Representative photomicrographs of sections labeled with H&E prepared 2 days after inoculation with WT and its derived mutants. The gray arrow indicates presence of bacteria whereas the black arrow indicates presence of PMN. (C) PMN depletion renders mice sensitive to the ΔscpA/ΔscpC mutant. Mice were injected subcutaneously with 1 × 108 CFU of ΔscpA/ΔscpC and survival was monitored daily in cyclophosphamide-treated ( n=7), RB6-8C5-treated (
n=7), RB6-8C5-treated ( n=10), or PBS-treated (
n=10), or PBS-treated ( n=17) mice. The Kaplan–Meier analysis shows statistically significant difference (P<0.001) in the survival of the three groups of mice (using the log rank (Mantel–Cox) test).
n=17) mice. The Kaplan–Meier analysis shows statistically significant difference (P<0.001) in the survival of the three groups of mice (using the log rank (Mantel–Cox) test).
Sections of epidermis, dermis, adipose-panniculus, subcutaneous fat-connective tissue, and skeletal muscle were examined histologically. Twenty-four hours after inoculation, with any of the three GAS strains, mice displayed a focal necrosis of the epidermis and dermis-panniculus with some edema. In subcutaneous fat-connective tissue, we observed focal necrosis, cell debris, many GAS bacteria, and some PMN infiltration. PMN infiltration was more pronounced in animals infected with the ΔscpA/ΔscpC mutant. No significant differences in skeletal muscle histology were observed among the three groups, with all mice showing superficial edema. Two days after inoculation, histological differences between the groups were evident. Extensive necrosis, vascular degeneration, bacterial spread, and a paucity or complete absence of PMNs were observed in the subcutaneous fat-connective tissue and skeletal muscles of mice challenged with either the WT or the ΔscpA mutant strains (Figure 5B). In comparison, tissue sections of mice inoculated with the ΔscpA/ΔscpC mutant exhibited necrosis and showed bacterial presence but revealed a marked PMN infiltration (Figure 5B).
To confirm that PMN infiltration plays a major role in rendering mice resistant to the infection by ΔscpA/ΔscpC mutant, we tested the virulence of the ΔscpA/ΔscpC mutant in PMN-depleted mice. PMN depletion was achieved by intraperitoneal injection of mice with either cyclophosphamide or the anti-GR1 antibody (RB6-8C5). Depletion of PMNs by both means rendered mice sensitive to the double mutant as compared to control mice, which were completely resistant (Figure 5C). The cyclophosphamide-treated mice were more sensitive than the RB6-8C5-treated mice, probably because the drug reduces also the populations of lymphocytes and monocytes (Braff et al, 2005). In conclusion, ScpC through its ability to degrade CXC chemokines impairs PMN recruitment to the site of infection, facilitating GAS survival and systemic spread.
Discussion
GAS NF is characterized by extensive local necrosis of subcutaneous soft tissues and skin and its microbiologic etiology is confirmed by isolation of the pathogen from a normally sterile body site. Because of the rapid progression of necrosis in GAS NF patients, medical treatment typically includes extensive debridement of soft tissues and occasionally necessitates amputation of extremities. Consequently, GAS has been coined by the public as ‘the flesh-eating bacterium'.
Impairment of PMN recruitment plays a central role in the pathogenesis of NF. A recent review of histopathological analysis of soft tissues debrided from human NF patients with highest disease severity revealed very few or absence of PMNs at the infection site (Bakleh et al, 2005). In the baboon model of human NF, surviving baboons have an intense PMN influx into the site of inoculation, whereas those that die have no PMN influx at all (Taylor et al, 1999). In our previous studies, we show that mice challenged with the WT GAS M14 strain develop a lethal infection that is typified by the absence of PMN migration to the initial site of infection, mirroring the pathological findings in the NF patient from which the bacterium was first isolated (Hidalgo-Grass et al, 2004). There, PMN infiltration was absent in necrotic tissues with high bacterial load but was clearly apparent in the non-necrotic surrounding tissues that were free of bacteria (Hidalgo-Grass et al, 2004). Thus, inhibition of PMN recruitment by GAS may be a local phenomenon evident only in the immediate vicinity of invading bacteria.
