

Abstract
Methylation of CpG dinucleotides is the major epigenetic modification of mammalian genomes, critical for regulating chromatin structure and gene activity. The mixed-lineage leukaemia (MLL) CXXC domain selectively binds nonmethyl-CpG DNA, and is required for transformation by MLL fusion proteins that commonly arise from recurrent chromosomal translocations in infant and secondary treatment-related acute leukaemias. To elucidate the molecular basis of nonmethyl-CpG DNA recognition, we determined the structure of the human MLL CXXC domain by multidimensional NMR spectroscopy. The CXXC domain has a novel fold in which two zinc ions are each coordinated tetrahedrally by four conserved cysteine ligands provided by two CGXCXXC motifs and two distal cysteine residues. We have identified the CXXC domain DNA binding interface by means of chemical shift perturbation analysis, cross-saturation transfer and site-directed mutagenesis. In particular, we have shown that residues in an extended surface loop are in close contact with the DNA. These data provide a template for the design of specifically targeted therapeutics for poor prognosis MLL-associated leukaemias.
Keywords: chromatin, CpG dinucleotide, CXXC domain, methylation, mixed lineage leukaemia
Introduction
In human leukaemia, the mixed-lineage leukaemia (MLL) gene is a frequent target for recurrent specific chromosomal translocations (Djabali et al, 1992; Gu et al, 1992; Tkachuk et al, 1992; Corral et al, 1993; Domer et al, 1993; Thirman et al, 1993) that result in the generation of novel chimaeric fusions between MLL and over 30 different partner genes (Daser and Rabbitts, 2005). MLL is the human homologue of the Drosophila trithorax gene, and is required for the maintenance of Hox gene expression during mammalian development for the establishment of body segment identity (Yu et al, 1995, 1998). MLL is required for Hox-dependent expansion of normal haematopoietic progenitors (Hess et al, 1997; Yagi et al, 1998; Ernst et al, 2004a, 2004b) and transformation of myeloid progenitors by MLL fusion proteins is dependent on specific Hoxa genes (Nakamura et al, 2002; Ayton and Cleary, 2003; Kumar et al, 2004; So et al, 2004; Zeisig et al, 2004; Wang et al, 2005).
The MLL protein is an SET domain-dependent histone H3 lysine 4 (K4)-specific methyltransferase that exists as part of a multiprotein supercomplex of at least 29 proteins (Milne et al, 2002; Nakamura et al, 2002). H3-K4 methylation status correlates with an active transcriptional state (Strahl et al, 1999; Noma et al, 2001), and provides the molecular basis whereby Hox gene expression is maintained by the MLL protein (Yu et al, 1998). However, the mechanisms by which wild type or oncogenic MLL fusion proteins are recruited to specific target genes in a chromatin context are poorly understood. The amino-terminal region of MLL contains a cysteine-rich CXXC domain (zf-CXXC; Pfam PF02008), characterised by two CGXCXXC repeats, which is also present in a number of other chromatin-associated proteins. These include the methyl-CpG binding domain protein (MBD1) (Cross et al, 1997), DNA methyltransferase 1 (DNMT1) (Bestor and Verdine, 1994), the major DNA maintenance DNA methyltransferase, CpG binding protein (CGBP), a component of the mammalian Set1 H3-K4 methyltransferase complex (Lee and Skalnik, 2005) and FBXL11, recently characterised as a histone demethylase that specifically demethylates histone H3 at lysine 36 (Tsukada et al, 2006). The CXXC domain is retained in all MLL fusion proteins and is essential for target gene recognition, transactivation and myeloid transformation (Ayton et al, 2004). The CXXC domain of several proteins, including MLL, has been shown to bind to nonmethyl-CpG dinucleotides (Lee et al, 2001; Birke et al, 2002; Ayton et al, 2004; Jorgensen et al, 2004). Cytosine methylation is the major epigenetic DNA modification in eukaryotes, and in vertebrates is found almost exclusively in a 5′ CpG context where it functions to maintain stable gene silencing through mitotic cell divisions. DNA methylated at the cytosine of CpG dinucleotides is found in transcriptionally inactive genes, whereas actively expressed genes are generally hypomethylated (Cross and Bird, 1995). The CXXC domain may therefore play an important role in directing MLL to transcriptionally active genes. To understand the molecular basis of nonmethyl-CpG DNA recognition, we have determined the solution structure of the MLL CXXC domain. By combining NMR spectroscopy with chemical shift perturbation analysis, cross-saturation transfer, site-directed mutagenesis and mass spectrometry, we have identified the DNA binding interface and revealed residues that are critical for DNA binding and maintaining the fold of the MLL CXXC domain. These studies provide a structural basis for understanding how vertebrates interpret the methylation status of CpG dinucleotides and provide a framework for the development of novel therapeutics for the treatment of the poor prognosis MLL-related leukaemias.
Results
Structure determination
The NMR spectra of residues V1146 to K1214 of MLL were assigned and the solution structure determined using standard techniques (Wüthrich, 1986; Bax, 1994). Residues R1150–P1201 adopt a well-defined tertiary structure with an r.m.s. deviation of 0.4 Å for backbone atoms. The N-terminal residues V1146–G1149 and C-terminal residues S1202–K1214 are unstructured as judged by a lack of long-range NOEs and negative heteronuclear NOE values (data not shown) and were excluded from the statistical analysis. Experimental restraints and structural statistics for the 20 accepted lowest energy structures are summarised in Table I. The coordinates for the structure are available from the Protein Data Bank (entry code 2j2s).
Table 1.
