

Abstract
Messenger RNA processing in trypanosomes by cis and trans splicing requires spliceosomal small nuclear ribonucleoproteins (snRNPs) U1, U2, U4/U6, and U5, as well as the spliced leader (SL) RNP. As in other eukaryotes, these RNPs share a core structure of seven Sm polypeptides. Here, we report that the identity of the Sm protein constituents varies between spliceosomal snRNPs: specifically, two of the canonical Sm proteins, SmB and SmD3, are replaced in the U2 snRNP by two novel, U2 snRNP-specific Sm proteins, Sm15K and Sm16.5K. We present a model for the variant Sm core in the U2 snRNP, based on tandem affinity purification-tagging and in vitro protein–protein interaction assays. Using in vitro reconstitutions with canonical and U2-specific Sm cores, we show that the exchange of two Sm subunits determines discrimination between individual Sm sites. In sum, we have demonstrated that the heteroheptameric Sm core structure varies between spliceosomal snRNPs, and that modulation of the Sm core composition mediates the recognition of small nuclear RNA-specific Sm sites.
Keywords: small nuclear RNA, snRNA, snRNP, splicing, trypanosomes
Introduction
Spliceosomal small nuclear ribonucleoproteins (snRNPs) U1, U2, U4/U6, and U5 are essential factors for splicing of all major, so-called U2-dependent introns. Their functions in splice-site recognition, spliceosome dynamics, and splicing catalysis have been studied in detail, primarily in the mammalian and yeast systems (reviewed by Krämer, 1996; Brow, 2002). Their biochemical composition is characterized very well in the mammalian system (Lührmann et al, 1990; Will and Lührmann, 2001). A highly conserved feature of all known spliceosomal snRNPs is that they share a common core of seven Sm proteins, SmB/B′, D1, D2, D3, E, F, and G, each carrying the characteristic bipartite Sm motif and all seven organized in a ring-like structure (Kambach et al, 1999b; reviewed by Kambach et al, 1999a; Khusial et al, 2005). This heteroheptameric Sm core assembles at the so-called Sm site, which occurs in the U1, U2, U4, and U5 small nuclear RNAs (snRNAs), usually as a single-stranded stretch flanked by stem loops; in mammalian and yeast snRNAs, the Sm site conforms to the consensus sequence 5′-RAU3−6GR-3′ (Branlant et al, 1982; Liautard et al, 1982; Guthrie and Patterson, 1988).
The U6 snRNA, in contrast, binds through its 3′ terminal uridine-rich sequence a set of structurally related proteins, the Sm-like (LSm) proteins, LSm2–8 (Séraphin, 1995; Achsel et al, 1999; Mayes et al, 1999; Salgado-Garrido et al, 1999). Interestingly, exchange of a single polypeptide, LSm8 by another LSm protein, LSm1, yields another seven-membered LSm ring, LSm1–7, that is involved in cytoplasmic RNA degradation (reviewed by He and Parker, 2000).
Compared with mammals and yeast, only a small subset of protein components of snRNPs have been identified in trypanosomes. Trypanosomes are particularly interesting as they process mRNAs through trans splicing, and in addition—at least in the case of the poly(A) polymerase gene—by cis splicing (reviewed by Liang et al, 2003): The U2, U4/U6, and U5 snRNPs are considered general and essential splicing factors, whereas the spliced leader (SL) RNP and the U1 snRNP represent trans- and cis-splicing-specific components, respectively.
An additional point of interest is the fact that trypanosomal snRNAs and their RNPs differ significantly from what we know in other systems, reflecting the large evolutionary distance and trypanosome-specific properties. For example, both the U1 and U5 snRNAs from trypanosomes represent the shortest known orthologs (Dungan et al, 1996; Xu et al, 1997; Schnare and Gray, 1999; Djikeng et al, 2001; Palfi et al, 2002). All five spliceosomal snRNPs have been characterized in trypanosomes to some extent; for example, the Trypanosoma brucei U1 snRNP shows an unusual protein composition (Palfi et al, 2005). From these studies, it is clear that trypanosome snRNPs share a common set of Sm proteins that are organized in the classical heptamer ring (Palfi et al, 2000). However, only five of these polypeptides, SmE/F/G and SmD1/D2, were identified directly by peptide sequences derived from affinity-purified T. brucei U2 snRNP; SmB and D3 came from database searches. Our conclusion that the T. brucei snRNPs share a common Sm core was based mainly on two lines of evidence: first, affinity-purified SL, U2, and U4/U6 snRNPs displayed a common set of at least five polypeptides (Palfi et al, 1991). Second, polyclonal antibodies raised against a mixture of four purified Sm polypeptides (SmE, SmF, SmG, and SmD1; Palfi and Bindereif, 1992) efficiently immunoprecipitated each of the known spliceosomal snRNPs, including the SL RNP (SL, U2, U4/U6: Palfi and Bindereif, 1992; U5: Lücke et al, 1997; U1: Palfi et al, 2002).
Here, we report the first evidence for a variation in the Sm core of the spliceosomal snRNPs, specifically in the U2 snRNP of T. brucei. Based on peptide sequences from affinity-purified U2 snRNPs, we have identified two new Sm proteins. Using in vivo tandem affinity purification (TAP) tagging and biochemical protein–protein interaction assays, we have established both of them as U2-specific Sm polypeptides, replacing the standard SmB/D3 dimer in the Sm heptamer ring. In addition, we have set up an in vitro reconstitution system of canonical and U2-specific Sm cores. Based on this, we conclude that variation in the Sm core composition is responsible for the discrimination between snRNA-specific Sm sites.
Results
T. brucei Sm15K and Sm16.5K, two novel Sm proteins
In our initial study (Palfi et al, 2000), we had identified a complete set of seven Sm polypeptides from trypanosomes: five of them, SmE, SmF, SmG, SmD1, and SmD2, were cloned on the basis of peptide sequences derived from affinity-purified T. brucei U2 snRNPs; the two others, SmB and SmD3, had been identified only by database searches. At that time, additional peptide sequences had been obtained from affinity-purified U2 snRNPs: first, from a protein with an apparent molecular mass of 16.5 kDa (16.5K), which appeared to be U2-specific (Palfi et al, 1991); second, from a mixture of proteins in the 15 kDa range, that could not be clearly assigned to any specific snRNP. With the progression of the T. brucei genome project, we were able to assign two of these peptides to the following two respective proteins: Sm16.5K protein (Tb10.70.2250; 14.7 kDa; 131 amino acids) and Sm15K protein (Tb927.6.4340; 12.7 kDa; 117 amino acids; Figure 1).
Figure 1.

Sequence alignments of two novel Sm proteins from T. brucei: Sm15K and Sm16.5K. (A, B) ClustalW alignment of the Sm15K (A) and Sm16.5K (B) protein sequences from T. brucei (Tb), T. cruzi (Tc), and L. major (Lm). In addition, alignments include sequences of the three trypanosomatid (Tb, Tc, Lm) and the human (Hs) SmB and SmD3 proteins, respectively. For human SmB, the C-terminal extension is not shown; the total numbers of amino acids are given on the right. Below the alignments, a consensus for Sm motifs 1 and 2 is given ($, hydrophobic residue; Séraphin, 1995). GeneDB and NCBI accession numbers: TbSm15K, Tb927.6.4340; TcSm15K, Tc00.1047053506943.114; LmSm15K, LmjF30.3015; TbSmB, Tb927.2.4540; TcSmB, Tc00.1047053507209.10; LmSmB, LmjF27.1970; HsSmB, S10594; TbSm16.5K, Tb10.70.2250; TcSm16.5K, Tc00.1047053506583.10; LmSm16.5K, LmjF36.0535; TbSmD3, Tb927.4.890; TcSmD3, Tc00.1047053508257.150; LmSmD3, LmjF34.3860; HsSmD3, NP_004166.