In this study, we used targeted mutagenesis to demonstrate that ScpC (also known as SpyCEP) is the GAS peptidase responsible for proteolysis of both human (IL-8) and the mouse (KC and MIP-2) CXC PMN chemokines. N- and C-terminal truncated LIX have been suggested to act as functional homologs of IL-8 in the mouse (Wuyts et al, 1999). LIX is a poor substrate of ScpC, as revealed by in vitro assays of degradation and by measuring LIX content in GAS-infected murine skin. Even so, LIX contribution to murine PMN recruitment seems to be negligible compared to that of MIP-2 or KC, as assessed in assays of PMN migration.
We show that IL-8 is not further degraded beyond its cleavage into two fragments, even following prolonged incubation with WT GAS culture supernatant. This strongly suggests that ScpC is solely responsible for inactivation of the host CXC chemokines. Indeed, it was reported that both IL-8 and MIP-2 are cleaved by partially purified ScpC at analogous positions, between Gln59 and Arg60 and Gln60 and Lys61, respectively (Edwards et al, 2005).
scpC transcription is negatively regulated by the SilCR CSP peptide in the M14 strain and this requires the SilA/B TCS. Blood isolates of GAS were better able to degrade human IL-8 than throat isolates, suggesting that ScpC expression is upregulated in invasive GAS isolates (Edwards et al, 2005). It would be interesting to decipher the mode of scpC regulation in these isolates, particularly in the highly invasive M1 and M3 serotype strains that lack the sil locus.
Here, we provide the first evidence that ScpC is an essential GAS virulence factor in the pathogenesis of invasive skin and soft tissue infection. This is best established by the innocuous phenotype of ΔscpA/ΔscpC mutant (compared to lethal phenotype of the WT) in untreated mice, in contrast to its lethal phenotype in PMN-depleted mice. The GAS C5a peptidase (ScpA) could also be speculated to interfere with PMN recruitment through inactivation of the chemotactic component C5a of the complement system (Wexler et al, 1985). However, our finding that ScpA does not contribute to GAS virulence in the murine NF model may reflect the observation that a homolog of ScpA, in Streptococcus agalactiae, has far lower activity against murine C5a than the human C5a (Bohnsack et al, 1993). ScpA has also been shown to have no effect on mortality in a mouse air sac model (Ji et al, 1996).
In summary, we provide the first direct in vivo evidence of a novel bacterial virulence mechanism: degradation of host CXC chemokines to prevent PMN recruitment with consequent systemic bacterial spread from the initial tissue focus of infection. The peptidase responsible for this virulence phenotype, ScpC, is highly conserved among all eight available genomes of GAS. Therapies to neutralize ScpC activity or to block its expression (e.g. through administration of SilCR CSP (Hidalgo-Grass et al, 2004) and see also Supplementary Figure 1Sb) can represent a novel strategy to enhance the host innate immune response to invasive GAS infection.
Materials and methods
Bacterial strains, growth conditions, and plasmids
Primers, bacterial strains, and plasmids used in the study are described in Supplementary Tables I and II. All experiments and strain construction employed GAS strain JS95 of M14 serotype (Hidalgo-Grass et al, 2002; Moses et al, 2003). Molecular cloning experiments utilized Escherichia coli JM109 strain, which was cultured in Laria-Bertani broth, Lennox (Becton, Dickinson, Sparks, MD, USA). For culturing of GAS, we employed Todd-Hewitt medium (Becton, Dickinson) supplemented with 0.2% yeast extract (Becton, Dickinson) (THY media) with incubation at 37°C in sealed tubes without agitation. To produce solid media, Bacto™ Agar (Becton, Dickinson) was added to a final concentration of 1.4%. Antibiotics were added at the following concentrations when necessary: for GAS: 250 μg/ml kanamycin (Km), 50 μg/ml spectinomycin (Spec), 1 μg/ml erythromycin (Erm), and 7 μg/ml chloramphenicol (Cm); for E. coli: 100 μg/ml ampicillin (Amp), 50 μg/ml Spec, 750 μg/ml Erm, and 10 μg/ml Cm. All the antibiotics were purchased from Sigma-Aldrich (St Louis, MO, USA).
Manipulation of DNA
Plasmid DNA was isolated by mini-preps (Roche Applied Science, Basel, Switzerland) or midi-preps (Promega, Madison, WI, USA) according to the manufacturer's instructions and used to transform E. coli by standard methods and to transform GAS by electroporation as described previously (Caparon and Scott, 1991). Restriction endonucleases, ligases, and polymerases were used according to the recommendations of the manufacturers. Chromosomal DNA was purified from GAS as described previously (Caparon and Scott, 1991) or by using the Wizard® Genomic DNA Purification Kit (Promega). Linear DNA fragments were purified using Certified™ low-melt agarose (Bio-Rad, Hercules, CA, USA) before electroporation into GAS strains. PCR products were purified using a commercial kit (QIAquick PCR Purification Kit, Qiagen, Hilden, Germany). All other procedures were conducted according to standard protocols (Sambrock and Maniatis, 1989).