Summary of conformational constraints and statistics for the 20 accepted structures of MLL CXXC domain
| Structural constraints | |
| Intraresidue | 607 |
| Sequential | 336 |
| Medium-range (2⩽∣i−j∣⩽4) | 210 |
| Long-range (∣i−j∣>4) | 284 |
| chi-1 angle constraints | 31 |
| TALOS constraints | 76 |
| Hydrogen bond constraints | 16 |
| Zinc co-ordination constraints | 20 |
| Total | 1580 |
| Statistics for accepted structures | |
| Statistics parameter (±s.d.) | |
| R.m.s. deviation for distance constraints | 0.014±0.001 Å |
| R.m.s. deviation for dihedral constraints | 0.27±0.02° |
| Mean X-PLOR energy term (kcal mol−1±s.d.) | |
| E (overall) | 137.3±17.4 |
| E (van der Waals) | 52.1±3.8 |
| E (NOE and hydrogen bond constraints) | 20.9±2.4 |
| E (chi-1 dihedral and TALOS constraints) | 1.0±0.2 |
| R.m.s. deviations from the ideal geometry (±s.d.) | |
| Bond lengths | 0.0024±0.0001 Å |
| Bond angles | 0.39±0.01° |
| Improper angles | 0.30±0.01° |
| Average atomic r.m.s. deviation from the mean structure (±s.d.) | |
| Residues 1150–1201 (N, Cα, C atoms) | 0.40±0.19 Å |
| Residues 1150–1201 (all heavy atoms) | 0.83±0.15 Å |
| Structural quality | |
| Residues in most favoured region of Ramachandran plot | 82.6% |
| Residues in additional allowed region of Ramachandran plot | 15.1% |
| Residues in disallowed region of Ramachandran plot | 0.0% |
Structure description
The CXXC domain adopts an extended crescent-like structure that incorporates two Zn ions (Figure 1A–C). The presence of zinc and the metal binding stoichiometry (zinc:protein 2:1) was established by induction coupled plasma mass spectrometry (data not shown) and mass spectrometry under native and denaturing conditions (Table II). Each of three cysteine residues in the two CGXCXXC motifs provides a ligand for the coordination of a Zn ion (Figure 1C). Both motifs adopt a very similar conformation in which the second and third cysteine residues lie within a small helix (residues T1171–L1174) or form part of a small helix-like turn (residues P1159–Q1162). The residue that follows the first cysteine has a positive phi angle, which accounts for the strong preference for glycine at this position. After the second motif the main chain changes direction by 180° to enable C1189 and C1194 to provide the additional fourth ligand for coordinating Zn, together with the three cysteines from the second and first CGXCXXC motifs, respectively.
Figure 1.
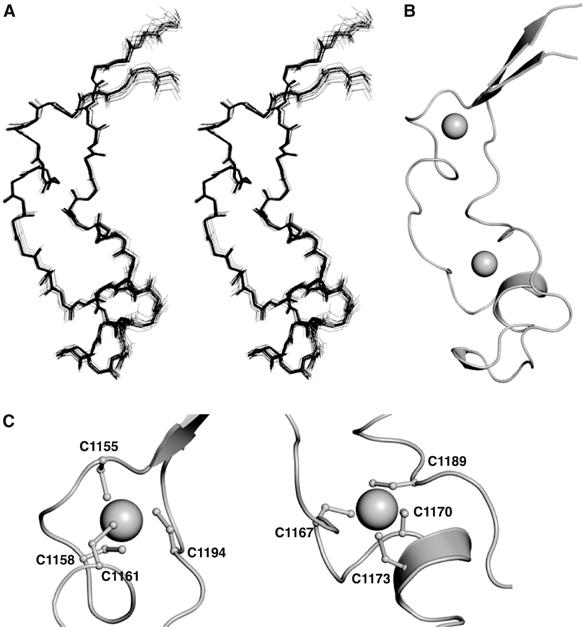
Solution structure of the MLL CXXC domain. (A) An overlay of the backbone atoms of the 20 lowest energy structures in stereo. (B) A ribbon representation of the lowest energy structure (same orientation as in (A)), prepared using the program PyMOL (http://www.pymol.org). Zn ions are shown as spheres. (C) A ribbon representation of the Zn coordination sites in MLL (PyMOL).
Table 2.
ESI-MS analysis of metal binding properties for wild type and mutant MLL CXXC domains
| Protein | Assay condition | ESI-MS |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mass proteina |
Zn2+ number |
||||||||||||||||
| Calculated | Measured | Difference in massb | No. of Zn2+ atoms | ||||||||||||||
| WT | Native | 9420.8 | 126.1 | 2 | |||||||||||||
| Denatured | 9294.9 | 9294.7 | |||||||||||||||
| C1155A | Native | 9255.6 | 0.2 | 0 | |||||||||||||
| Denatured | 9262.8 | 9255.4c | |||||||||||||||
| C1158A | Native | 9255.2 | −0.4 | 0 | |||||||||||||
| Denatured | 9262.8 | 9255.6c | |||||||||||||||
| C1161A | Native | 9254.8 | −0.4 | 0 | |||||||||||||
| Denatured | 9262.8 | 9255.2c | |||||||||||||||
| C1167A | Native | 9255.8 | 0.8 | 0 | |||||||||||||
| Denatured | 9262.8 | 9255.0 | |||||||||||||||
| C1170A | Native | 9257.2 | −1.4 | 0 | |||||||||||||
| Denatured | 9262.8 | 9258.6c | |||||||||||||||
| C1173A | Native | 9256.6 | −1.8 | 0 | |||||||||||||
| Denatured | 9262.8 | 9258.4c | |||||||||||||||
| C1188A | Native | 9389.0 | 127.0 | 2 | |||||||||||||
| Denatured | 9262.8 | 9262.0 | |||||||||||||||
| C1189A | Native | 9257.4 | 0.4 | 0 | |||||||||||||
| Denatured | 9262.8 | 9257.0c | |||||||||||||||
| C1194A | Native | 9315.4 | 60.4 | 1 | |||||||||||||
| Denatured | 9262.8 | 9255.0c | |||||||||||||||
| R1151A | Native | 9336.8 | 127.4 | 2 | |||||||||||||
| Denatured | 9209.8 | 9209.4 | |||||||||||||||
| R1153A | Native | 9336.8 | 127.2 | 2 | |||||||||||||
| Denatured | 9209.8 | 9209.6 | |||||||||||||||
| R1154A | Native | 9334.8 | 125.4 | 2 | |||||||||||||
| Denatured | 9209.8 | 9209.4 | |||||||||||||||
| Q1162A | Native | 9361.6 | 125.0 | 2 | |||||||||||||
| Denatured | 9237.8 | 9236.6 | |||||||||||||||
| D1166A | Native | 9247.0 | 0 | 0 | |||||||||||||
| Denatured | 9250.9 | 9247.0 | |||||||||||||||
| N1172A | Native | 9380.1 | 128.4 | 2 | |||||||||||||
| Denatured | 9251.8 | 9251.7 | |||||||||||||||
| D1175A | Native | 9378.0 | 127.2 | 2 | |||||||||||||
| Denatured | 9250.9 | 9250.8 | |||||||||||||||
| K1176A | Native | 9363.5 | 126.9 | 2 | |||||||||||||
| Denatured | 9237.8 | 9236.6 | |||||||||||||||
| K1178A | Native | 9363.8 | 126.4 | 2 | |||||||||||||
| Denatured | 9237.8 | 9237.4 | |||||||||||||||
| F1179A | Native | 9344.8 | 126.2 | 2 | |||||||||||||
