

Abstract
CapG is the only member of the gelsolin family unable to sever actin filaments. Changing amino acids 84–91 (severing domain) and 124–137 (WH2-containing segment) simultaneously to the sequences of gelsolin results in a mutant, CapG-sev, capable of severing actin filaments. The gain of severing function does not alter actin filament capping, but is accompanied by a higher affinity for monomeric actin, and the capacity to bind and sequester two actin monomers. Analysis of CapG-sev crystal structure suggests a more loosely folded inactive conformation than gelsolin, with a shorter S1–S2 latch. Calcium binding to S1 opens this latch and S1 becomes separated from a closely interfaced S2–S3 complex by an extended arm consisting of amino acids 118–137. Modeling with F-actin predicts that the length of this WH2-containing arm is critical for severing function, and the addition of a single amino acid (alanine or histidine) eliminates CapG-sev severing activity, confirming this prediction. We conclude that efficient severing utilizes two actin monomer-binding sites, and that the length of the WH2-containing segment is a critical functional determinant for severing.
Keywords: actin-regulatory proteins, CapG, gelsolin, severing, WH2
Introduction
Nonmuscle cells are able to change shape and migrate during embryonic development, wound healing and chemotaxis. Shape change also enables phagocytic cells to ingest particles and platelets to spread during coagulation. The actin filament cytoskeleton is primarily responsible for maintaining nonmuscle cell shape, and a myriad of actin-binding proteins regulate its architecture (Stossel, 1993; dos Remedios et al, 2003). One class of actin-binding proteins, the actin filament severing proteins are particularly efficient at quickly altering the length of actin filaments. In mammalian cells, gelsolin and villin primarily accomplish this task (Kwiatkowski, 1999). These proteins share sequence homology with Acanthamoeba severin and Dictyostelium fragmin, and this family of proteins has been called the gelsolin/villin family (Hartwig and Kwiatkowski, 1991). These proteins possess a series of homologous 12 kDa subunits. Gelsolin and villin contain 6 repeat subunits while severin and fragmin possess 3 repeats. In addition to severing, all members of this family also cap or block actin monomer exchange at the barbed-ends or fast-growing ends of actin filaments. All members of the gelsolin/villin family are calcium-sensitive, requiring micromolar Ca2+ to bind actin.
Severing proteins allow the cell to quickly disassemble orthogonal actin filament networks found in the peripheral cytoplasm, and transform the cytoplasm from a gel-like consistency to a less viscous solution. Gel–sol transformations are thought to play an important role in cell shape change (Stossel et al, 1999). Evidence supporting the importance of severing proteins is provided by the phenotypic changes found in gelsolin-null mice (Witke et al, 1995). Loss of gelsolin is associated with impaired neutrophil chemotaxis, defective platelet spreading and prolonged bleeding times, and the formation of thickened actin stress fibers in fibroblasts. Overexpression of gelsolin in NIH 3T3 fibroblasts has the opposite effects, enhancing cell motility (Cunningham et al, 1991).
The mechanism by which gelsolin and other severing proteins cut actin filaments has not been fully delineated; however, considerable progress has been made over the past decade by investigating the tertiary structure of specific domains of gelsolin (McGough et al, 2003). One region of interest is the amino-acid sequence linking the first two domains of gelsolin, G1 and G2. This region contains the consensus sequence FKHVXPN known as the WH2 domain that is found in a number of actin regulatory proteins, including thymosin-β4, NWASP, WASP, WIP and WAVE (Irobi et al, 2003). The functional importance of this conserved domain is not fully understood, and its role in actin filament severing has not been defined.