The analysis of their domain structure revealed that both of them carry the bipartite Sm motif. This was surprising, as we had previously reported a complete set of seven canonical Sm proteins in trypanosomes. Therefore, several questions were raised: first, are the two new Sm proteins U2-specific, and, if there is also a heteroheptameric Sm core in the U2 snRNP, which Sm subunits do they replace? Second, what is the snRNA specificity of the SmB and SmD3 proteins identified previously by us through database search (Palfi et al, 2000)? Third, is there at least a common subcore of Sm polypeptides?
Initial evidence of the nature of the two new Sm proteins came from Blast searches and sequence alignments. Blast searches yielded SmB and SmD3, which are known to interact with each other (Camasses et al, 1998), as the closest homologs of Sm15K and Sm16.5K, respectively. This suggested that these two new Sm proteins might replace SmB and SmD3 in the U2 Sm core (see Figure 1 for a ClustalW alignment of these two Sm protein sequences from T. brucei, T. cruzi, and Leishmania major, together with SmB and SmD3, respectively, from the three trypanosomatid species and the human system, as well as Supplementary Figure S1 for a phylogenetic tree diagram).
T. brucei Sm15K and Sm16.5K are U2-specific Sm proteins
To clarify these questions, we used a TAP procedure, followed by the analysis of copurifying snRNAs. Each of the two new Sm proteins, Sm15K and Sm16.5K, as well as all the seven known Sm proteins (SmE, F, G, D1, D2, B, and D3; Palfi et al, 2000) were TAP-tagged at their C-terminus. These constructs were integrated into the T. brucei genome, and cell lines were generated that stably express the tagged Sm proteins (Figure 2A). As shown for Sm16.5K, for which specific antibodies were available (Palfi et al, 1992), the tagged Sm protein and the endogenous protein are incorporated into U2 snRNPs at a ratio of approximately 1:3 (Supplementary Figure S2). Cell lysates were prepared, tagged complexes were purified on IgG-Sepharose, and copurifying snRNAs were detected by Northern blotting (Figure 2B–D). To unequivocally identify each of the known snRNAs, we analyzed on separate Northern blots the U2 snRNA (panel B), U4 and U6 snRNAs (panel C), and SL, U1, and U5 snRNAs (panel D).
Figure 2.
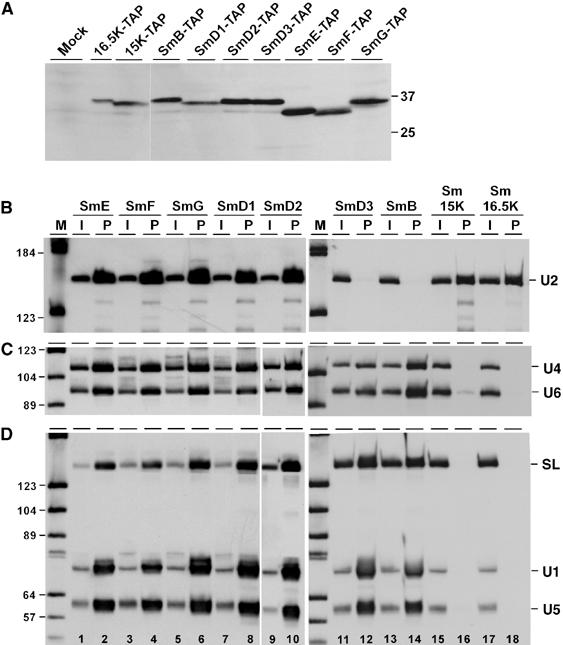
Both Sm15K and 16.5K proteins are U2-specific. (A) Stable expression of TAP-tagged Sm proteins. Lysates were prepared from the wild-type strain (lane mock) and all T. brucei cell lines stably expressing TAP-tagged Sm proteins (as indicated above the lanes). Protein was analyzed by SDS–polyacrylamide gel electrophoresis and Western blotting with peroxidase anti-peroxidase (PAP) soluble complex (Sigma). The positions of marker proteins are given on the right (sizes in kDa). (B, C, D) Extract was prepared from T. brucei cell lines, which stably express TAP-tagged versions of either of the seven canonical Sm proteins (SmE, SmF, SmG, SmD1, SmD2, SmD3, SmB), or of Sm15K or Sm16.5K proteins (as indicated above the lanes). TAP-tagged complexes were affinity-purified from each cell line, and copurifying RNAs were analyzed by Northern blotting, using a U2-specific probe (B), or mixed probes detecting U4 and U6 snRNAs (C), or SL RNA, U1, and U5 snRNAs (D). The snRNA positions are indicated on the right. The input in panel B represents 5% of the total material, in panels C and D 1% (see lanes I); all of the precipitated material is shown (see lanes P). Note that the probe mixture used for the SmD3, SmB, Sm15K, and Sm16.5K pull-downs contains relatively more SL probe than the probe mixture used for the other Sm polypeptides (e.g., compare input lanes 1 and 11, panel D). M, DIG marker V (Roche).
This analysis demonstrated, first, that there is a general subcore of five Sm proteins (SmE/F/G/D1/D2) present in each of the snRNPs U2, U4/U6, U1, and U5, as well as in the SL RNP (Figure 2, panels B–D, lanes 1–10). Comparing the different snRNAs and Sm proteins, the selection efficiencies ranged between 12 and 16% for U2 snRNA, and between 3 and 7% for the other snRNAs, which most likely reflects differential accessibilities of Sm proteins in each of the core complexes. Note that we had previously obtained direct biochemical evidence, based on peptide data, for the presence of these five Sm proteins in purified U2 snRNP (Palfi et al, 2000).
Second, in addition to this common five-membered subcore of SmE/F/G/D1/D2, we analyzed the snRNA association of SmD3 and SmB: The Sm proteins SmB and SmD3 are clearly present in the SL, U1, and U5 snRNPs (panel D, lanes 11–14; selection efficiencies: 2–6%). SmB and SmD3 also exist in the U4/U6 snRNP (panel C, lanes 11–14; selection efficiencies: 2–4%), although with SmD3, the pull-down efficiencies of U4 and U6 were consistently lower than with SmB (see Discussion). In contrast, there was no detectable U2 snRNA association with SmD3 and SmB (panel B, lanes 11–14).
Third, both the new Sm proteins Sm15K and Sm16.5K showed a highly selective snRNA association. Specifically, selection through either Sm15K or Sm16.5K resulted in high levels of U2 snRNA (10 and 7%, respectively; see panel B, lanes 15–18), but no precipitation of the other snRNAs (except for very minor levels of U6, which most likely reflect coprecipitation with U2 and a low-abundance post-spliceosomal U2/U6 complex; see Figure 2C, lane 16). In sum, this establishes Sm15K and Sm16.5K as two U2-specific Sm proteins, replacing the canonical SmB and D3 subunits.
The set of pull-down assays through SmE, F, G, D1, and D2 were performed under low-stringency conditions (150 mM KCl washes), whereas the SmD3, B, 15K, and 16.5K pull-down assays required more stringent conditions (500 mM KCl washes). When the latter set of reactions was performed under low-stringency conditions, low levels of U2 snRNA precipitations were observed also for SmD3 and SmB, as well as low levels of U6 and U5 in the case of Sm15K (data not shown). Most likely this is due to co-precipitation effects caused by larger snRNP complexes, that were disrupted at higher ionic strength. This difference in the stringency of the assays explains also why for SmD3, B, 15K, and 16.5K (lanes 11–18) the pull-down efficiencies were consistently lower than for the other Sm polypeptides (lanes 1–10).
We note that the T. brucei Sm15K protein corresponds to a protein recently identified by genome database screening and homology searching (Liu et al, 2004). This protein termed LSm5 (AY551263) had been suggested to be a component of the U6-specific LSm core. In contrast to that study, we have clearly demonstrated here—using a TAP tagging strategy—that Sm15K associates specifically with the U2 snRNA.