| Denatured | 9218.8 | 9218.6 | |||||||||||||||
| N1183A | Native | 9379.8 | 128.3 | 2 | |||||||||||||
| Denatured | 9251.8 | 9251.5 | |||||||||||||||
| K1185A | Native | 9365.1 | 127.4 | 2 | |||||||||||||
| Denatured | 9237.8 | 9237.7 | |||||||||||||||
| K1186A | Native | 9363.8 | 126.2 | 2 | |||||||||||||
| Denatured | 9237.8 | 9237.6 | |||||||||||||||
| Q1187A | Native | 9365.6 | 127.8 | 2 | |||||||||||||
| Denatured | 9237.8 | 9237.8 | |||||||||||||||
| R1192A | Native | 9206.0 | −2.0 | 0 | |||||||||||||
| Denatured | 9209.8 | 9208.0 | |||||||||||||||
| K1193A | Native | 9365.2 | 127.6 | 2 | |||||||||||||
| Denatured | 9237.8 | 9237.6 | |||||||||||||||
| Q1195A | Native | 9364.6 | 127.8 | 2 | |||||||||||||
| Denatured | 9237.8 | 9236.8 | |||||||||||||||
| N1196A | Native | 9379.4 | 127.9 | 2 | |||||||||||||
| Denatured | 9251.8 | 9251.5 | |||||||||||||||
| KFGG/AAAA | Native | 9181.8 | −0.8 | 0 | |||||||||||||
| Denatured | 9189.7 | 9182.6 | |||||||||||||||
| Masses are given in daltons. | Even if the average mass of the divalent bound Zn2+ is 65.4, the loss of two or more protons upon protein binding gives an apparent mass of 63.4. | The difference between the calculated and measured mass for the metal binding cysteine mutants in the denatured state is due to the formation of disulphide bonds resulting in proton loss. The molecular weight measurement for each species was accurate to within 100 p.p.m. | |||||||||||||||
The topology of the fold is primarily dictated by the pattern of Zn coordination and contains little regular secondary structure. Residues R1151–R1154 and L1197–M1200 form a short two-stranded antiparallel β-sheet that places the N and C termini close together in an arrangement seen in many protein modules. Following the small helix in the second CGXCXXC motif is a 310 helix (residues P1177–F1179), in which F1179 packs onto G1168 of the second CGXCXXC motif (Figure 2). The gamma and delta carbon atoms of the K1178 pack onto the aromatic ring of F1179, while the zeta nitrogen is close to the carboxyl group of D1166. The packing of these helices dictates the overall structure of the turn and acts as the scaffold for an extended surface loop. This loop begins with the break in structure caused by the sequential glycines G1180 and G1181 and ends with the distal Zn ligand residue, C1189. There are several charge–charge interactions, the most notable being a salt bridge between D1166 and R1192. D1166 is situated between the two CGXCXXC motifs while R1192 lies in the helix-like turn (residues 1190–1192) between the two cysteine residues that provide the fourth zinc ligands for each motif. Hydrogen exchange experiments showed that amide protons are protected only in the CGXCXXC motifs and the helix-like turn (residues K1190–R1192) between the fourth zinc ligands. Amide protons in more peripheral parts of the structure, such as the helix-like turn (residues P1159–Q1162) and the β-sheet, exchange rapidly with the solvent.
Figure 2.

Ribbon representation of the elaborate turn in the CXXC domain of MLL showing the side chains of the residues from the KFGG motif and the second Zn coordination site (PyMOL). An extended loop is formed between residues G1181 and C1189.
The structure of the CXXC domain differs from that of other Zn-dependent binding motifs (Krishna et al, 2003) and a search for structurally similar proteins using the program DALI produced no hits (Holm and Sander, 1995). However, this type of Zn ligation has been seen previously in the CGXCXXC motif of the RecQ family of helicases (Bernstein et al, 2003). However, in RecQ, a cysteine residue N-terminal to the motif provides the fourth Zn ligand.
A structure-based sequence alignment of CXXC domains is shown in Figure 3. The CXXC domain is highly conserved in MLL proteins from different species. For example, there is only one amino-acid change between the Homo sapiens and Fugu rubripes proteins (Caldas et al, 1998). The CXXC domain of the human MLL paralogue MLL4 is less well conserved (FitzGerald and Diaz, 1999; Huntsman et al, 1999). Other CXXC domains are more diverse in sequence. The residues that provide the ligands for the Zn ions are, however, strictly conserved, and it is likely that all the other CXXC domains have a similar overall fold to that of MLL. Residue R1192 is invariant among all CXXC domains, and D1166 is highly conserved or shows conservative substitution to glutamate. These residues form the salt bridge described above and their strict conservation would seem to indicate importance to the structure. Comparison of the three CXXC domains within the MBD1 protein (only one of which, CXXC-3, shown as MBD1c in Figure 3, binds DNA) suggests residues that may be critical for CpG recognition (Jorgensen et al, 2004). Residues K1178–G1181, discussed above, comprise a KFGG motif that is conserved in other CXXC domains known to bind DNA. Q1187 follows an identical pattern of conservation to that of the KFGG motif suggesting functional importance. This is particularly apparent in the MBD1 protein in which the only CXXC domain to maintain both the KFGG motif and residue Q1187 is the one that binds DNA.
Figure 3.