We have investigated severing by utilizing another member of the gelsolin/villin family, CapG (previously called macrophage capping protein, mbh1, and gCap39) (Southwick and DiNubile, 1986; Yu et al, 1990; Prendergast and Ziff, 1991; Dabiri et al, 1992). Like fragmin and severin, CapG possesses three repeat subunits. Unlike other members of the gelsolin/villin family, this protein caps the barbed ends of actin filaments, but does not sever them. CapG represents 1% of the total protein in the cytoplasm of macrophages and dendritic cells (Parikh et al, 2003). When CapG is knockedout, these cells fail to ruffle in response to cytokines, while loss of gelsolin has no effect on ruffling (Witke et al, 2001). Several unique characteristics of CapG, as compared to gelsolin, may explain its central role in ruffling. Ruffling is associated with rapid shifts in actin filament length, and the ability of CapG to reversibly cap the barbed-ends of actin filaments in response to fluctuations in Ca2+ could mediate these changes in length (Southwick and DiNubile, 1986; Young et al, 1994). Calcium-activated capping by gelsolin is not reversed by lowering calcium levels (Kurth et al, 1983). Secondly, as compared to gelsolin, CapG requires lower calcium concentrations for activation (Yu et al, 1990). Finally, the affinity of CapG for actin is considerably lower than gelsolin (KD nM to μM range as compared to pM for gelsolin), and this characteristic allows accurate assessment of changes in function utilizing standard actin assembly and binding assays (usual sensitivity range nM–μM).
We have previously shown that a few amino-acid mutations convert CapG from a strictly capping protein to a capping and severing protein (Southwick, 1995). We have now made additional changes to the WH2-containing segment of CapG to match the sequence of gelsolin, and created a protein with enhanced severing capacity. We have explored the functional effects of switching from a capping to capping and severing protein, and have solved the structure of this gain-of-function CapG mutant. Our modeling suggests significant differences in the inactive conformation of CapG as compared to gelsolin, as well as differences in calcium activation. Gain of severing function is associated with increased actin monomer binding affinity and the addition of a second actin monomer binding site. Structural analysis suggests that severing should be particularly sensitive to alterations in the length of the WH2 containing S1–S2 linking region, and we find that the addition of a single amino acid can eliminate severing activity.
Results
Generation of a gain-of-function mutant with enhanced severing activity
Amino-acid sequence comparisons revealed two regions in the wild-type (WT) CapG sequence (Figure 1A) that diverged from all the other gelsolin/villin family members. These differences served as the basis for the production of the gain-of-function mutations in our original studies (Southwick, 1995). We converted CapG cDNA codons in two small regions to those of gelsolin, from amino-acids 84LNTLLGE90 to 84LDDYLGG90 and 124AFHKTS129 to 124GFKHV128. Functional studies revealed a gain of function, that is, CapG was converted from a strictly capping protein to a severing and capping protein. Under the same experimental conditions, the mutant CapG required 50 times the concentration of gelsolin to achieve the same rate of cleavage (Southwick, 1995).
Figure 1.
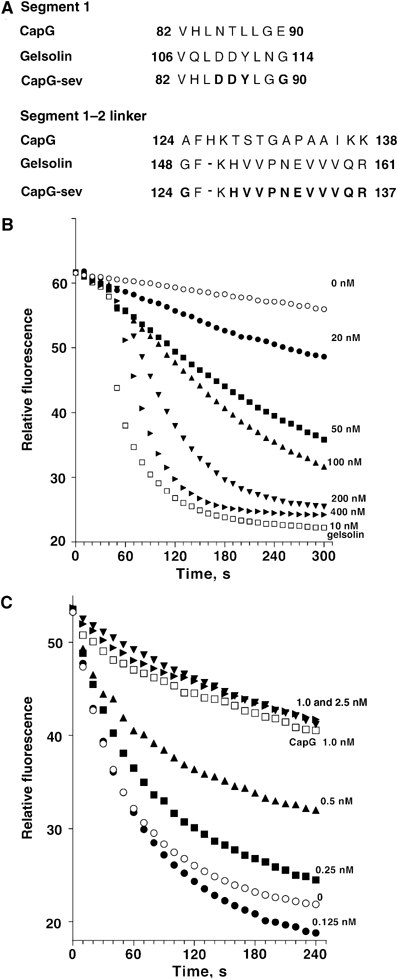
Generation of a gain-of-function CapG severing and capping mutant protein. (A) Sequence comparisons of CapG, gelsolin and CapG-sev. Bold letters indicate the amino acids changed from the native CapG sequence to the sequences of gelsolin. (B) Severing activity of the CapG-sev. A final concentration of 2 μM gelsolin-capped pyrene actin filaments (see Materials and methods) was diluted to 100 nM in S2 buffer containing various concentrations of the CapG-sev, and the rate of depolymerization measured by monitoring fluorescence intensity over time. Using a final concentration of 200 nM CapG-sev, dilution of gelsolin-capped filaments into buffer containing a final concentration 1 μM thymosin β4 demonstrated identical disassembly curves to actin filaments diluted into buffer alone. (C) Barbed filament capping of CapG-sev. A final concentration of 2 μM pyrene actin filaments was diluted to 100 nM in S2 buffer in the presence of increasing concentrations of the CapG-sev (filled symbols). The disassembly rates were monitored by fluorescence intensity. The final concentrations of CapG-sev are indicated above each curve. Open circles show the disassembly rate of the same concentration of pyrene actin in buffer and the open squares show the disassembly of the same concentration of pyrene actin diluted into a final concentration of 1 nM CapG.