RNA isolation and real-time RT–PCR
For RNA preparations, WT or derivative mutants were grown in THY in the absence or presence of SilCR (10 μg/ml) to an OD600 of ∼0.4. SilCR was synthesized and purified to 96% purity by BioSight (Karmiel, Israel). RNA was prepared by hot acidic phenol extraction as described previously (Ravins et al, 2000). Representative samples were assessed for RNA integrity by electrophoretic analysis and measurement of the A260/A280 ratio was used to determine the RNA concentration and purity (accepted if >1.8). Contaminating DNA was removed by DNase treatments according to the manufacturer's instructions (RQ1 RNase free DNase, Promega). Samples were rejected if PCR amplification preformed with RNA templates (primers V-sra-f and V-sra-r; Supplementary Table I) indicated the presence of contaminating DNA. Total RNA (5 μg) was used for cDNA synthesis using M-MLV reverse transcriptase (Promega), according to the manufacturer's protocol. For real-time RT–PCR, primers (Supplementary Table I) were designed using Primer Express™ software v2.0 (Applied Biosystems, Foster City, CA). SYBR-green mix (Applied Biosystems) was used for fluorescence detection with the ABI Prism 7000 SDS real-time PCR system (Applied Biosystems) according to the manufacturer's protocol. The level of transcription of gyrase subunit A (gyrA) was used to normalize expression data for each target gene. Transcription of gyrA is constant under a variety of in vitro experimental conditions (Graham et al, 2002). Each assay was performed in duplicate with at least three RNA templates prepared from bacteria from independent cultures on different days. The data were analyzed according to the standard curve method (Applied Biosystem support) and are presented as abundance of transcript relative to that of gyrA. Tissue samples were isolated from lesional (GAS) and control (PBS) mice using 6 mm punch biopsy (Acuderm Inc., USA) 24 h after inoculation. The samples were homogenized by a Polytron (Kinematica AG, Lucerne, Switzerland) in the presence of guanidine thiocyanate and β-mercaptoethanol to prevent degradation by ribonucleases. Total RNA was isolated using SV Total RNA Isolation System (Promega) according to the manufacturer's recommendations. Quantitative real-time PCRs were performed as described above. Primer sequences for KC, IL-6, IL-1β, and the normalizer, β-actin, are provided in Supplementary Table I.
Strategy of mutants' construction
Mutant strains of the genotypes silA−, Δemm, ΔscpA, and ΔscpA/ΔscpC were derived from strain WT by insertion inactivation or by replacing the corresponding chromosomal genes with either aad9 or Ωkm2. Δemm was constructed as described previously (Hidalgo-Grass et al, 2002). The deletion mutant ΔscpA gene was constructed by replacing 1911 bp of the scpA gene with aad9. This was carried out by cloning a fragment containing 453 bp of the upstream region of ScpA, aad9, and 509 bp of the 3′ region of ScpA. The upstream region contained the intragenic region between mga and ScpA and 21 bp of the 5′ end of scpA and was PCR amplified using the primers M-scpC5′-f and M-scpA5′-r (Supplementary Table I). aad9 was PCR amplified using the plasmid pFW11 as a template and the primers M-aad9NcoI-f and M-aad9PstI-r (Supplementary Tables I and II). The downstream region of scpA was amplified using the primers M-scpA3′-f and M-scpA3′-r (Supplementary Table I). aad9 and its upstream and downstream flanking fragments were cloned into the temperature-sensitive E. coli-streptococcal shuttle vector pJRS233 yielding the plasmid pJscpAaad9 (Supplementary Table II). The plasmid was electroporated into WT, and Erm- and Spec-resistant transformants were isolated at the permissive temperature (30°C). Growth of transformants at the non-permissive temperature (37°C) resulted in the integration of the plasmid. For the second recombination event, one of the single recombination mutants was further passaged for 8 days at 30°C without Erm and colonies were replica plated on THY plates containing either Erm or Spec. Growth of transformants in the presence of Spec but not in the presence of Erm indicated that a second recombination event occurred resulting in scpA exchange with the aad9 and excision of pJRS233 yielding the strain ΔscpA. The replacement was verified using the primers M-scpA5′-f and M-scpA3′-r (Supplementary Table I). The loss of C5a peptidase expression was confirmed by dot blot analysis (Supplementary Figure 2S).