T. brucei Sm15K and Sm16.5K interact with SmD1 and SmG, respectively
The data described above on snRNA associations of the seven canonical and the two U2-specific Sm proteins indicate that in the U2 snRNP SmB and SmD3 are replaced by Sm15K and Sm16.5K, respectively. Next, we needed to establish the overall topology of the newly identified U2-specific Sm core. To obtain biochemical evidence for how the two new U2-specific Sm proteins are arranged in the Sm core, we performed protein–protein interaction assays, using GST pull-down in combination with purified recombinant FLAG-tagged proteins (Figure 3A): GST-Sm15K, GST-Sm16.5K, and as a control, GST alone were immobilized on glutathione-Sepharose, followed by incubation with purified FLAG-tagged SmG or SmD1 proteins, the two potential neighboring Sm proteins. After washing, bound proteins were eluted and analyzed by sodium dodecyl sulfate (SDS)-gel electrophoresis and Western blotting, using anti-FLAG antibodies. This provided clear evidence for specific interactions between GST-Sm15K and SmD1, between GST-Sm16.5K and SmG (lanes 3, 6). There was insignificant or no binding in the other combination (lanes 4, 5), and background binding to GST alone and another Sm protein, GST-SmF, was also very low or undetectable (lanes 7, 8, and data not shown).
Figure 3.
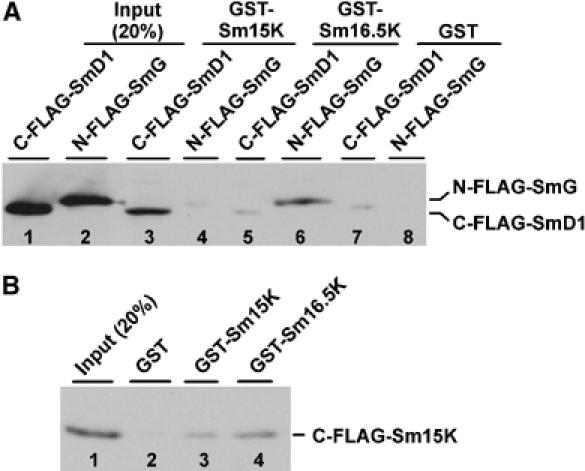
Protein–protein interactions of Sm15K and Sm16.5K proteins. (A) Immobilized proteins GST-Sm15K (lanes 3, 4), GST-Sm16.5K (lanes 5, 6), and as a control, GST (lanes 7, 8) were incubated with purified FLAG-tagged SmG or SmD1 proteins (C-FLAG-SmD1 and N-FLAG-SmG; see 20% of the input, lanes 1 and 2). After washing, bound proteins were eluted and analyzed by SDS-gel electrophoresis and Western blotting, using anti-FLAG antibodies. The positions of the FLAG-tagged SmG and SmD1 proteins are marked on the right. (B) Immobilized proteins GST-Sm15K (lane 3), GST-Sm16.5K (lane 4), and as a control, GST (lane 2) were incubated with purified FLAG-tagged Sm15K protein (C-FLAG-Sm15K; see 20% of the input, lane 1). After washing, bound proteins were eluted and analyzed by SDS-gel electrophoresis and Western blotting, using anti-FLAG antibodies. The position of the FLAG-tagged Sm15K is marked on the right.
In addition, we have obtained direct evidence for Sm15K and Sm16.5K interacting with each other (Figure 3B): Immobilized GST protein, GST-Sm15K, and GST-Sm16.5K were incubated with purified FLAG-tagged Sm15K, followed by the elution of bound proteins and their analysis by SDS-gel electrophoresis and Western blotting, using anti-FLAG antibodies. Although there was a low level of self-interaction for Sm15K (lane 3), the interaction was much stronger between GST-Sm16.5K and FLAG-tagged Sm15K (lane 4), and there was no detectable signal for the control combination FLAG-Sm15K and GST (lane 2). Additional support for an Sm15K/16.5K interaction is based on the coexpression of both Sm15K and Sm16.5K proteins in Escherichia coli: first, only by coexpressing both proteins we could obtain high yields of recombinant proteins. Second, recombinant Sm15K and Sm16.5K proteins were purified after coexpression in E. coli, with only one of them, Sm16.5K, carrying a His-tag, yet resulting in Sm15K/16.5K heterodimer in equimolar stoichiometry (as assessed by Coomassie staining; data not shown).
In sum, these interaction assays clearly established the topology of the newly identified U2-specific Sm core: Sm15K and Sm16.5K interact with each other and substitute for SmB and SmD3, respectively, bridging between SmD1 and SmG.
Sm core variation in the U2 snRNP: model and correlation with special Sm binding site in the U2 snRNA
Figure 4A summarizes our model of the U2 snRNP-specific Sm core variation. We note that this remarkable feature of Sm core organization is paralleled only in one other RNP complex, the U7 snRNP, which functions in histone 3′ end processing: in the U7 snRNP, SmD1 and D2 are exchanged by two U7-specific LSm proteins, LSm10 and LSm11 (Pillai et al, 2001, 2003; reviewed by Schümperli and Pillai, 2004). It is noteworthy that in both cases a stable heterodimer unit is replaced, which also acts as an assembly intermediate (Raker et al, 1996): SmD1/D2 in the U7 snRNP, and SmB/D3 in the trypanosome U2 snRNP. We note that replacing two neighboring Sm polypeptides—as opposed to two separate ones—requires fewer switches in specific intersubunit contacts and may therefore be evolutionarily favorable. In contrast, in the LSm heptamer ring only a single subunit is exchanged, LSm1 versus LSm8 (see Introduction for references).
Figure 4.
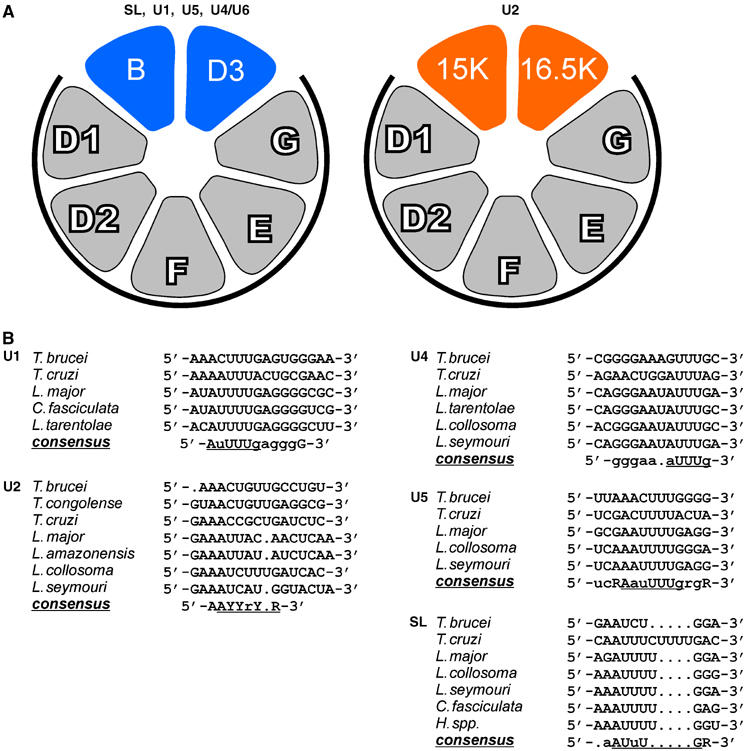
Model of Sm core variation in the U2 snRNP (A) and sequence comparison of Sm sites in trypanosomid snRNAs (B). (A) The arrangement of Sm proteins in the standard core (left model) is according to Palfi et al (2000). This composition of Sm polypeptides has been experimentally verified for the SL RNP, and the U1, U5, and U4/U6 snRNPs from T. brucei (this study; see Figure 2). The U2 snRNP, however, contains a variant Sm core, in which the canonical SmB and SmD3 proteins are replaced by Sm15K and Sm16.5K (right model). The relative orientation of the latter two Sm proteins is based on their homology to SmB and SmD3, respectively, and on protein–protein interaction data (see Figure 3). The common subcore of SmD1/D2/F/E/G is marked by heavy lines. (B) Sequence alignment of the Sm sites of U1, U2, U4, U5 snRNAs as well as of the SL RNA from the following trypanosomatid species: T. brucei; T. cruzi, T. congolense, L. major, L. amazonensis, Leptomonas collosoma, Leptomonas seymouri, Crithidia fasciculata, Herpetomonas species. For U1, U4, U5, and SL RNAs, sequences from only a few representative species are shown, for U2 all available sequences. Nucleotides in large letters indicate absolute conservation for the sequences shown, small letters indicate at most one deviation. The consensus sequences of the Sm sites are underlined. R, purine, Y, pyrimidine nucleotide.