Using a 1/4 length gelsolin truncate (aa 1–160), the minimal length known to support severing (Kwiatkowski et al, 1989), we next produced a reverse gelsolin mutant in an attempt to mimic CapG. Specifically, the sequences 108LDDYLGG114 and 148GFKHV152 in gelsolin were converted to the CapG sequences 108LNTLLGE114 and 148AFHKTS153. However, the new gelsolin mutant still severed effectively (Supplementary Figure S1A and B), significant severing being observed at similar concentrations to the unmodified truncate (final concentrations of 10–20 nM). This observation emphasized the limitations of structure–function analysis of gelsolin, and prompted us to further investigate the structural determinants of mutant CapG proteins.
To more closely recapitulate the severing activity of gelsolin, further mutations were made in the region of amino acids of 130–138 in CapG, from 124AFHKTSTGAPAAIKK138 to 124GFKHVVPNEVVVQR137, where additional sequences diverged from the other members of the gelsolin/villin family (Figure 1A). A final concentration of 50 nM of the new mutant resulted in comparable severing activity to 200 nM of our original gain-of-function mutant (Figure 1B), with no detectable effects on capping (the Kapp=0.5 nM, being identical to native CapG) (Figure 1C). This more potent severing mutant, designated as CapG-sev, was used for our subsequent functional and structural studies.
Comparisons of actin monomer binding affinity of CapG and CapG-sev
Next, we determined whether the conversion from an actin capping to an actin capping and severing protein was associated with a change in stoichiometry and/or binding affinity of CapG-sev for actin monomers. We first examined the effects of native CapG and CapG-sev on the fluorescence of NBD-conjugated actin. We, and others, have observed that when gelsolin or CapG binds actin monomers, the fluorescence intensity of NBD-conjugated actin (Bryan and Kurth, 1984) as well as pyrene-conjugated actin increases (Southwick and DiNubile, 1986). Using a concentration of NBD-actin below the critical concentration for actin filament assembly (100 nM final concentration), we observed a concentration-dependent rise in actin fluorescence that plateaued at approximately 100 nM CapG (Figure 2A). CapG-sev displayed a distinctly different binding curve. The initial rise in fluorescence reached a half-maximal intensity at a CapG-sev concentration of 15–20 nM and peak intensity at 40–50 nM. Addition of higher concentrations of the severing mutant to the NBD-actin solution resulted in a progressive decrease in fluorescence intensity that reached a nadir at 100–120 nM (Figure 2A). Fluorescence intensity did not change further following the addition of higher concentrations CapG-sev. These findings suggested the existence of two actin monomer-binding sites on CapG-sev. Assuming that binding was noncooperative and that the decrease in NBD-actin fluorescence above 50 nM CapG-sev was the consequence of dissociation of an NBD-actin monomer from the lower affinity binding site, we were able to generate a curve that closely approximated the actual values (compare filled circles to the solid line, Figure 2A). The KD values used for this approximation were 0.1 nM for the first monomer-binding site and 220 nM for the second binding site. Applying the same assumptions, we were also able to predict the concentration dependent fluorescence curve of CapG using a KD of 25 nM for the first binding site and a KD 1 μM for the second site. Under the conditions of our kinetic and ultracentrifugation experiments (see below), the second site on CapG would be expected to be essentially unbound. These experiments suggest that the CapG-sev high-affinity binding site has a 250 × higher affinity (KD 0.1 nM for CapG-sev versus 25 nM for CapG) and the low-affinity site >4 × higher affinity (KD 0.22 μM versus 1 μM) than CapG for monomeric actin. Similar experiments using pyrene actin yielded qualitatively and quantitatively comparable results (Figure 2B). Our modeling suggested that the differences in the fluorescence intensity profiles of NBD-actin and pyrene actin were the consequence of differences in the fluorescence changes induced by binding, but did not reflect significant differences in their binding affinity or stoichiometry.