To construct the silA-disrupted mutant, the internal region of the gene was PCR amplified using the primers M-silA-f and M-silA-r (Supplementary Table I), cloned into the pJRS233 vector and transformed into WT, and mutants resistant to Erm were isolated. The fidelity of the single integration was verified by two sets of primers: V-silA5′-f/V-M13-r and V-M13-f/V-silB3′-r (Supplementary Table I).
To construct the double deletion mutant ΔscpA/ΔscpC, a DNA fragment of 5313 bp containing scpC, 326 bp upstream and 42 bp downstream, was PCR amplified with the high-fidelity polymerase Pwo (Roche) using the primers M-scpC-f and M-scpC-r (Supplementary Table I). Adenine was added to the ends of the PCR product by Klenow polymerase in the presence of dATP and the DNA fragment was then cloned into pGEM-T-easy vector (Promega) according to the manufacturer's instructions to yield pGscpC (Supplementary Table II). An EcoRV restriction released a 753 bp from within the scpC including the serine 617 required for proteolysis (see Figure 1B). The EcoRV fragment was replaced with SmaI-digested Ωkm2 (2043 bp) derived from pBRΩkm2 (Perez-Casal et al, 1991), yielding the plasmid pGscpCΩkm2 (Supplementary Table II). The fidelity of the construction was verified by sequencing using the primers V-scpC-f and V-scpC-r (Supplementary Table I). The scpCΩkm2 fragment was released by digestion of pGscpCΩkm2 with EcoRI and the linear fragment of 6623 bp was electroporated into ΔscpA and transformants were selected for double homologous recombination on plates containing Km and Spec. The fidelity of scpC replacement with scpCΩkm2 allele in ΔscpA/ΔscpC was verified by PCR with the primers V-scpC-f and V-scpC-r (Supplementary Table II).
For the complementation of ΔscpA/ΔscpC with scpC, the latter was released from pGscpC by digestion with EcoRI and cloned into pLZ12 (Supplementary Table II), which was digested with the same enzyme and dephosphorylated before ligation. The E. coli colonies resistant to Km and Cm were screened for scpC presence using the primers V-scpC-f and V-scpC-r (Supplementary Table I). The plasmid pLscpC was electroporated into ΔscpA/ΔscpC and transformants were selected on plates containing Km and Cm.
Sequence analyses
Serine peptidases of GAS were retrieved from MEROPS database (Rawlings et al, 2004) using Streptococcus pyogenes as the key word for an organism. Sequences of scpC in the GAS genomes were identified by database homology search using BLAST (BLAST with microbial genomes, NCBI) (Altschul et al, 1997). The complete aa sequences of ScpC from the various GAS genomes (S. pyogenes M1 strain SF370 (Spy0416, NP_268723), M3 strain MGAS315 (SpyM3_0298, NP_664102), M3 SSI-1 (SPs1559, BAC64654), M5 strain MGAS5005 (M5005_Spy0341, AAZ50960), M6 strain MGAS10394 (M6_Spy0367, AAT86502), M18 strain MGAS8232 (spyM18_0464, NP_606696), M28 strain MGAS6180 (M28_Spy0329, AAX71443), and M49 strain M49 591 (SpyoM01000941, ZP_00365806)), CspA from GBS (AAN85092), and PrtS from Streptococcus thermophilus (AAG09771) were analyzed and compared by multiple sequence alignment using VNTI (Vector NTI 9.1.0 2004; Invitrogen Corporation, Carlsbad, CA, USA) and ClustalX (Thompson et al, 1997). Domains were identified and analyzed using NCBI Conserved Domain Database Search CDD http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi and by Conserved Domain Architecture Retrieval Tool (CDART) http://www.ncbi.nlm.nih.gov/Structure/lexington/lexington.cgi?cmd=rps. Motifs were identified using ProSite (Motif Scan) (Falquet et al, 2002) and by Block Search http://blocks.fhcrc.org/blocks/blocks_search.html. We used SignalP 3.0 Server (Bendtsen et al, 2004) to predict the presence and location of signal peptide cleavage sites.