Structure based sequence alignment of representative CXXC domains. Sequences were aligned using Jalview (Clamp et al, 2004) and are shaded in blue according to the degree of amino-acid sequence identity. All sequences are from Homo sapiens except where indicated. The secondary structure elements are labelled α for helix and β for strands, as calculated by DSSP (Kabsch and Sander, 1983). Filled circle (•) and filled diamonds (⧫) indicate cysteines involved in the first and second Zn coordination sites, respectively. NCBI accession numbers are as follows: MLL gi:56550039; MLL (F. rubripes) gi:3309542; MLL4 gi:7662046; DNMT1 gi:4503351; FLX10 gi:54112382; CXCC1/CGBP gi:7656975; MBD1 gi:21464117; LCX gi:33859755.
DNA binding
A series of isothermal titration calorimetry (ITC) experiments was conducted to measure binding of the CXXC domain to DNA palindromes of 12, 16 and 20 base pairs (bp), all with a unique and centrally positioned CpG dinucleotide. No significant difference in binding affinity was observed for different length of DNA (data not shown) and subsequent work was carried out using 12 bp duplexes. The CXXC domain binds CpG 12-mer DNA with a Kd of 4.3 μM (standard error of 0.4 μM from three independent determinations) and an enthalpy of complex formation of 1.4 kcal mol−1 at 22°C under the conditions of the ITC buffer (Figure 4). Binding to CpG 12-mer DNA was measured at a series of temperatures to determine the ΔCp for binding. A value of −0.3 kcal mol−1 K−1 was determined (data not shown), consistent with other known protein–DNA interactions (Peters et al, 2004). There was no evidence of binding to the same DNA containing a central methyl-CpG under these conditions. A 12 bp DNA containing a central GpC showed only minor heat effects above the baseline, possibly indicating significantly weaker binding for this sequence, but this was too small to be analysed. These findings are consistent with the results obtained in previous studies on DNA binding by this domain using other techniques (Birke et al, 2002; Ayton et al, 2004).
Figure 4.

ITC analysis of DNA binding by the MLL CXXC domain. Typical ITC data are shown for the endothermic binding of the CXXC domain to a 12-mer CpG-containing DNA oligonucleotide at 22°C in 20 mM MES, pH 6.5, 250 mM NaCl, 5 mM β-mercaptoethanol. Upper panel: (A) CXXC domain (1.3 mM) into the calorimetric cell (1.4 ml) containing CpG 12-mer DNA (49 μM). (B) CXXC domain (970 μM) into ITC buffer. (C) ITC buffer into CpG 12-mer DNA (49 μM). Lower Panel: Integrated heat pulses, normalised per mole of injectant, giving a differential binding curve that is adequately described by a one-site binding model.
The DNA binding site was localised by monitoring the changes in the 2D 1H-15N-HSQC spectra of the MLL CXXC domain upon the addition of a 12-bp DNA duplex containing a central CpG dinucleotide. DNA binding significantly alters the NMR spectrum of the domain (Figure 5A) with many residues undergoing large changes in chemical shift (Figure 5B). The majority of these residues are located on one face of the CXXC domain (Figure 6A). This region of the protein contains many positively charged amino acids (Figure 7A) consistent with it being the DNA binding site. Cross-saturation transfer experiments, which provide direct information on through space interactions (Ramos et al, 2000; Lane et al, 2001), were also employed to precisely identify residues at the DNA binding surface. Specific saturation of the imino protons of the DNA resulted in an attenuation of peak intensity in the 1H-15N HSQC spectrum for residues R1182–C1188 (Ramos et al, 2000). These residues lie within an extended surface loop (compare Figures 2 and 6B).
Figure 5.

Chemical shift perturbation analysis of DNA binding by the MLL CXXC domain. (A) An overlay of the 1H–15N HSQC spectra of the CXXC domain in the absence (black contour levels) or the presence (red contour levels) of an equimolar concentration of the palindromic 12-mer oligonucleotide GTATCCGGATAC. This is a combined 15N 1H chemical shift perturbation map as defined by Δ1H+(Δ15N/5) (Hajduk et al, 1997). Chemical shift perturbations greater than 0.3 p.p.m. are indicated as lines connecting the amide resonances in the free and bound states. (B) A plot of chemical shift due to DNA binding for residues R1150–P1201, with a cutoff at 0.3 p.p.m.
Figure 6.

Mapping the DNA binding surface of the MLL CXXC domain. (A) Representations of the molecular surface of the MLL CXXC domain, with two views related by a rotation of 120° about the vertical axis. Residues that undergo significant (>0.3 p.p.m.) chemical shift upon binding DNA are coloured orange. (B) Representations of the molecular surface of the MLL CXXC domain. The two views are related by a rotation of 120° about the vertical axis. Residues that show a decrease in peak intensity of >15% upon saturation of the imino protons of the DNA and a mixing time of 1.44 s are coloured pink.
Figure 7.

Mutational analysis of DNA binding by the MLL CXXC domain. (A) Representation of the electrostatic surface potential of the CXXC domain as calculated by the program APBS (Baker et al, 2001) and coloured using a linear colour ramp from −25.0 kT (red) to +25.0 kT (blue). Residues that are functionally implicated in DNA binding in gel shift assays are indicated. (B) Gel shift assays. Purified 6xHis-tagged proteins were incubated with 12-mer dsDNA carrying a methyl- or nonmethyl-CpG pair and electrophoresed in agarose gel shift assays as shown. The wild type and mutant proteins utilised are indicated.
Mutagenesis
A series of mutations was devised to identify the role that individual residues play in the function and stability of the domain. DNA binding was tested by gel-shift assays (Figure 7B). Mutations that disrupted the native fold of the CXXC domain were detected by performing mass spectrometry under native conditions (Table II).