Figure 2.
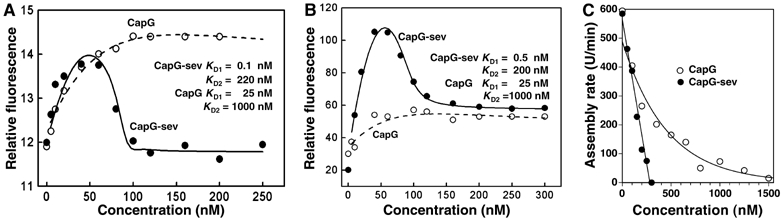
CapG and CapG-sev binding to actin monomers. (A) Binding to NBD-conjugated monomeric actin. NBD-actin monomers (final concentration 100 nM) were mixed with increasing concentrations of CapG and CapG-sev in modified S2 buffer (see Materials and methods) and the steady-state fluorescence measured. The lines represent the calculated curves for CapG (dashed line) and CapG-sev (solid line). The fitting parameters assumed two non-cooperative binding sites for both CapG and CapG-sev. The dashed line for CapG was fitted using a KD of 25 nM for the first binding site and 1 μM for the second binding site. The solid line for CapG-sev was fitted using a KD of 0.1 nM for the first site and a KD of 220 nM for second site. (B) Binding to pyrene-conjugated monomeric actin. The identical conditions described in (A) were used, except the studies were performed with pyrene actin monomers. The solid line connecting the data points for CapG-sev and the dashed line for CapG were fitted using the parameters shown in the figure, and are almost identical to NBD-actin. (C) Pointed end actin filament assembly in the presence of increasing concentrations of CapG and CapG-sev. Increasing concentrations of the two proteins were added to a 1.5 μM pyrene actin solution. Actin filament assembly was then stimulated by the addition of a final concentration of 40 nM gelsolin capped filamentous actin (see Materials and methods). The rate of rise of fluorescence intensity was monitored over 15 min and these rates plotted versus the final concentrations of CapG and CapG-sev. Note the steep reduction in pointed-end growth rate in the presence of CapG-sev.
To examine actin monomer binding in more detail, gelsolin–actin filament seeds (40 nM actin containing 2 nM gelsolin, 1:20 gelsolin/actin molar ratio) were added to a final concentration of monomeric actin of 1.5 μM, and the assembly rates determined in the presence of increasing concentrations of CapG or CapG-sev. As shown in Figure 2C, the rate of actin assembly demonstrated a linear decrease as the concentration of CapG-sev increased, reflecting the ability of the protein to sequester actin and prevent actin monomers from adding to the free pointed ends. Complete inhibition occurred with the addition of 300–400 nM CapG-sev, indicating sequestration of all actin monomers above the critical concentration of the pointed end (the critical concentration of the pointed end was determined to be 800 nM in these experiments, leaving 700 nM of monomers that could be added to the filament). Thus, one CapG-sev molecule was able to sequester two actin monomers. The linear relationship between CapG-sev concentration and the reduction in actin filament assembly rate was consistent with a high-affinity interaction (KD in nanomolar range) between monomeric actin and CapG-sev. CapG also progressively slowed the actin filament assembly rate; however, the binding curve was consistent with a 1 to 1 stoichiometry for CapG binding to monomeric actin with an estimated Kapp of 0.25 μM, similar to previously reported values (Young et al, 1994).