What are the functional consequences of the altered core in the trypanosome U2 snRNP? Comparing the Sm site sequences in the U2 snRNA with those in SL RNA, U1, U4, and U5 snRNAs revealed a striking difference (Figure 4B): with the exception of U2, a consensus can be derived for each of the other spliceosomal snRNAs that closely resembles the Sm site consensus 5′-RAU3−6GR-3′. In contrast, in the loose consensus, 5′-AAYYrY(U)R-3′, derived from the trypanosomatid U2 snRNAs, an unusual purine position interrupts the central pyrimidine stretch in the middle (see Discussion).
In vitro assembly of reconstituted canonical and U2-specific Sm cores: Sm proteins confer specificity for U2 Sm site
To address the question of whether the Sm core composition is required and sufficient for snRNA binding specificity, we established an in vitro reconstitution approach. Similarly as previously performed for the mammalian Sm and LSm heptamers (Zaric et al, 2005), each of the three subcomplexes of the trypanosome Sm cores were coexpressed in E. coli and purified, each with a single His6-tag per subcomplex (see Materials and methods for details): for the canonical Sm core, SmD1/D2, SmB/D3, and SmE/F/G; for the U2 Sm core, SmD1/D2, Sm15K/16.5K, and SmE/F/G.
In principle, reconstitutions were carried out by incubating equimolar amounts of the three respective subcomplexes together with RNAs to be tested for binding. Reconstituted Sm cores were recovered by His tag pull-down, using Ni-NTA agarose beads, and co-precipitated RNAs were released and analyzed by denaturing polyacrylamide gel electrophoresis. Initially, to test RNA binding specificity, we have used mixtures of in vitro transcribed snRNAs from T. brucei (Figure 5A), then short RNA oligonucleotides covering the U1 and U2 Sm sites (Figure 5B; for quantitations, see Supplementary Figure S3).
Figure 5.
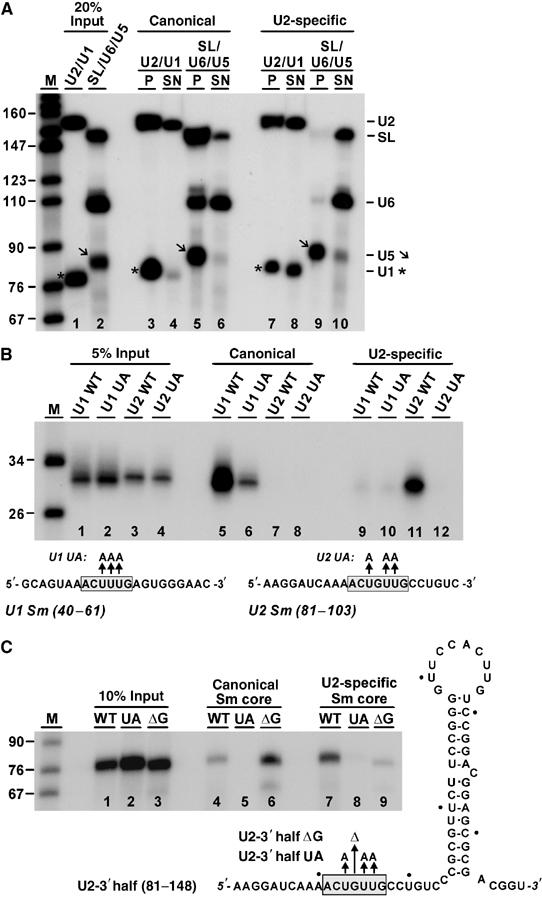
snRNA specificity of reconstituted canonical and U2 Sm cores. (A) Binding of in vitro transcribed snRNAs. 32P-labeled snRNA from T. brucei were prepared by transcription (20% of U2/U1 and SL/U6/U5 input shown in lanes 1 and 2, respectively). After incubation with recombinant canonical (lanes 3–6) or U2-specific Sm cores (lanes 7–10), co-precipitated RNAs were recovered by His-tag pull-down with Ni-NTA agarose beads and analyzed by denaturing gel electrophoresis (snRNA positions marked on the right; U5 bands denoted by an arrow, U1 bands by asterisks). For each reconstitution reaction, both the pull-down material (P; lanes 3, 5, 7, and 9) and the supernatant fractions (SN; lanes 4, 6, 8, and 10; 20% shown) were analyzed. M, markers (sizes in nucleotides). (B) Binding specificity of Sm site RNA oligonucleotides. 32P-labeled short RNAs were prepared by T7 transcription, containing the U1 and U2 Sm sites, both as wild-type and mutant versions (U1 WT and U1 UA, U2 WT and U2 UA; RNA sequences shown below, with the Sm sites boxed). Each of them was reconstituted in vitro with recombinant canonical (lanes 5–8) or U2-specific Sm cores (lanes 9–12). Co-precipitated RNAs were recovered by His-tag pull-down with Ni-NTA agarose beads and analyzed by denaturing gel electrophoresis. For comparison, 5% of the input RNAs are shown (lanes 1–4). M, markers (sizes in nucleotides). (C) Sequence requirements for binding canonical and U2-specific Sm cores. 32P-labeled RNAs derived from the T. brucei U2 snRNA 3′ half (nucleotides 81–148) were in vitro transcribed: WT, wild type; UA, U93A U95A U96A; ΔG, ΔG94 (sequences and secondary structure model shown on the right; 10% of the input RNAs in lanes 1–3). Following reconstitution in vitro with recombinant canonical (lanes 4–6) or U2-specific Sm cores (lanes 7–9), co-precipitated RNAs were recovered by His-tag pull-down with Ni-NTA agarose beads and analyzed by denaturing gel electrophoresis. M, markers (sizes in nucleotides).
First, 32P-labeled U2, SL, U6, U5, and U1 snRNAs from T. brucei were prepared by SP6/T7 transcription and reconstituted as separate mixtures of SL, U6, and U5 snRNAs (Figure 5A, lane 2), as well as U2 and U1 snRNAs (lane 1), followed by heparin treatment. For each reconstitution, both the His-tag pull-down (lanes P) and the supernatant fractions (lanes SN) were analyzed. The canonical Sm core associated with SL, U5, and U1 snRNAs more efficiently than the U2-specific Sm core did (SL: 32 versus 1%; U5: 44 versus 30%; U1: 27 versus 7%; compare input lanes 1–2 with lanes 3–10). U6 snRNA, which is expected to interact with neither of the Sm cores, associated efficiently with the canonical Sm core (12%), but not significantly with the U2-specific Sm core (12 versus 1%; compare lanes 5/6 and 9/10). Finally, U2 snRNA interacted efficiently with both canonical (21%) and U2-specific Sm cores (21 versus 14%; compare lanes 3/4 and 7/8). Based on these in vitro reconstitutions with full-length snRNA transcripts, we conclude that the canonical and U2-specific Sm core show only limited specificity for their respective snRNAs under these conditions.