Mutation of any of the cysteine residues involved in Zn ligation led to unfolding of the domain. Furthermore, disruption of the conserved salt bridge between D1166 and R1192 also unfolded the protein. Mutations R1153A, Q1162A, N1172A, Q1195A and N1196A had no effect on either stability or DNA binding. These residues are on the opposite face of the CXXC domain to that implicated in DNA binding. Mutation of residues R1151, R1154, D1175, K1176, K1178, F1179, K1185, K1186, Q1187 and K1193 abolished or significantly decreased DNA binding, but had no effect on the global fold. For most of these mutations, the lack of DNA binding activity is likely to be solely the result of the removal of a functionally important side chain. Furthermore, most of these residues localise to the binding face of the domain as discussed above (see Figure 7A). Other mutations may, however, perturb local structure that is important for binding without the residues themselves playing a direct role. For example, as discussed above, the conserved K1178 and F1179 are packed in such a manner that they significantly contribute to the local structure (see Figure 2) and mutation of either residue is found to impair DNA binding. Furthermore, mutation of the KFGG motif (residues K1178–G1181) to alanine destabilises the protein sufficiently to cause it to unfold (see Table II).
Discussion
Residues important for DNA binding
Taken together, our NMR binding and mutagenesis data clearly delineate the DNA binding interface of the MLL CXXC domain (Figures 6A, B, 7A and B). In particular, there are two distinctive features of the domain with potential relevance to binding. A positively charged groove runs along the DNA binding face of the domain consisting of residues shown to abolish or significantly decrease DNA binding upon mutation (R1154, K1176, K1178, K1186, K1193) (Figure 7A and B). There is also a surface patch at the tip of the domain corresponding to residues R1182–C1188 of the extended loop that are all shown to make direct contact with protons in the DNA by cross-saturation experiments. Methylation of cytosine at the 5′ position places a methyl group in the major groove of the DNA. One could envisage a model whereby the positively charged groove on the binding surface interacts with the DNA phosphate backbone, while the residues of the extended loop insert into the major groove to probe the methylation state of the CpG dinucleotide.
The MBD1 transcriptional repressor is unique in that it contains both a methyl-CpG binding domain and a CXXC domain that binds specifically to nonmethyl-CpG. This allows MBD1 to interpret the CpG dinucleotide as a repressive signal in vivo regardless of its methylation status (Jorgensen et al, 2004). Taken together, the architectures of the MLL CXXC domain and the MBD domain of the MBD1 protein provide a structural basis for understanding how vertebrates interpret the methylation status of DNA, the major epigenetic DNA modification in eukaryotes. Although the CXXC domain of MLL is known to be required for the transforming activity of MLL fusion proteins, the biochemical role it plays in this process has not been fully defined. The mutation of several residues in the CXXC domain has been shown to abolish both DNA binding and prevent myeloid transformation (Ayton et al, 2004). To date, however, these mutations have either removed zinc ligands, which would perturb the structure of the domain, or have involved changes in multiple residues that we have shown result in unfolding of the protein (Table II). It is also possible that these types of mutation could affect other activities of the domain. In addition to DNA binding, the CXXC domain of MLL also recruits the polycomb repressor proteins HPC2 and BMI-1, and the corepressor CTBP (Xia et al, 2003) and forms part of a low-affinity binding site for the menin tumour suppressor oncogenic cofactor (Yokoyama et al, 2005). With the structure of the CXXC domain now available, it will be possible to design nondisruptive mutations that can help to define the role of individual residues in these activities, and thus promoting a deeper understanding of the role of the CXXC domain in transformation by MLL fusion proteins. The CXXC domain is retained in all forms of leukaemogenic MLL fusion proteins, including partial tandem duplications (Lochner et al, 1996) and internal PHD finger 1 deletions (Chaplin et al, 2001; von Bergh et al, 2001; Deveney et al, 2003; Morel et al, 2003). Thus, novel approaches to the treatment of MLL-associated leukaemias might involve addressing the continued occupancy of key target genes by MLL fusion proteins by disrupting the interaction between the CXXC domain and nonmethyl-CpG DNA. Our structure provides a potential template for the development of such novel reagents for the treatment of poor prognosis MLL-related leukaemias.
Materials and methods
Preparation of the protein and the DNA
The CXXC domain of the MLL protein used for NMR and calorimetry, corresponding to residues V1146–K1214, was cloned into a modified pET24a plasmid (Novagen) that expresses proteins fused to the lipoyl domain of Bacillus stearothermophilus dihydrolipoamide acetyltransferase. The fusion protein was expressed in the E. coli strain Tuner [DE3] (Novagen). For isotope labelling, K-MOPS minimal medium containing 15N-NH4Cl and/or 13C-glucose was used. The fusion protein was initially purified by Ni2+-chelating sepharose affinity chromatography. Subsequent TEV protease digestion and Ni2+-chelating sepharose affinity chromatography removed the lipoyl domain fusion-tag. The CXXC domain was concentrated and then gel-filtered through a Superdex 75 (Amersham) column and the fractions containing the CXXC domain pooled. All DNA was supplied as an HPLC purified powder by Operon Biotechnologies, Inc. DNA was dissolved in buffer (20 mM MES pH 6.5, 250 mM NaCl, 5 mM β-mercaptoethanol) before being annealed by heating to 90°C for 10 min and cooled slowly to room temperature.
MLL CXXC domain used for gel shift assays (residues T1136–K1208) was expressed with an N-terminal His6-tag in Escherichia coli strain C41 (DE3). The protein was purified using Ni2+-NTA affinity resin (Qiagen) and resource S ion-exchange (Amersham) and dialysed into 10 mM Tris, 150 mM NaCl, 1 mM DTT, pH 7.4.