Actin monomer binding was also analyzed by analytical ultracentrifugation. Sedimentation equilibrium runs of CapG or CapG-sev alone, at a concentration of 12 μM were consistent with a monomeric state, the partial specific volume being 0.73 for both proteins (data not shown). Samples containing a ratio of 2:1, actin to CapG or CapG-sev caused a shift in the gradient of pyrene actin to a steeper slope, implying complexation between actin and CapG or CapG-sev (Supplementary Figure S2A). The sedimentation equilibrium data for WT CapG could be fitted assuming high-affinity binding of a single actin monomer (KD⩽100 nM), reflecting a stable mixture of 50% actin monomer and 50% actin bound to CapG. The gradient slope for the CapG-sev data was steeper than that for CapG, and could only be fitted reasonably under the assumption of two actin-binding sites, the first being a high-affinity site similar to native CapG, and the second a lower-affinity site with a KD of 0.4 μM. This consideration does not rule out other possible and perhaps more complicated binding schemes.
The sedimentation coefficient for the CapG-sev-2 actin complex was consistent with a spherical structure (Supplementary Figure S2B). The expected ratio of the sedimentation coefficients for CapG-sev complexed with two actin monomers versus monomeric actin (∼124 versus 42 kDa) would be 2.08 assuming a spherical geometry, similar to the experimentally determined ratio 2.03 (s20,w 6.9 S for the CapG-sev-2 actin complex and 3.4 S for monomeric actin). Assuming uniform hydration of 0.31, the results for actin alone and the CapG-sev-2 actin complex can both be modeled in the shape of oblate ellipsoids with axial ratios of 3.1±0.1. In contrast, the sedimentation coefficient for actin-CapG (s20,w of 4.8 S) was consistent with a relatively elongated complex that formed an oblate ellipsoid with an axial ratio of 5.0±0.2.
Structural analysis of CapG-sev crystals
Although unsuccessful in crystallizing native CapG despite multiple attempts using varying conditions, we successfully produced diffraction quality crystals from CapG-sev. Supplementary Table II describes the outcome of data collection, phasing, and refinement. The CapG-sev structure reveals 3 homologous domains that exhibit the same overall topology present in the related gelsolin molecule, consisting of a central five-stranded β-sheet sandwiched between a long α-helix running approximately parallel to the strands and a short α-helix running almost perpendicular to the strands. In the crystalline state, the CapG-sev molecule is remarkably extended with domains 2 and 3 packing to form a compact globular unit, while domain 1 projects away from domains 2 and 3. In this extended organization, domains 1 and 2 are connected by segment 118KGGVESGFKHVVPNEVVVQR137, which includes the second set of mutations and spans 36 Å between the two domains (Figure 3A). The first set of gain of function mutations in S1, 84LDDYLGG90, maps to the long α-helix that contributes to the putative binding interface with actin. Two cation-binding sites were identified by soaking crystals in EuCl3 as described in the Materials and methods. One site is located in segment 1 at Gly45 and Asp46 and the second in segment 3 consisting of Asp279, Asp280, and Glu304 (Figure 3A, inset).
Figure 3.
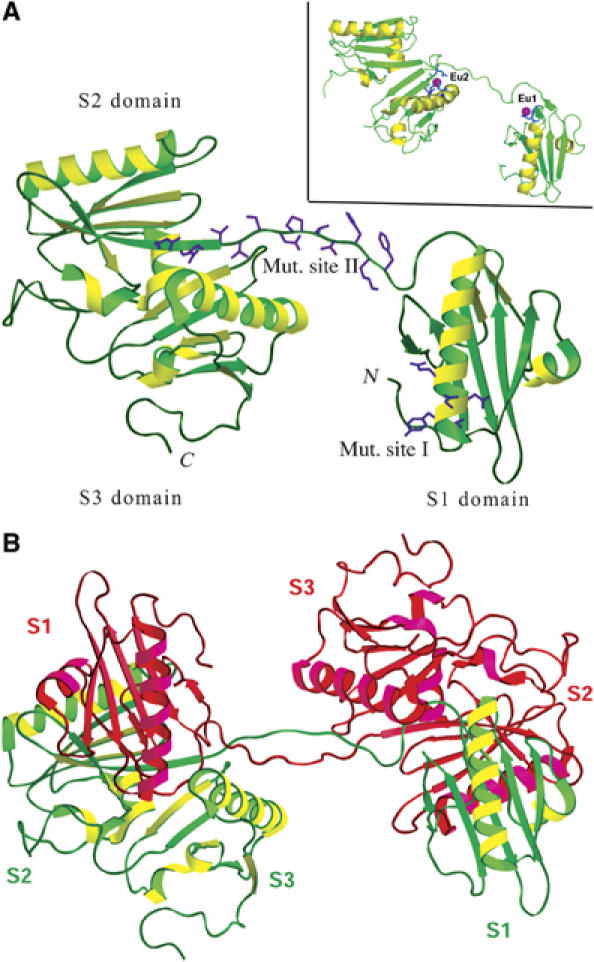