Second, we did reconstitutions with short RNA oligonucleotides comprised of the Sm site sequence of U1 (nucleotides 40–61) or U2 (nucleotides 81–103). To test whether reconstitution depends on an intact Sm site sequence, we also assayed mutant derivatives, where three uridine residues were replaced by adenosines; these three-nucleotide substitutions should inactivate the Sm site (Figure 5B; Sm site sequences below). Each of these 32P-labeled RNA oligonucleotides was generated by in vitro transcription (see lanes 1–4 for 5% input) and reconstituted with the canonical or the U2-specific Sm proteins (lanes 5–8 and 9–12, respectively). In contrast to the previous assays, specificity with these short RNA oligonucleotides was very high: first, U1 and U2 Sm site sequences efficiently assembled only with the canonical and U2-specific Sm cores, respectively (compare lanes 5, 7, 9, and 11). Second, the mutated Sm sites did not significantly (lanes 8, 10, and 12) or only to a very low extent (lane 6) reconstitute with the Sm core complexes.
Third, we further investigated the specificity issue and tested by mutational analysis what in the U2 Sm site is required for discriminating between the U2-specific and the canonical Sm core (Figure 5C). Because the context around the Sm site may be important for snRNA specificity (see Jarmolowski and Mattaj, 1993), we used a T. brucei U2 snRNA derivative containing nucleotides 81–148, including the 3′ terminal stem loop. Besides the wild-type RNA (U2-3′ half), two mutant derivatives were tested: U2-3′ half UA, which should inactivate the Sm site by substitution of the three uridine residues by adenosines, and U2-3′ half ΔG, in which G94 is deleted. Note that the latter single-nucleotide deletion makes the U2 Sm site identical to the core of the U1 Sm site. Each of these 32P-labeled, transcribed RNAs (see lanes 1–3 for 10% input) was reconstituted with the canonical or the U2-specific Sm proteins (lanes 4–6 and 7–9, respectively). As a result, the U2-specific Sm core associated efficiently with U2-3′ half RNA (see panel C, lane 7), but not detectably or only very weakly with the UA and the ΔG mutant RNAs, respectively (lanes 8–9), consistent with the analysis of the short RNA oligonucleotides (see panel B) and indicating that the unusual guanosine in the U2 Sm site is essential for recognition by the U2 Sm core. Furthermore, in this extended context, the canonical Sm core associated with the wild-type U2-3′ half RNA, although less efficiently than the U2-specific Sm core did (compare lanes 4 and 7); again, this interaction was abolished by the UA mutation (lane 5). Importantly, however, deleting G94 in the U2 Sm site strongly increased association with the canonical Sm core (compare lanes 4 and 6), underlining the importance of the central guanosine position. In sum, a single point mutation (ΔG94) in the U2 Sm site converted the U2 Sm site into an Sm site recognized efficiently by the canonical core (Figure 5, panel C, compare lanes 6 and 9). This demonstrates that G94 provides an important determinant for discriminating between the U2-specific and the canonical Sm core.
Discussion
We have identified two novel Sm proteins from T. brucei that specifically associate with the U2 snRNP: Sm15K substitutes for SmB, whereas Sm16.5K replaces SmD3. By in vitro reconstitution, we demonstrated that these two novel and U2-specific Sm proteins are able to confer specificity for Sm core assembly on the Sm site of the U2 snRNA. This shows for the first time that a variation within the Sm core of a spliceosomal snRNP can change the RNA binding specificity. The altered Sm core composition correlates with a striking difference of the U2 Sm binding site from the otherwise well-conserved consensus: in contrast to the standard 5′-RAU3−6GR-3′ consensus, the Sm site of U2 snRNA contains a highly unusual purine nucleotide interrupting the central pyrimidine stretch (5′-AAYYrY(U)R-3′; see Figure 4B).
In this context, it is interesting to note that Jarmolowski and Mattaj (1993) had already found that the short single-stranded Sm site is not always sufficient to determine Sm protein binding; also, the Sm sites themselves are not always interchangeable between snRNAs, further arguing for additional, complex determinants for Sm protein binding on specific snRNAs. For example, in the T. brucei U2 snRNP such additional sequence or structural determinants may be involved in transient contacts with the U2-specific Sm polypeptides.
Significantly, Urlaub et al (2001) had mapped an RNA–protein contact within an Sm site-containing RNA oligonucleotide (5′-AAU5GA-3′) to human SmB/B′, which in the trypanosome U2 core is replaced by Sm15K; on the RNA level, they had mapped this contact to the third uridine position of the pyrimidine stretch, where in the trypanosome U2 Sm site the unusual purine resides. Most likely therefore, the special Sm site of U2 reflects binding of the U2-specific Sm core. In fact, already during the initial characterization of the trypanosome U2 snRNP we had inferred that—in contrast to the other spliceosomal snRNPs—it carries a ‘special core structure' (Cross et al, 1991; Günzl et al, 1993), based on the instability of the U2 snRNP under the high-stringency conditions of CsCl gradient centrifugation. In sum, the Sm core variation described here clearly modulates the RNA binding specificity; moreover, it may also affect other aspects of U2 snRNP biogenesis and function, such as unique protein–protein interactions. Finally, these differences may also be relevant for the special requirements of the U2 snRNP during branch point/3′ splice site recognition (Lücke et al, 2005).
When we assessed RNA binding specificity in detail, we compared the in vitro assembly of Sm cores with different RNAs: with mixtures of snRNA transcripts (Figure 5A) and with short RNA oligonucleotides carrying the Sm sites of U1 or U2 snRNAs (Figure 5B). As a result, a high degree of specificity could be achieved only with short oligonucleotides. In more complex mixtures, however, such as RNA transcripts, only a limited degree of specificity was observed; in particular, the canonical Sm core was unable to discriminate against U2 snRNA. This apparent lack of discrimination with longer RNAs was also demonstrated by comparing the Sm core binding behavior between RNA oligonucleotides and the U2-3′ half RNAs (Figure 5, panels B and C). Finally, we were able to demonstrate that the unusual purine residue in the U2 Sm site (G94) is important for discriminating between the U2-specific and the canonical Sm core.
Why do longer RNAs show reduced specificity? Most likely, aberrant or U2-like Sm sites are recognized in many RNAs to some degree, so that in vivo, additional specificity and discriminatory activities may be required. This situation is reminiscent of the role of the SMN complex in mammalian cells (Pellizzoni et al, 2002; reviewed by Yong et al, 2004).
What are the more general implications of these findings? Although our study represents to our knowledge the first case of spliceosomal core variation, it raises the possibility that this may occur also in other trypanosome snRNPs or also in other biological systems. As shown in Figure 2B–D (lanes 11–12), TAP tagging of SmD3 recovered U4/U6 snRNPs at relatively low efficiency, compared with SL, U1, and U5 snRNPs, suggesting that in the trypanosome U4/U6 snRNP, SmD3 and SmB may be replaced by another, unknown Sm protein; there may be a mixed population of U4/U6 snRNPs, carrying different sets of Sm proteins. We would like to point out that careful inspection of our initial protein identification in the T. brucei snRNPs had revealed subtle differences in the low-molecular-weight range (see Figure 4 in Palfi et al, 1991). One could even envision a switch of certain Sm polypeptides during the life cycle of snRNPs. Finally, although no experimental evidence is available for Sm core differences in the mammalian spliceosomal snRNPs, Sm core variations may exist only in certain minor snRNP fractions or be restricted to certain tissues or developmental stages.