Isothermal titration calorimetry
Experiments to determine the DNA binding characteristics of the CXXC domain utilised a number of palindromic DNA sequences of differing length and composition:
- CpG 12-mer:
5′-GTATCCGGATAC-3′
5′-CAGTATCCGGATACTG-3′
5′-GTCAGTATCCGGATACTGAC-3′
5′-GTATGGCCATAC-3′
5′-GTATC (5MeC)GGATAC-3′
- CpG 16-mer:
5′-CAGTATCCGGATACTG-3′
5′-GTCAGTATCCGGATACTGAC-3′
5′-GTATGGCCATAC-3′
5′-GTATC (5MeC)GGATAC-3′
- CpG 20-mer:
5′-GTCAGTATCCGGATACTGAC-3′
5′-GTATGGCCATAC-3′
5′-GTATC (5MeC)GGATAC-3′
- GpC DNA:
5′-GTATGGCCATAC-3′
5′-GTATC (5MeC)GGATAC-3′
- Methyl-CpG DNA:
5′-GTATC (5MeC)GGATAC-3′
Samples of CXXC domain and DNA were dialysed extensively against ITC buffer (20 mM MES pH 6.5, 250 mM NaCl, 5 mM β-mercaptoethanol) prior to the experiment. DNA was concentrated to ∼50 μM dsDNA prior to dialysis. Final DNA concentrations were determined spectroscopically after filtering and loading of the calorimetric cell by measuring absorbance at 260 nm, assuming 50 μg−1 ml−1 A260 unit−1. Final concentrations of DNA ranged from 47–54 μM except for methyl-CpG DNA, which for reasons of solubility, was 30 μM. CXXC domain was concentrated to ∼1.2 mM prior to dialysis. After filtering it was loaded into the syringe and the final CXXC domain concentration was determined spectroscopically at 280 nm, given an extinction coefficient ɛ280 of 6990 cm−1 M−1. Final CXXC domain concentrations ranged from 1.0–1.3 mM. ITC experiments were performed at 22°C using a high-precision VP-ITC system (Microcal Inc.).
Experiments were conducted such that the heat change was measured over 250 s following a 10 μl injection for either 20 or 25 injections. Included was an initial preinjection of 3 μl, according to the manufacturer's recommendation to counter diffusion of samples during the thermal equilibration. Analysis was carried out using Microcal Origin Software. Individual injections were integrated following manual adjustment of the baselines. Heats of dilution and mixing were determined from separate control experiments or from the end point of the titration. This value was subtracted prior to curve fitting using a one-site model.
Spectroscopic measurements
The NMR spectra were recorded on Bruker Advance-800, Advance-600 and AMX-500 spectrometers. 2D NOESY, TOCSY, DQF-COSY, 15N-HSQC, constant-time 13C-HSQC and 3D HNCACB, CBCACONH, HNCO, HNCACO HNHB, 15N-NOESY, 15N-TOCSY were recorded at 290 K. The mixing times chosen were 55 ms for TOCSY, and 120 ms for NOESY. Spectra were referenced relative to external sodium 2,2-dimethyl-2-silapentane-5-sulfonate, for signals of proton and carbon, or liquid ammonium for that of nitrogen. Approximately half the Hβ resonances were assigned stereospecifically using a combination of HNHB and DQF-COSY spectra. All the Val Hγ and Leu Hδ resonances were assigned stereospecifically using a 10% 13C-labelled sample of CXXC domain (Neri et al, 1989). All the NMR spectra were analysed with ANSIG v3.3 (Kraulis et al, 1994).
All NMR sample concentrations were 1.0 mM and were prepared in 20 mM MES pH 6.5, 250 mM NaCl, 10% D2O. The CXXC domain was concentrated to 1.5 mM for spectroscopic measurements of the free form of the domain. For binding studies, protein–dsDNA complexes were made with a stoichiometry of 1:1 (protein: dsDNA).
For hydrogen exchange experiments, the 15N-labelled CXXC domain was exchanged into NMR buffer containing 100% D2O using a NAP-10 column (Amersham) and a series of 1H-15N-FHSQC spectra (Mori et al, 1995) were recorded over the course of 24 h.
Cross-saturation transfer
Resonances of the bound form of the CXXC domain were reassigned using standard triple resonance techniques (Wüthrich, 1986; Bax, 1994). A cross-saturation transfer period similar to that described by Ramos et al (2000) was incorporated immediately prior to the first 1H pulse of an FHSQC sequence (Mori et al, 1995). Saturation of the DNA imino proton resonances was achieved via a pulse train of 15 ms hyperbolic secant inversion pulses, which were centreed at δ1H=13 p.p.m. Saturation transfer periods were 0.360, 0.720 and 1.440 s, and the overall relaxation delay was kept constant at 1.94 s. To avoid sample heating effects, the pulse sequence contained an identical train of compensation pulses centreed at δ1H=−5 p.p.m., which were executed so that the overall number of pulses was kept constant. Attenuation was measured relative to control experiments executed at the beginning and end of the series of experiments with errors extracted from these controls.
Structure determination
The distance constraints derived from the NOESY spectra were classified into four categories corresponding to inter-proton distance constraints of 1.8–2.8, 1.8–3.5, 1.8–4.75 and 1.8–6.0 Å, respectively. Hydrogen bond constraints of 1.8–2.1 Å were imposed on the distance between the hydrogen and the acceptor oxygen, while another constraint of 2.7–3.1 Å was imposed on the distance between the donor nitrogen and the acceptor oxygen. Artificial restraints were added to represent the constraints imposed by coordination of the zinc ions. Six sulphur–sulphur distance constraints of 3.55–3.95 Å and four zinc–sulphur distance constraints of 2.25–2.35 Å were incorporated for each of the zinc–cysteine clusters. Torsion angle constraints were obtained from stereo-specific assignment of residue side chains and incorporated in the structure calculation, along with the backbone φ and ψ angle constraints determined with the program TALOS (Cornilescu et al, 1999). The structures were calculated using a standard torsion angle dynamics simulated annealing protocol with the program CNS (Brunger et al, 1998). Twenty structures were accepted where no distance violations were greater than 0.25 Å and no angle violations were greater than 5.0°.
Electrospray ionisation mass spectrometry (ESI-MS)
Mass spectra of wild type or mutant proteins were generated on an LCT time-of-flight mass spectrometer with electrospray ionisation (ESI) (Micromass, Altrincham, UK). Before MS analysis, the protein samples were desalted by dialysis against water. For analysis in denaturing conditions, samples were diluted to 2 pmol ml−1 in 50% (v/v) methanol and 1% (v/v) formic acid. For analysis in native conditions, samples were diluted to 10 pmol ml−1 in 20 mM ammonium acetate buffer. The samples were infused into the ESI source at a flow rate of 10 ml min−1 using a Harvard Model 22 syringe infusion pump (Harvard Apparatus, Harvard, MA, USA) and calibration was performed in the positive ion mode using horse heart myoglobin. Typically, 60–80 scans were acquired and added to yield a mass spectrum. Molecular masses were obtained by deconvoluting the multiply charged protein mass spectra using the software package, MassLynxTM Version 4.0 (Micromass). Theoretical molecular masses of wild type and mutant proteins were calculated using Protparam (//us.expasy.org/tools/protparam.html). The zinc content of each protein was derived from the difference in mass between the native and denatured proteins.