Structures of CapG-sev. (A) Structure of activated CapG-sev, showing the location of two Eu2+. Domain 1 extends away from domains 2 and 3 and is connected to them by the extended S1–S2 WH2 linker domain (estimated length 36 Å). This is the region where CapG was converted to the gelsolin WH2 sequence (labeled mut. site II). The other mutation is in S1 (labeled mut. site I) and is located at one end of the long α-helix. This region in gelsolin is responsible for dissociating the second domain of the actin monomer from it adjacent monomer in the actin filament. Inset: The same structure showing the locations of the two Eu2+ sites on S1 and S3. These sites are likely to represent the two Ca2+ binding sites (PDB 1jhw). The linker between S2 and S3 is not shown, due to the fact that this linker is not ordered in the EU crystal structure. (B) Structure of the CapG-sev dimer. S1 of each molecule interacts with S2 of the other monomer creating a symmetry-related dimer. The positioning of S1 in relation to S2 and S3 of the other monomer is likely to reflect the conformation of the CapG-sev and CapG in the inactive state.
As shown in Figure 3B, CapG-sev formed an unexpected domain-swapped dimer, with domain 1 interacting with domains 2 and 3 of the symmetry-related molecule forming a compact, but elongated overall structure. We propose that the compact organization associated with domain 1 and the symmetry-related domains 2 and 3 represent the inactive conformation of CapG-sev and by extension WT CapG (Figure 3B and Supplementary Figure S3B). The interface between domain 1 and the symmetry-related domain 2 in this proposed structure is very similar to those observed in the Ca2+-free gelsolin monomer (Supplementary Figure S3A–C). However, the domain 2–domain 3 interface is novel (Supplementary Figure S3B and Supplementary Table III). In the inactive state, the first three segments of gelsolin display an organization in which G1 makes significant contacts with both G2 (1765 Å2 of buried surface area) and G3 (2160 Å2 of buried surface area) (Supplementary Figure S3A). In contrast to gelsolin G1–G3, the proposed inactive conformation of CapG-sev supports a significant interface between S1 and S2 (2144 Å2 of buried surface area), but only a modest interface between S1 and S3 (1189 Å2 of buried surface area) (Supplementary Figure S3B and Supplementary Table III). The interface between S1 and S2 is formed by a tight antiparallel association of two β-sheets in S2 (Supplementary Figure S3B and Supplementary Table III). This interaction would be expected to cover up the S1 actin monomer binding site as well as the LDDYL severing loop. The interaction of S1 and S3 is weak and consists of interactions between the side chains of two amino acids (Supplementary Table III).
Upon activation, the CapG molecule has been shown to expose new sites for proteolysis consistent with unfolding (Young et al, 1994). Our structure-based model suggests that activation of CapG-sev causes S1 to dissociate from S2, extending the S1–S2 linker to a length of approximately 36 Å (Figures 3A and 4B). Similar to the activated gelsolin G1–G3 domain arrangement, S2 and S3 in CapG-sev form an interface. However, the sites of interaction are distinctly different from gelsolin and consist of interfaces between two antiparallel β-sheets (S2: Glu132–Gln136; S3: Gln259–Val265) as well as a loop in S2 (Glu151–Trp156) that interacts with the same S3 β-sheet (Supplementary Table III). Furthermore, the CapG-sev S2:S3 interaction (1252 Å2 of buried surface area) is tighter than the gelsolin G2:G3 interaction (644 Å2 of buried surface area).
Figure 4.