Materials and methods
Cell culture, transfection, extract preparation
Cell culture of the procyclic form of T. brucei strain 427 and of stably transfected cell lines was as described (Cross et al, 1991). For transfection, 10 μg of Sm-TAP plasmids were linearized inside the open reading frames and electroporated, using approximately 3 × 108 T. brucei cells (Schimanski et al, 2004). Afterwards, transfected cells were selected in medium containing 40 μg/ml of G418 (Geneticin, Gibco-BRL). Total cell extract was prepared in IPP-150 buffer (150 mM KCl; 20 mM Tris–Cl, pH 7.7; 3 mM MgCl2; 0.5 mM dithiothreitol (DTT)), containing a Complete Mini, ethylene diaminetetra acetic acid (EDTA)-free protease inhibitor cocktail tablet (Roche), by using a Polytron PT 3100 cell homogenizer (Kinematica AG, Switzerland). Homogenized cells were then centrifuged at 14 000 r.p.m. for 15 min to remove aggregates. One milliter of total cell extract corresponds to ∼2 × 109 cell equivalents.
Database analysis
The accession numbers of the trypanosomatid genes are annotations of GeneDB (Hertz-Fowler et al, 2004; http://www.genedb.org/). Protein sequence alignments were performed by ClustalW (Thompson et al, 1994).
Protein sequence analysis
The Sm15K and Sm16.5K proteins were initially obtained by affinity purification of U2 snRNPs from T. brucei based on a biotinylated antisense 2′-O-methyl RNA oligonucleotide (Palfi et al, 1991), electroblotted onto nitrocellulose and digested with trypsin. The resulting peptide mixture was separated by narrow bore high-performance liquid chromatography (HPLC) using a Vydac C18 2.1 × 150-mm reverse-phase column on a Hewlett-Packard 1090 HPLC with 1040 diode array detector. Optimum fractions from this chromatogram were chosen based on differential UV absorbance at 210, 277, and 292 nm and molecular weight information by matrix-assisted laser desorption time of flight mass spectrometry. Selected fractions were submitted to automated Edman degradation on an Applied Biosystems 477A Protein Sequencer using a microcartridge and cycles optimized for a 30-min cycle time. The following peptide sequences were determined:
15K protein: VSVDLDDGSTLVG, LVSFSPTSNLILTDAER, NECYNCVLFVR, GSSVLSVK, HSSGVTTDATVIDSITGR and TIQAASQSLDTPLR;
16.5K protein: VETTDGSVYDGK, DSSLSVEGR, RAPFLDW and NLSIQK.
Cloning of Sm15K and Sm16.5K genes from T. brucei; plasmid constructs
The open reading frames coding for the Sm15K (Tb927.6.4340) and Sm16.5K (Tb10.70.2250) proteins were identified, based on mass-spectrometric analysis of affinity-purified T. brucei U2 snRNP proteins. Open reading frames were cloned into the pCR2.1-TOPO vector (Invitrogen), using polymerase chain reaction (PCR) products generated from genomic DNA as template (DNAzol reagent, Invitrogen), and sequenced in both directions. For the PTP-tagged Sm15K and 16.5K constructs, the full open reading frames were inserted in-frame into the pC-PTP-NEO vector upstream of the PTP tag sequence, using the ApaI and NotI restriction sites. The PTP tag is a modified TAP tag consisting of two protein A domains, a TEV protease cleavage site and the protein C epitope (Schimanski et al, 2005a, 2005b).
The open reading frames of each of the seven canonical Sm proteins (Palfi et al, 2000) were also generated from T. brucei genomic DNA by PCR: SmB (Tb927.2.4540), SmD1 (Tb927.7.3120), SmD2 (Tb927.2.5850), SmD3 (Tb927.4.890), SmE (Tb927.6.2700), SmF (Tb09.211.1695), and SmG (Tb11.01.5915). Open reading frames were cloned—in analogy to the Sm15K/16.5K-PTP constructs—into the TAP vector, which contains instead of the PTP, the standard TAP tag (Rigaut et al, 1999).
FLAG-tagged and GST fusion proteins
For the FLAG-tagged proteins, the open reading frames of SmD1, SmG, and Sm15K were cloned into the pQE30 vector (Qiagen), with a FLAG tag at the C- or N-terminus (C-FLAG-SmD1, N-FLAG-SmG, and C-FLAG-Sm15K), and then overexpressed in E. coli strain M15 cells. Total cell extract was prepared by sonication in lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 100 mM imidazole, and 1.25 mg/ml lysozyme), containing a Complete Mini, EDTA-free protease inhibitor cocktail tablet (Roche). Lysed cells were centrifuged at 14 000 r.p.m. for 15 min to remove aggregates.
For the purification of C-FLAG-SmD1 and N-FLAG-SmG proteins, cell lysates were incubated with 20 μl of packed bead volume of anti-FLAG M2 affinity beads (Sigma) equilibrated with 1 × wash buffer (50 mM Tris–Cl, pH 7.4, 150 mM NaCl) at 4°C for 4 h, and then washed with 1 × wash buffer. Afterwards, bound proteins were eluted with 10 bead volumes of 1 M glycine, pH 2.5, immediately neutralized with 1/10 volume of 100 mM Tris–Cl, pH 8.5. Eluted proteins were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
For the purification of C-FLAG-Sm15K protein through its His-tag, 1 ml of the cell lysate was incubated with 30 μl of Ni-NTA Agarose beads (Qiagen) equilibrated with 1 × wash buffer (50 mM NaH2PO4, pH 8.0; 300 mM NaCl; 20 mM imidazole) for 4 h at 4°C, followed by elution under native conditions (50 mM NaH2PO4, pH 8.0; 300 mM NaCl; 300 mM imidazole).
For the GST fusion proteins, the open reading frames of Sm15K and Sm16.5K gene were cloned into pGEX-2TK vector (Pharmacia) and overexpressed in E. coli BL-21 stain. Cell lysates were prepared as described above. Proteins were purified through glutathione-Sepharose 4B beads (Amersham) and washed with 1 × PBS containing 0.05% NP40.
Western blot analysis
For analyzing the expression of PTP- and TAP-tagged proteins in T. brucei, 10 μl of cell lysates from stably transfected or untransfected (negative control) cell lines were separated by SDS polyacrylamide gel electrophoresis (15%) and detected with the PAP reagent (Sigma).
TAP tag pull-down assays and Northern analysis
For pull-down assays via TAP- or PTP-tag, cell extract from stably transfected cell lines was incubated at 4°C with 50 μl packed IgG Sepharose 6 Fast Flow beads (Invitrogen) equilibrated with IPP-150 buffer. After washing with the same buffer (or, as specified, IPP-500, which contains 500 mM KCl), coselected RNAs were released by proteinase K buffer treatment and analyzed by denaturing polyacrylamide gel electrophoresis, followed by Northern blotting, using DIG-labeled probes (Bell and Bindereif, 1999) and signal quantitation (Tina Version 2.07d software).
Protein–protein interaction assays by GST pull-down
For assaying protein–protein interactions, 6 μg of a GST-tagged Sm15K, Sm16.5K protein, or GST protein alone (as negative control) were immobilized on 25 μl packed glutathione-Sepharose 4B beads (Amersham), incubated with ∼500 ng of FLAG-tagged SmD1 or SmG in 900 μl of binding buffer (50 mM Tris–Cl, pH 7.5, 300 mM KCl, 5 mM MgCl2, 0.05% NP-40, 0.5 mM DTT). After a 4-h incubation at 4°C, beads were washed with the same buffer, but containing 500 mM KCl. Bound proteins were released from the beads, and analyzed by SDS polyacrylamide gel electrophoresis (15%) and Western blotting with anti-FLAG M2 monoclonal antibody (Sigma).
To detect the interaction between Sm15K and Sm16.5K protein, the same GST pull-down assay was performed: 800 ng of C-terminally FLAG-tagged Sm15K was added to 2 μg of GST-Sm15K, GST-Sm16.5K, or GST protein in 900 μl of binding buffer, washed with the same buffer containing 300 mM KCl.