Gel-shift assays
Palindromic olignucleotides PALCpG (5′-GTATCCGGATAC-3′), PALGpC (5′-GTATGGCCATAC-3′) and PALmeCpG (5′-GTATCmCGGATAC-3′) were annealed in 10 mM Tris pH 7.4, 1 mM EDTA, 100 mM NaCl and buffer exchanged into 10 mM Tris pH 7.4 using a Microspin G-25 column (Amersham). DNA binding reactions were carried out in 20 μl of binding buffer (10 mM Tris pH 7.4, 1 mM DTT, 150 mM NaCl) for 30 min at room temperature. Binding reactions contained a final dsDNA concentration of 10 μM and a two-fold molar excess of purified protein. Binding reaction mixtures were electrophoresed in 0.7% agarose in TB (89 mM Tris-borate, pH 8.3) buffer at 4°C and DNA was visualised by ethidium bromide staining.
Acknowledgments
We are grateful to Sew Peak-Chew for help with the ESI-MS; Dr Keith Sinclair, Elsie Widdowson Laboratory, MRC Human Nutrition Research Laboratory for ICP-MS studies, Dr A Andreeva for help with sequence alignment. This study was supported by the Leukaemia Research Fund, the Kay Kendall Leukaemia Fund and an MRC Senior Clinical Fellowship.
References
- Ayton PM, Cleary ML (2003) Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev 17: 2298–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton PM, Chen EH, Cleary ML (2004) Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol Cell Biol 24: 10470–10478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax A (1994) Multidimensional nuclear-magnetic-resonance methods for protein studies. Curr Opin Struct Biol 4: 738–744 [Google Scholar]
- Bernstein DA, Zittel MC, Keck JL (2003) High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J 22: 4910–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH, Verdine GL (1994) DNA methyltransferases. Curr Opin Cell Biol 6: 380–389 [DOI] [PubMed] [Google Scholar]
- Birke M, Schreiner S, Garcia-Cuellar MP, Mahr K, Titgemeyer F, Slany RK (2002) The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res 30: 958–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54 (Part 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Caldas C, Kim MH, MacGregor A, Cain D, Aparicio S, Wiedemann LM (1998) Isolation and characterization of a pufferfish MLL (mixed lineage leukemia)-like gene (fMll) reveals evolutionary conservation in vertebrate genes related to Drosophila trithorax. Oncogene 16: 3233–3241 [DOI] [PubMed] [Google Scholar]
- Chaplin T, Jones L, Debernardi S, Hill AS, Lillington DM, Young BD (2001) Molecular analysis of the genomic inversion and insertion of AF10 into MLL suggests a single-step event. Genes Chromosomes Cancer 30: 175–180 [PubMed] [Google Scholar]
- Clamp M, Cuff J, Searle SM, Barton GJ (2004) The Jalview Java alignment editor. Bioinformatics 20: 426–427 [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13: 289–302 [DOI] [PubMed] [Google Scholar]
- Corral J, Forster A, Thompson S, Lampert F, Kaneko Y, Slater R, Kroes WG, van der Schoot CE, Ludwig WD, Karpas A, Pocock C, Cotter F, Rabbitts TH (1993) Acute leukemias of different lineages have similar MLL gene fusions encoding related chimeric proteins resulting from chromosomal translocation. Proc Natl Acad Sci USA 90: 8538–8542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SH, Bird AP (1995) CpG islands and genes. Curr Opin Genet Dev 5: 309–314 [DOI] [PubMed] [Google Scholar]
- Cross SH, Meehan RR, Nan X, Bird A (1997) A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nat Genet 16: 256–259 [DOI] [PubMed] [Google Scholar]
- Daser A, Rabbitts TH (2005) The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol 15: 175–188 [DOI] [PubMed] [Google Scholar]
- Deveney R, Chervinsky DS, Jani-Sait SN, Grossi M, Aplan PD (2003) Insertion of MLL sequences into chromosome band 5q31 results in an MLL-AF5Q31 fusion and is a rare but recurrent abnormality associated with infant leukemia. Genes Chromosomes Cancer 37: 326–331 [DOI] [PubMed] [Google Scholar]
- Djabali M, Selleri L, Parry P, Bower M, Young BD, Evans GA (1992) A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat Genet 2: 113–118 [DOI] [PubMed] [Google Scholar]
- Domer PH, Fakharzadeh SS, Chen CS, Jockel J, Johansen L, Silverman GA, Kersey JH, Korsmeyer SJ (1993) Acute mixed-lineage leukemia t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc Natl Acad Sci USA 90: 7884–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst P, Fisher JK, Avery W, Wade S, Foy D, Korsmeyer SJ (2004a) Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev Cell 6: 437–443 [DOI] [PubMed] [Google Scholar]
- Ernst P, Mabon M, Davidson AJ, Zon LI, Korsmeyer SJ (2004b) An Mll-dependent Hox program drives hematopoietic progenitor expansion. Curr Biol 14: 2063–2069 [DOI] [PubMed] [Google Scholar]
- FitzGerald KT, Diaz MO (1999) MLL2: a new mammalian member of the trx/MLL family of genes. Genomics 59: 187–192 [DOI] [PubMed] [Google Scholar]
- Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, Croce CM, Canaani E (1992) The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 71: 701–708 [DOI] [PubMed] [Google Scholar]
- Hajduk PJ, Dinges J, Miknis GF, Merlock M, Middleton T, Kempf DJ, Egan DA, Walter KA, Robins TS, Shuker SB, Holzman TF, Fesik SW (1997) NMR-based discovery of lead inhibitors that block DNA binding of the human papillomavirus E2 protein. J Med Chem 40: 3144–3150 [DOI] [PubMed] [Google Scholar]
- Hess JL, Yu BD, Li B, Hanson R, Korsmeyer SJ (1997) Defects in yolk sac hematopoiesis in Mll-null embryos. Blood 90: 1799–1806 [PubMed] [Google Scholar]
- Holm L, Sander C (1995) Dali: a network tool for protein structure comparison. Trends Biochem Sci 20: 478–480 [DOI] [PubMed] [Google Scholar]