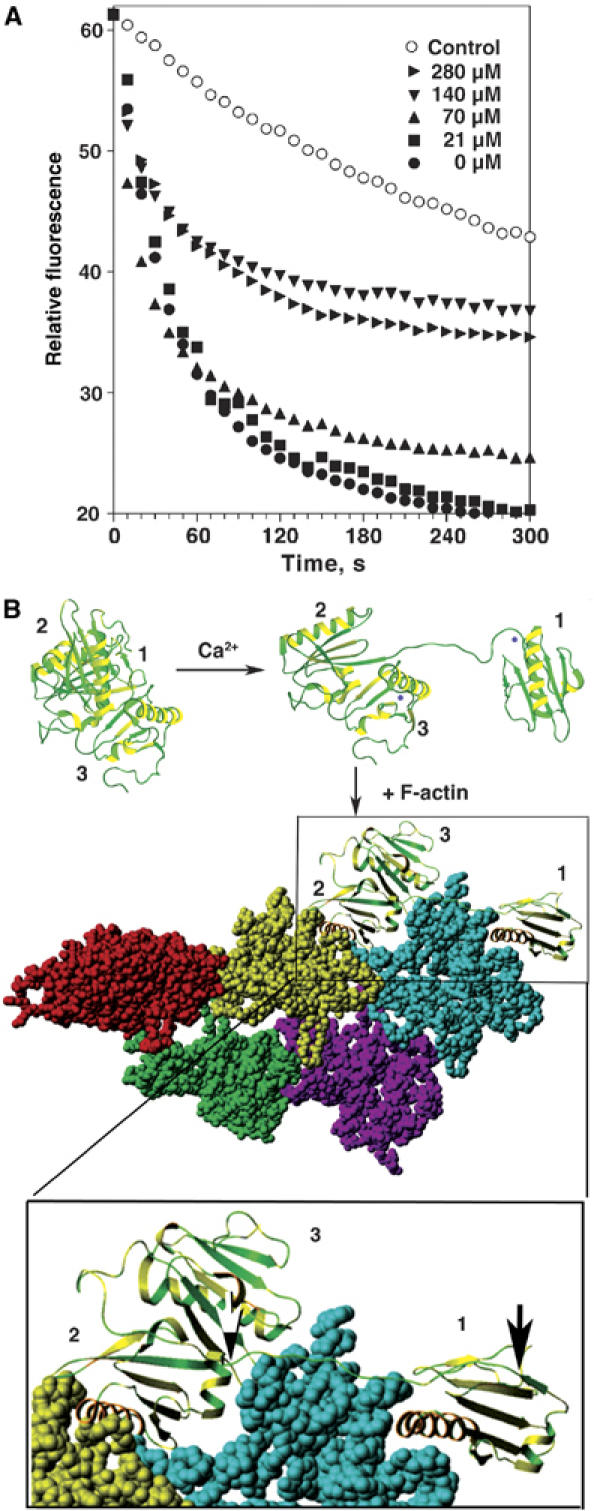
The role of S1–S2 WH2 linker domain in CapG-sev actin filament severing. (A) Effects of the synthetic peptide 118-KGGVESGFKHVVPNEVVVQR-137 on actin filament severing by CapG-sev. The exact conditions described in Figure 1B were used to assess the actin filament severing of CapG-sev in the presence of increasing concentrations of the synthetic peptide representing the WH2 linking arm between domains 1 and 2 of CapG-sev. (B) Model for CapG-sev activation and actin filament severing and capping. All three models are based on the CapG-sev crystal structure. In the inactive state, CapG-sev is compact in conformation, and the actin binding and severing sites of S1 would be expected to be covered up by S2. On the addition of μM Ca2+ S1 dissociates from S2 and extends away from S2 and S3. The lower drawing depicts a model for CapG-sev interaction with F-actin. Starting with the Holmes model of the actin filament (Holmes et al, 1990), S1 was docked to the barbed end, according to the features of the gelsolin S1/G-actin crystal structure (McLaughlin et al, 1993). Following the approach of Puius et al (2000), S2 was docked to the subsequent monomer in the filament in an S1-like binding mode. In order to achieve these two binding interfaces, rotation about the S1–S2 linker was required. This model shows the CapG-sev S1–S2 linking segment is fully extended across the longitudinal axis of one actin monomer, linking the actin-bound S2 domain to the S1 domain. This region is enlarged (lower box). The black–white arrow marks the beginning and the black arrow the end of the S1–S2 linker. The extended arm would be expected to allow exact insertion of S1 at the barbed end of the next actin monomer causing a steric clash that dissociates this monomer from adjacent monomer to the right (not shown) resulting in severing of the filament. This arm would also allow CapG-sev to continue to sterically hinder addition of new actin monomers at the barbed end. Our models for activation and binding to F-actin represent minimal models, and there are no data relevant to the placement of S3 when CapG-sev is bound to the actin filament.