Analysis of ScpA expression
To analyze the expression of ScpA on GAS surface, strains were cultured overnight in THY, washed in PBS by repeated centrifugation, and resuspended in PBS to an OD600 ∼ 0.8. Bacterial suspensions were diluted 1:50 and then diluted serially by two-fold and 3 μl was spotted on two membranes of nitrocellulose. After blocking (10% non-fat milk), the membranes were incubated with anti-ScpA (a generous gift from P Cleary, Minnesota, USA and Patrick Trieu-Cuot, Paris, France) diluted (1:1000) or with anti-GAS (Fitzgerald, USA) diluted (1:3000) antibodies for 2 h. Then, the membranes were washed three times with Tris-buffered saline supplemented with 0.03% Tween 20 (TBS-T), followed by 1 h incubation with a secondary goat anti-rabbit IgG HRP-conjugated (Promega) at a dilution of 1:20 000. Membranes were washed four times in TBS-T and incubated for 5 min with chemiluminescent substrate Super Signal®WestPico (Pierce, Rockford, IL, USA) and dots were visualized on X-ray film after 1 min of exposure.
Blood survival, mouse infection assays, and histology
The ability of GAS to survive in non-human blood as well as the murine model soft tissue infection both were performed as detailed previously (Hidalgo-Grass et al, 2002, 2004). For histological analysis, skin samples including underlying bone (lumbar area) were analyzed. Central full-thickness specimens were made and fixed in formalin (10%). Two apposing halves were placed in the block to be processed, so that the examined tissue consisted of the central area of the skin sample. Tissues were decalcified for 4 h and placed in formalin, dissected, and embedded in paraffin. Hematoxylin and eosin (H&E) staining was performed on the paraffin-coated sections. All procedures were carried out by Diagnostic Veterinary Pathology Services (PathoVet, Kfar Bilu, Israel). For analysis, we examined the following sections: epidermis, dermis, adipose-panniculus, subcutaneous fat-connective tissue, and skeletal muscle surrounding vertebrae. Measurements of total lesion size (cm2) were made by analyzing digital photographs (Nikon Coolpix 5700) of mice taken every day between days 1 and 4 after inoculation and at day 8 for mice inoculated with the ΔscpA/ΔscpC double mutant. Analysis was performed with the software program ‘Image J' (NIH Research Service Branch http://rsbweb.nih.gov/ij/) and a millimeter ruler as a reference. The Institutional Ethics Committee for animal care approved all animal procedures.
Assay of chemokine degradation in vitro
IL-8, KC, and MIP-2 degradation was performed and quantified by ELISA using the Quantikine kit (R&D Systems, Minneapolis, MN, USA) as detailed previously (Hidalgo-Grass et al, 2004). To follow the kinetics of IL-8 degradation by SDS–PAGE, the concentration of IL-8 in the proteolysis reaction (Hidalgo-Grass et al, 2004) was increased from 1 to 8 μg/ml and the fetal calf serum in the bacterial supernatant was lowered to 0.1% to enable detection of IL-8 on 17.5% SDS–PAGE. Samples of 24 μl from proteolysis reaction were withdrawn after 30 min, 2 h, and 16 h, then 6 μl of 5 × SDS–PAGE sample buffer was added and samples were boiled for 2 min. Samples were subjected to electrophoresis and protein bands were visualized by silver staining (silver stain kit (Bio-Rad)). To follow the kinetics of IL-8, KC, MIP-2, and RANTES degradation, the chemokines were incubated at an initial concentration of 150 ng/ml and samples of 0.1 ml were withdrawn from the reaction at the desired time points. The amount of the relevant chemokine was determined by ELISA (R&D Systems) as described above.
Assay of chemokine degradation in vivo
The WT and its derived mutants ΔscpA and ΔscpA/ΔscpC were grown and prepared for subcutaneous inoculation exactly as for evaluating mice survival in the soft tissue model of human NF (Hidalgo-Grass et al, 2002, 2004). At specific times after injection, mice were killed and various tissues were isolated. Tissue sections surrounding the lesion were incised and minced with scissors. The chemokines were extracted from the disrupted tissues by incubation for 1 h at room temperature with lysis buffer containing 10 mM Tris–HCl pH 7.8, supplemented with 1% NP-40, 150 mM NaCl, and 40 mM EDTA. The presence or absence of complete Minimix protease inhibitors (Roche) did not affect the content of the extracted chemokines. The extracts were spun at 17 000 g for 5 min at room temperature. The supernatants were stored at −80°C until all samples were collected. The amounts of KC, MIP-2, and LIX were determined by ELISA (R&D Systems) and normalized according to the protein content of the corresponding samples, which was measured by BIO-RAD protein assay (Bio-Rad Laboratories). Isolated spleens were gently disrupted and suspended in Dulbecco's PBS (Sigma), containing complete minimix protease inhibitors (Roche). Supernatants of spleen suspensions were accumulated and the amounts of KC and MIP-2 were determined as described above. Blood samples were obtained by cardiac puncture. After coagulation, sera were collected and the amounts of KC and MIP-2 were determined as described.