- Huntsman DG, Chin SF, Muleris M, Batley SJ, Collins VP, Wiedemann LM, Aparicio S, Caldas C (1999) MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene 18: 7975–7984 [DOI] [PubMed] [Google Scholar]
- Jorgensen HF, Ben-Porath I, Bird AP (2004) Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol Cell Biol 24: 3387–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637 [DOI] [PubMed] [Google Scholar]
- Kraulis PJ, Domaille PJ, Campbell-Burk SL, Van Aken T, Laue ED (1994) Solution structure and dynamics of ras p21.GDP determined by heteronuclear three- and four-dimensional NMR spectroscopy. Biochemistry 33: 3515–3531 [DOI] [PubMed] [Google Scholar]
- Krishna SS, Majumdar I, Grishin NV (2003) Structural classification of zinc fingers: survey and summary. Nucleic Acids Res 31: 532–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AR, Hudson WA, Chen W, Nishiuchi R, Yao Q, Kersey JH (2004) Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood 103: 1823–1828 [DOI] [PubMed] [Google Scholar]
- Lane AN, Kelly G, Ramos A, Frenkiel TA (2001) Determining binding sites in protein–nucleic acid complexes by cross-saturation. J Biomol NMR 21: 127–139 [DOI] [PubMed] [Google Scholar]
- Lee JH, Skalnik DG (2005) CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem 280: 41725–41731 [DOI] [PubMed] [Google Scholar]
- Lee JH, Voo KS, Skalnik DG (2001) Identification and characterization of the DNA binding domain of CpG-binding protein. J Biol Chem 276: 44669–44676 [DOI] [PubMed] [Google Scholar]
- Lochner K, Siegler G, Fuhrer M, Greil J, Beck JD, Fey GH, Marschalek R (1996) A specific deletion in the breakpoint cluster region of the ALL-1 gene is associated with acute lymphoblastic T-cell leukemias. Cancer Res 56: 2171–2177 [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL (2002) MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 10: 1107–1117 [DOI] [PubMed] [Google Scholar]
- Morel F, Le Bris MJ, Douet-Guilbert N, Duchemin J, Herry A, Le Calvez G, Marion V, Berthou C, De Braekeleer M (2003) Insertion of chromosome 11 in chromosome 4 resulting in a 5′MLL-3′AF4 fusion gene in a case of adult acute lymphoblastic leukemia. Cancer Genet Cytogenet 145: 74–77 [DOI] [PubMed] [Google Scholar]
- Mori S, Abeygunawardana C, Johnson MO, van Zijl PC (1995) Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J Magn Reson B 108: 94–98 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E (2002) ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 10: 1119–1128 [DOI] [PubMed] [Google Scholar]
- Neri D, Szyperski T, Otting G, Senn H, Wuthrich K (1989) Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry 28: 7510–7516 [DOI] [PubMed] [Google Scholar]
- Noma K, Allis CD, Grewal SI (2001) Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293: 1150–1155 [DOI] [PubMed] [Google Scholar]
- Peters WB, Edmondson SP, Shriver JW (2004) Thermodynamics of DNA binding and distortion by the hyperthermophile chromatin protein Sac7d. J Mol Biol 343: 339–360 [DOI] [PubMed] [Google Scholar]
- Ramos A, Kelly G, Hollingworth D, Pastore A, Frenkiel TA (2000) Mapping the interfaces of protein–nucleic acid complexes using cross-saturation. J Am Chem Soc 122: 11311–11314 [Google Scholar]
- So CW, Karsunky H, Wong P, Weissman IL, Cleary ML (2004) Leukemic transformation of hematopoietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood 103: 3192–3199 [DOI] [PubMed] [Google Scholar]
- Strahl BD, Ohba R, Cook RG, Allis CD (1999) Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc Natl Acad Sci USA 96: 14967–14972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirman MJ, Gill HJ, Burnett RC, Mbangkollo D, McCabe NR, Kobayashi H, Ziemin-van der Poel S, Kaneko Y, Morgan R, Sandberg AA, Chaganti RSK, Larson RA, Le Beau MM, Diaz MO, Rowley JD (1993) Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. N Engl J Med 329: 909–914 [DOI] [PubMed] [Google Scholar]
- Tkachuk DC, Kohler S, Cleary ML (1992) Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 71: 691–700 [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439: 811–816 [DOI] [PubMed] [Google Scholar]
- von Bergh A, Gargallo P, De Prijck B, Vranckx H, Marschalek R, Larripa I, Kluin P, Schuuring E, Hagemeijer A (2001) Cryptic t(4;11) encoding MLL-AF4 due to insertion of 5′ MLL sequences in chromosome 4. Leukemia 15: 595–600 [DOI] [PubMed] [Google Scholar]
- Wang J, Iwasaki H, Krivtsov A, Febbo PG, Thorner AR, Ernst P, Anastasiadou E, Kutok JL, Kogan SC, Zinkel SS, Fisher JK, Hess JL, Golub TR, Armstrong SA, Akashi K, Korsmeyer SJ (2005) Conditional MLL-CBP targets GMP and models therapy-related myeloproliferative disease. EMBO J 24: 368–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K (1986) NMR of Proteins and Nucleic acid. New York: Wiley J [Google Scholar]
- Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ (2003) MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci USA 100: 8342–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Deguchi K, Aono A, Tani Y, Kishimoto T, Komori T (1998) Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood 92: 108–117 [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML (2005) The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123: 207–218 [DOI] [PubMed] [Google Scholar]
- Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ (1998) MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci USA 95: 10632–10636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ (1995) Altered Hox expression segmental identity in Mll-mutant mice. Nature 378: 505–508 [DOI] [PubMed] [Google Scholar]
- Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R, Hess JL, Slany RK (2004) Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol 24: 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]