Oligonucleotides, mutagenesis, and in vitro transcription
Sequences of DNA oligonucleotides are available upon request. Full-length, α-32P-UTP-labeled, m7G-capped trypanosome snRNAs SL, U1, U2, U5, and U6 were transcribed in vitro by T7 (U2, U5 and U6 snRNA) or SP6 RNA polymerase (SL and U1 snRNA), using as templates PCR fragments generated from trypanosome genomic DNA (SL, U1, U5), or plasmid DNA linearized by XbaI (U2, U6; Cross et al, 1991).
α-32P-ATP-labeled, uncapped TbU1 (40–61) wild type, which contains nucleotides 40–61 of the T. brucei U1 snRNA with the Sm site sequence, was transcribed by T7 RNA polymerase, using a double-stranded DNA oligonucleotide as a template. Similarly, α-32P-CTP-labeled TbU2 Sm (81–103) wild type was generated, which contains nucleotides 81–103 of the T. brucei U2 snRNA with the Sm site sequence. In TbU1 (40–61) mutant, the Sm site ACUUUG was changed to ACAAAG, in TbU2 (81–103) mutant the Sm site AACUGUUG to AACAGAAG (mutated positions underlined).
α-32P-ATP or -UTP-labeled, uncapped TbU2-3′ half wild type, which contains nucleotides 81–148 of the T. brucei U2 snRNA with the Sm site sequence, was transcribed by T7 RNA polymerase, using as a template two overlapping DNA oligonucleotides filled-in by Taq DNA polymerase. In the mutant derivative TbU2-3′ half UA, the Sm site ACUGUUG was changed to ACAGAAG (mutated positions underlined); in TbU2-3′half ΔG, nucleotide G94 was deleted.
Expression and purification of His-tagged Sm proteins and subcomplexes
T. brucei cDNAs for SmD1, D2, D3, B, E, F, G, 16.5K, and 15K were subcloned as di- (SmD1/D2, SmD3/B, Sm16.5K/15K) or tri-cistrons (SmE/F/G) by PCR into the pQE30 expression vector (Qiagen). In each case, the first cistron bears an N-terminal His6-tag followed by a Tobacco Etch Virus (TEV) cleavage site. Protein subcomplexes were expressed in SG13009[pREP4] cells (Qiagen) for 16 h at 25°C, inducing with 1 mM IPTG. Cells were lysed on ice in 20 mM Tris–Cl, pH 8.0, 0.5 M NaCl, 10% glycerol, 5 mM β-mercaptoethanol, 10 mM imidazole-Cl, pH 8.0 supplemented with Complete™ (Roche) protease inhibitor, using four cycles on an Emulsiflex-C3 (Avestin Inc., Ottawa, Ontario, Canada) cell disruptor, followed by sonication for 5 min (Sonics Vibracell). Lysates were cleared by ultracentrifugation (1 h, 40 000 r.p.m., 4°C), 0.22 μm-filtered, and purified via Ni-IMAC followed by gel filtration on an ÄKTAxpress (GE Healthcare) FPLC system. IMAC elution buffer contained 0.5 M imidazole-Cl, pH 8.0 (otherwise as lysis buffer); samples were eluted from the Superdex 200 16/60 column (GE Healthcare), using 20 mM Tris–Cl, pH 8.0, 200 mM NaCl, 10% glycerol, 5 mM β-mercaptoethanol, concentrated in this buffer to between 2 and 25 mg/ml, and frozen in liquid N2.
Reconstitution of recombinant Sm core and His-tag pull down assays
First, 400 μmol of purified Sm subcomplexes (for canonical Sm core: SmB/D3, SmD1/D2, and SmE/F/G; for U2 Sm core: Sm16.5K/15K, SmD1/D2, and SmE/F/G) were mixed in equimolar amounts in 10 μl of 5 × reconstitution buffer (100 mM Tris–Cl, pH 7.5, 1 M NaCl, 25 mM MgCl2, 5 mM DTT). Second, ∼30 ng (1.5 × 106 c.p.m.) of full-length snRNA transcripts (U2, SL, U6, U5, U1), or ∼5 ng (2.7 × 105 c.p.m.) of short RNA transcripts [U1 Sm (40–61), U2 Sm (81–103), each as wild-type or UA mutant], or 13 ng U2-3′ half (81–148) wild-type or mutant derivatives UA and ΔG (4 × 105 c.p.m.) were added to the reaction, to give a final volume of 50 μl. The reconstitution reactions were incubated at 30°C for 30 min and then at 37°C for 15 more minutes. To block unspecific binding, heparin was added to a final concentration of 1 mg/ml after reconstitution with full-length snRNA transcripts (Figure 5A) and incubated at 30°C for 10 min. For the short RNAs (Figure 5B and C), reconstitutions were carried out without heparin.
For the pull-down assays, the reconstituted Sm complexes were incubated with 25 μl of packed Ni-NTA agarose beads (Qiagen) in 1 × reconstitution buffer (20 mM Tris–Cl, pH 7.5, 200 mM of NaCl, 5 mM MgCl2, 1 mM DTT, 0.05 % NP40) at 4°C. After washing with the same buffer, bound RNAs were released from the Ni-NTA agarose beads by treatment with proteinase K buffer and analyzed by denaturing polyacrylamide gel electrophoresis and autoradiography; signals were quantitated using Tina Version 2.07d software.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Acknowledgments
We are grateful to Arthur Günzl for the pC-PTP-NEO vector and advice on TAP-tagging. This work was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 535; Graduiertenkolleg Biochemie von Nukleoproteinkomplexen; to AB), the Fonds der Chemischen Industrie (to AB), and by the German-Israeli Foundation (GIF, to AB).