MPO assay
Lesional 6 mm punch biopsy (Acuderm) specimens were incised and then homogenized for 30 s at 0°C by Polytron (Kinematica AG, Lucerne, Switzerland). MPO activity was determined with an MPO assay kit according to the manufacturer's recommendations (Cytostore, Calgary, Alberta, Canada). The MPO units of activity were normalized according to the protein content present in the corresponding samples, which was measured by BIO-RAD protein assay (Bio-Rad Laboratories).
Neutrophil isolation and transwell migration assays
Bones from BALB/c mice were splashed with PBS and cells were treated with RBC lysing solution (0.155 M NH4Cl, 0.01 M KHCO3, 0.01 mM EDTA, pH 7.4), washed, and then re-suspended in RPMI (600 μl) supplemented with 1% FCS. The assay of PMN migration to the indicated concentrations (2, 10, and 100 ng/ml) of KC, MIP-2, LIX74, and LIX93 (R&D Systems and Cytolab/Peprotech Asia) was performed as described previously (Beider et al, 2003). Briefly, RPMI (600 μl) supplemented with 1% FCS containing the chemokines at the indicated concentrations was placed into the lower chamber of a Costar 24-well transwell (Corning, NY). Cells (2 × 105) in 100 μl medium were placed into the upper chamber (pore size 5 μm) and cells were collected from both chambers after 4 h of migration at 37°C and counted by flow cytometry (FACSsort, Becton Dickinson, San Jose, CA) after labeling with anti-GR1 antibody. Percentage of migrating PMN was calculated by dividing the number of migrating PMNs by the total number of PMNs present.
PMN depletion
PMN depletion was achieved by administration of either cyclophosphamide or the monoclonal antibody RB6-8C5 (R&D) into the BALB/c mice used for the model of human NF, according to the procedure described previously (Braff et al, 2005) with the following modifications. Mice were injected with ΔscpA/ΔscpC mutant 48 h after cyclophosphamide treatment with 3 μg/mouse. After 48 h, the number of circulating PMNs in the peripheral blood dropped from 6% to less than 0.6% as determined by flow cytometry (FACSsort, Becton Dickinson, San Jose, CA), following labeling with anti-GR1 antibody (R&D). The amount of administered monoclonal antibody RB6-8C5 was 0.1 μg/mouse. Twenty-four hours after treatment, the number of circulating PMNs in the peripheral blood dropped from 9 to 2%.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Pat Cleary (Department of Microbiology, University of Minnesota, Minneapolis, MN, USA) and Patrick Trieu-Cuot (Unit of Gram-positive Bacterial Pathogens, Institute Pasteur, Paris, France) for providing us with the anti-ScpA antibody and Miriam Ravins (Institute of Microbiology, The Hebrew University-Hadassah Medical School) for participating in the construction of the scpA-deficient mutant and for critical reading of the manuscript. We thank Hana Wald from the Goldyne Savad Institute of Gene Therapy for technical assistance. We thank Nahum Shpigel for encouragement and for valuable suggestions concerning the MPO assay. This work was supported by grants from the USA–Israel Binational Science Foundation (to EH and VN), the Center for the Study of Emerging Diseases (to EH), and The Israeli Science Foundation administered by the Israel Academy of Science and Humanities (to EH). EH is an international research scholar from the Howard Hughes Medical Institute.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM (2005) Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis 40: 410–414 [DOI] [PubMed] [Google Scholar]
- Beider K, Nagler A, Wald O, Franitza S, Dagan-Berger M, Wald H, Giladi H, Brocke S, Hanna J, Mandelboim O, Darash-Yahana M, Galun E, Peled A (2003) Involvement of CXCR4 and IL-2 in the homing and retention of human NK and NK T cells to the bone marrow and spleen of NOD/SCID mice. Blood 102: 1951–1958 [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: signalP 3.0. J Mol Biol 340: 783–795 [DOI] [PubMed] [Google Scholar]
- Bisno AL, Brito MO, Collins CM (2003) Molecular basis of group A streptococcal virulence. Lancet Infect Dis 3: 191–200 [DOI] [PubMed] [Google Scholar]
- Bohnsack JF, Chang JK, Hill HR (1993) Restricted ability of group B streptococcal C5a-ase to inactivate C5a prepared from different animal species. Infect Immun 61: 1421–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff MH, Zaiou M, Fierer J, Nizet V, Gallo RL (2005) Keratinocyte production of cathelicidin provides direct activity against bacterial skin pathogens. Infect Immun 73: 6771–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparon MG, Scott JR (1991) Genetic manipulation of pathogenic streptococci. Methods Enzymol 204: 556–586 [DOI] [PubMed] [Google Scholar]
- Clark-Lewis I, Schumacher C, Baggiolini M, Moser B (1991) Structure–activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J Biol Chem 266: 23128–23134 [PubMed] [Google Scholar]
- Claverys JP, Havarstein LS (2002) Extracellular-peptide control of competence for genetic transformation in Streptococcus pneumoniae. Front Biosci 7: d1798–d1814 [DOI] [PubMed] [Google Scholar]
- Cleary PP, Kaplan EL, Handley JP, Wlazlo A, Kim MH, Hauser AR, Schlievert PM (1992) Clonal basis for resurgence of serious Streptococcus pyogenes disease in the 1980s. Lancet 339: 518–521 [DOI] [PubMed] [Google Scholar]
- Cunningham MW (2000) Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13: 470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies HD, McGeer A, Schwartz B, Green K, Cann D, Simor AE, Low DE (1996) Invasive group A streptococcal infections in Ontario, Canada. Ontario Group A Streptococcal Study Group. N Engl J Med 335: 547–554 [DOI] [PubMed] [Google Scholar]
- DeMaster E, Schnitzler N, Cheng Q, Cleary P (2002) M(+) group A streptococci are phagocytized and killed in whole blood by C5a-activated polymorphonuclear leukocytes. Infect Immun 70: 350–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RJ, Taylor GW, Ferguson M, Murray S, Rendell N, Wrigley A, Bai Z, Boyle J, Finney SJ, Jones A, Russell HH, Turner C, Cohen J, Faulkner L, Sriskandan S (2005) Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J Infect Dis 192: 783–790 [DOI] [PubMed] [Google Scholar]
- Falquet L, Pagni M, Bucher P, Hulo N, Sigrist CJ, Hofmann K, Bairoch A (2002) The PROSITE database, its status in 2002. Nucleic Acids Res 30: 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillitzer R, Goebeler M (2001) Chemokines in cutaneous wound healing. J Leukoc Biol 69: 513–521 [PubMed] [Google Scholar]
- Graham MR, Smoot LM, Migliaccio CA, Virtaneva K, Sturdevant DE, Porcella SF, Federle MJ, Adams GJ, Scott JR, Musser JM (2002) Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc Natl Acad Sci USA 99: 13855–13860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson M, Olsson I, Nauseef WM (2006) Biosynthesis, processing, and sorting of human myeloperoxidase. Arch Biochem Biophys 445: 214–224 [DOI] [PubMed] [Google Scholar]
- Hassell M, Fagan P, Carson P, Currie BJ (2004) Streptococcal necrotising fasciitis from diverse strains of Streptococcus pyogenes in tropical northern Australia: case series and comparison with the literature. BMC Infect Dis 4: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo-Grass C, Dan-Goor M, Maly A, Eran Y, Kwinn LA, Nizet V, Ravins M, Jaffe J, Peyser A, Moses AE, Hanski E (2004) Effect of a bacterial pheromone peptide on host chemokine degradation in group A streptococcal necrotising soft-tissue infections. Lancet 363: 696–703 [DOI] [PubMed] [Google Scholar]
- Hidalgo-Grass C, Ravins M, Dan-Goor M, Jaffe J, Moses AE, Hanski E (2002) A locus of group A Streptococcus involved in invasive disease and DNA transfer. Mol Microbiol 46: 87–99 [DOI] [PubMed] [Google Scholar]
- Husmann LK, Scott JR, Lindahl G, Stenberg L (1995) Expression of the Arp protein, a member of the M protein family, is not sufficient to inhibit phagocytosis of Streptococcus pyogenes. Infect Immun 63: 345–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, McLandsborough L, Kondagunta A, Cleary PP (1996) C5a peptidase alters clearance and trafficking of group A streptococci by infected mice. Infect Immun 64: 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DR, Wotton JT, Shet A, Kaplan EL (2002) A comparison of group A streptococci from invasive and uncomplicated infections: are virulent clones responsible for serious streptococcal infections? J Infect Dis 185: 1586–1595 [DOI] [PubMed] [Google Scholar]