References
- Achsel T, Brahms H, Kastner B, Bachi A, Wilm M, Lührmann R (1999) A doughnut-shaped heteromer of human Sm-like proteins binds to the 3′-end of U6 snRNA, thereby facilitating U4/U6 duplex formation in vitro. EMBO J 18: 5789–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell M, Bindereif A (1999) Cloning and mutational analysis of the Leptomonas seymouri U5 snRNA gene: function of the Sm site in core RNP formation and nuclear localization. Nucleic Acids Res 27: 3986–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branlant C, Krol A, Ebel JP, Lazar E, Haendler B, Jacob M (1982) U2 RNA shares a structural domain with U1, U4, and U5 RNAs. EMBO J 1: 1259–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brow DA (2002) Allosteric cascade of spliceosome activation. Annu Rev Genet 36: 333–360 [DOI] [PubMed] [Google Scholar]
- Camasses A, Bragado-Nilsson E, Martin R, Séraphin B, Bordonné R (1998) Interactions within the yeast Sm core complex: from proteins to amino acids. Mol Cell Biol 18: 1956–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross M, Günzl A, Palfi Z, Bindereif A (1991) Analysis of small nuclear ribonucleoproteins (RNPs) in Trypanosoma brucei: structural organization and protein components of the spliced leader RNP. Mol Cell Biol 11: 5516–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A, Ferreira L, D'Angelo M, Dolezal P, Lamb T, Murta S, Triggs V, Ulbert S, Villarino A, Renzi S, Ullu E, Tschudi C (2001) Characterization of a candidate Trypanosoma brucei U1 small nuclear RNA gene. Mol Biochem Parasitol 113: 109–115 [DOI] [PubMed] [Google Scholar]
- Dungan JM, Watkins KP, Agabian N (1996) Evidence for the presence of a small U5-like RNA in active trans-spliceosomes of Trypanosoma brucei. EMBO J 15: 4016–4029 [PMC free article] [PubMed] [Google Scholar]
- Günzl A, Cross M, Palfi Z, Bindereif A (1993) Assembly of the U2 small nuclear ribonucleoprotein from Trypanosoma brucei. A mutational analysis. J Biol Chem 268: 13336–13343 [PubMed] [Google Scholar]
- Guthrie C, Patterson B (1988) Spliceosomal snRNAs. Annu Rev Genet 22: 387–419 [DOI] [PubMed] [Google Scholar]
- He W, Parker R (2000) Functions of Lsm proteins in mRNA degradation and splicing. Curr Opin Cell Biol 12: 346–350 [DOI] [PubMed] [Google Scholar]
- Hertz-Fowler C, Peacock CS, Wood V, Aslett M, Kerhornou A, Mooney P, Tivey A, Berriman M, Hall N, Rutherford K, Parkhill J, Ivens AC, Rajandream MA, Barrell B (2004) GeneDB: a resource for prokaryotic and eukaryotic organisms. Nucleic Acids Res 32 (Database issue): D339–D343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmolowski A, Mattaj IW (1993) The determinants for Sm protein binding to Xenopus U1 and U5 snRNAs are complex and non-identical. EMBO J. 12: 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambach C, Walke S, Nagai K (1999a) Structure and assembly of the spliceosomal small nuclear ribonucleoprotein particles. Curr Opin Struct Biol 9: 222–230 [DOI] [PubMed] [Google Scholar]
- Kambach C, Walke S, Young R, Avis JM, de la Fortelle E, Raker VA, Lührmann R, Li J, Nagai K (1999b) Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell 96: 375–387 [DOI] [PubMed] [Google Scholar]
- Khusial P, Plaag R, Zieve GW (2005) LSm proteins form heptameric rings that bind to RNA via repeating motifs. Trends Biochem Sci 30: 522–528 [DOI] [PubMed] [Google Scholar]
- Krämer A (1996) The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem 65: 367–409 [DOI] [PubMed] [Google Scholar]
- Liang XH, Haritan A, Uliel S, Michaeli S (2003) Trans and cis splicing in trypanosomatids: mechanism, factors, and regulation. Eukaryot Cell 2: 830–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liautard JP, Sri-Widada J, Brunel C, Jeanteur P (1982) Structural organization of ribonucleoproteins containing small nuclear RNAs from HeLa cells. Proteins interact closely with a similar structural domain of U1, U2, U4 and U5 small nuclear RNAs. J Mol Biol 162: 623–643 [DOI] [PubMed] [Google Scholar]
- Liu Q, Liang XH, Uliel S, Belahcen M, Unger R, Michaeli S (2004) Identification and functional characterization of Lsm proteins in Trypanosoma brucei. J Biol Chem 279: 18210–18219 [DOI] [PubMed] [Google Scholar]
- Lücke S, Jürchott K, Hung LH, Bindereif A (2005) mRNA splicing in Trypanosoma brucei: branch-point mapping reveals differences from the canonical U2 snRNA-mediated recognition. Mol Biochem Parasitol 142: 248–251 [DOI] [PubMed] [Google Scholar]
- Lücke S, Klockner T, Palfi Z, Boshart M, Bindereif A (1997) Trans mRNA splicing in trypanosomes: cloning and analysis of a PRP8-homologous gene from Trypanosoma brucei provides evidence for a U5-analogous RNP. EMBO J 16: 4433–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lührmann R, Kastner B, Bach M (1990) Structure of spliceosomal snRNPs and their role in pre-mRNA splicing. Biochim Biophys Acta 1087: 265–292 [DOI] [PubMed] [Google Scholar]
- Mayes AE, Verdone L, Legrain P, Beggs JD (1999) Characterization of Sm-like proteins in yeast and their association with U6 snRNA. EMBO J 18: 4321–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi Z, Bindereif A (1992) Immunological characterization and intracellular localization of trans-spliceosomal small nuclear ribonucleoproteins in Trypanosoma brucei. J Biol Chem 267: 20159–20163 [PubMed] [Google Scholar]
- Palfi Z, Günzl A, Cross M, Bindereif A (1991) Affinity purification of Trypanosoma brucei small nuclear ribonucleoproteins reveals common and specific protein components. Proc Natl Acad Sci USA 88: 9097–9101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi Z, Lane WS, Bindereif A (2002) Biochemical and functional characterization of the cis-spliceosomal U1 small nuclear RNP from Trypanosoma brucei. Mol Biochem Parasitol 121: 233–243 [DOI] [PubMed] [Google Scholar]
- Palfi Z, Lücke S, Lahm HW, Lane WS, Kruft V, Bragado-Nilsson E, Séraphin B, Bindereif A (2000) The spliceosomal snRNP core complex of Trypanosoma brucei: cloning and functional analysis reveals seven Sm protein constituents. Proc Natl Acad Sci USA 97: 8967–8972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi Z, Schimanski B, Günzl A, Lücke S, Bindereif A (2005) U1 small nuclear RNP from Trypanosoma brucei: a minimal U1 snRNA with unusual protein components. Nucleic Acids Res 33: 2493–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L, Yong J, Dreyfuss G (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science 298: 1775–1779 [DOI] [PubMed] [Google Scholar]
- Pillai RS, Grimmler M, Meister G, Will CL, Lührmann R, Fischer U, Schümperli D (2003) Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev 17: 2321–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Will CL, Lührmann R, Schümperli D, Müller B (2001) Purified U7 snRNPs lack the Sm proteins D1 and D2 but contain Lsm10, a new 14 kDa Sm D1-like protein. EMBO J 20: 5470–5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raker VA, Plessel G, Lührmann R (1996) The snRNP core assembly pathway: identification of stable core protein heteromeric complexes and an snRNP subcore particle in vitro. EMBO J 15: 2256–2269 [PMC free article] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B (1999) Ageneric protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Salgado-Garrido J, Bragado-Nilsson E, Kandels-Lewis S, Séraphin B (1999) Sm and Sm-like proteins assemble in two related complexes of deep evolutionary origin. EMBO J 18: 3451–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski B, Laufer G, Gontcharova L, Günzl A (2004) The Trypanosoma brucei spliced leader RNA and rRNA gene promoters have interchangeable TbSNAP50-binding elements. Nucleic Acids Res 32: 700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski B, Nguyen TN, Günzl A (2005a) Characterization of a multisubunit transcription factor complex essential for spliced-leader RNA gene transcription in Trypanosoma brucei. Mol Cell Biol 25: 7303–7313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski B, Nguyen TN, Günzl A (2005b) Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell 4: 1942–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnare MN, Gray MW (1999) A candidate U1 small nuclear RNA for trypanosomatid protozoa. J Biol Chem 274: 23691–23694 [DOI] [PubMed] [Google Scholar]
- Schümperli D, Pillai RS (2004) The special Sm core structure of the U7 snRNP: far-reaching significance of a small nuclear ribonucleoprotein. Cell Mol Life Sci 61: 2560–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B (1995) Sm and Sm-like proteins belong to a large family: identification of proteins of the U6 as well as the U1, U2, U4 and U5 snRNPs. EMBO J 14: 2089–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlaub H, Raker VA, Kostka S, Lührmann R (2001) Sm protein-Sm site RNA interactions within the inner ring of the spliceosomal snRNP core structure. EMBO J 20: 187–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Lührmann R (2001) Spliceosomal UsnRNP biogenesis, structure and function. Curr Opin Cell Biol 13: 290–301 [DOI] [PubMed] [Google Scholar]
- Xu Y, Ben-Shlomo H, Michaeli S (1997) The U5 RNA of trypanosomes deviates from the canonical U5 RNA: the Leptomonas collosoma U5 RNA and its coding gene. Proc Natl Acad Sci USA 94: 8473–8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong J, Wan L, Dreyfuss G (2004) Why do cells need an assembly machine for RNA-protein complexes? Trends Cell Biol 14: 226–232 [DOI] [PubMed] [Google Scholar]
- Zaric B, Chami M, Remigy H, Engel A, Ballmer-Hofer K, Winkler FK, Kambach C (2005) Reconstitution of two recombinant LSm protein complexes reveals aspects of their architecture, assembly, and function. J Biol Chem 280: 16066–16075 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3