- Kaul R, McGeer A, Low DE, Green K, Schwartz B (1997) Population-based surveillance for group A streptococcal necrotizing fasciitis: clinical features, prognostic indicators, and microbiologic analysis of seventy-seven cases. Ontario Group A Streptococcal Study. Am J Med 103: 18–24 [DOI] [PubMed] [Google Scholar]
- Miller LS, O'Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, Thirumala A, Cheung AL, Cheng G, Modlin RL (2006) MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 24: 79–91 [DOI] [PubMed] [Google Scholar]
- Monnickendam MA, McEvoy MB, Blake WA, Gaworzewska ET, Hallas G, Tanna A, Efstratiou A, George RC (1997) Necrotising fasciitis associated with invasive group A streptococcal infections in England and Wales. Adv Exp Med Biol 418: 87–89 [DOI] [PubMed] [Google Scholar]
- Morrison DA, Lee MS (2000) Regulation of competence for genetic transformation in Streptococcus pneumoniae: a link between quorum sensing and DNA processing genes. Res Microbiol 151: 445–451 [DOI] [PubMed] [Google Scholar]
- Moses AE, Hidalgo-Grass C, Dan-Goor M, Jaffe J, Shetzigovsky I, Ravins M, Korenman Z, Cohen-Poradosu R, Nir-Paz R (2003) emm typing of M nontypeable invasive group A streptococcal isolates in Israel. J Clin Microbiol 41: 4655–4659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien KL, Beall B, Barrett NL, Cieslak PR, Reingold A, Farley MM, Danila R, Zell ER, Facklam R, Schwartz B, Schuchat A (2002) Epidemiology of invasive group a streptococcus disease in the United States, 1995–1999. Clin Infect Dis 35: 268–276 [DOI] [PubMed] [Google Scholar]
- Perez-Casal J, Caparon MG, Scott JR (1991) Mry, a trans-acting positive regulator of the M protein gene of Streptococcus pyogenes with similarity to the receptor proteins of two-component regulatory systems. J Bacteriol 173: 2617–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravins M, Jaffe J, Hanski E, Shetzigovski I, Natanson-Yaron S, Moses AE (2000) Characterization of a mouse-passaged, highly encapsulated variant of group A streptococcus in in vitro and in vivo studies. J Infect Dis 182: 1702–1711 [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Tolle DP, Barrett AJ (2004) MEROPS: the peptidase database. Nucleic Acids Res 32: D160–D164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrock JFFF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Press [Google Scholar]
- Sharkawy A, Low DE, Saginur R, Gregson D, Schwartz B, Jessamine P, Green K, McGeer A (2002) Severe group a streptococcal soft-tissue infections in Ontario: 1992–1996. Clin Infect Dis 34: 454–460 [DOI] [PubMed] [Google Scholar]
- Siezen RJ (1999) Multi-domain, cell-envelope proteinases of lactic acid bacteria. Antonie Van Leeuwenhoek 76: 139–155 [PubMed] [Google Scholar]
- Taylor FB Jr, Bryant AE, Blick KE, Hack E, Jansen PM, Kosanke SD, Stevens DL (1999) Staging of the baboon response to group A streptococci administered intramuscularly: a descriptive study of the clinical symptoms and clinical chemical response patterns. Clin Infect Dis 29: 167–177 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton-That H, Marraffini LA, Schneewind O (2004) Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim Biophys Acta 1694: 269–278 [DOI] [PubMed] [Google Scholar]
- Wexler DE, Chenoweth DE, Cleary PP (1985) Mechanism of action of the group A streptococcal C5a inactivator. Proc Natl Acad Sci USA 82: 8144–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuyts A, Govaerts C, Struyf S, Lenaerts JP, Put W, Conings R, Proost P, Van Damme J (1999) Isolation of the CXC chemokines ENA-78, GRO alpha and GRO gamma from tumor cells and leukocytes reveals NH2-terminal heterogeneity. Functional comparison of different natural isoforms. Eur J Biochem 260: 421–429 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information