Assessment of the functional importance of the CapG-sev S1–S2, WH2 containing linker for actin filament severing
The S1–S2 linker of CapG-sev, as well as the G1–G2 linker of gelsolin, share a WH2 domain. To assess the functional importance of this region for actin filament severing, we synthesized the polypeptide representing the full length of S1–S2 linker arm in the CapG-sev containing the WH2 sequence (118KGGVESGFKHVVPNEVVVQR137) and determined its effects on CapG-sev filament severing. We consistently observed that addition of final peptide concentrations of 140–200 μM significantly inhibited the severing activity of CapG-sev (Figure 4A).
We have modeled the proposed active CapG-sev structure with F-actin (Figure 4B) as previously described for gelsolin (Puius et al, 2000; Irobi et al, 2003; Burtnick et al, 2004). The S1–S2 linker of CapG-sev is stretched tightly along the filament, and appears to be exactly positioning the LDDYL loop between two actin monomers in the filament to allow efficient severing (McLaughlin et al, 1993) (see the expanded image, bottom of Figure 4B). This structural arrangement suggested to us that a lengthening of this segment might lead to inaccurate positioning of the LDDYL loop, and interfere with severing. Therefore, we first chose to add a single amino acid to the S1–S2 linker. Addition of either a histidine (S123_G124insH) or an alanine (S123_G124insA) to the S1–S2 linker region of CapG-sev profoundly impaired severing (Figure 5A). A final concentration of 0.2 μM of CapG-sev markedly accelerated the disassembly rate of gelsolin-capped actin filaments, indicating severing activity (solid triangles), whereas 20-fold higher concentrations of the either CapG-sev-S123_G124insH (solid squares) or -S123_G124insA (solid circles) failed to sever. Loss of severing was associated with minimal changes in barbed end capping activity (Supplementary Figure S4A and B) and no detectable alterations in actin monomer binding affinity, as assessed by the pyrenyl actin subcritical concentration assay (Table I), or stoichiometry, as assessed by assembly rates of gelsolin–actin nuclei (Supplementary Figure S4C). We chose to insert the H and A at position 124 so as not to interfere with the juxtaposition of a hydrophobic amino acid (F125) and charged amino acid (K126) within the WH2 consensus sequence. We subsequently inserted an alanine between these two amino acids (CapG-sev-F125_K126 insA) and this mutation also reduced, but did not eliminate severing activity; 1 μM of this mutant demonstrated equivalent severing activity to 30–50 nM CapG-sev (Supplementary Figure S5).
Figure 5.
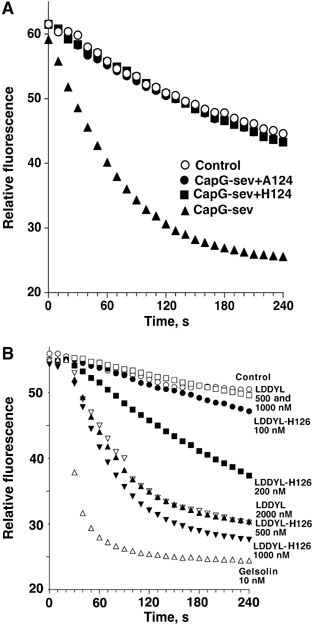
Effects of single amino-acid mutations in S1–S2 linker on actin filament severing. (A) Severing activity of CapG-sev before and after addition of A or H at position 124. The assay described in Figure 1B was performed to compare the CapG-sev severing to CapG-sev mutants containing a single amino acid addition at position 124 (CapG-sev+A124 and CapG-sev+H124). Note the absence of any severing activity after addition of a final concentration of 4 μM of either protein containing an amino-acid insertion, while under the same conditions addition of a final concentration of 200 nM CapG-sev markedly accelerated depolymerization. Insertion of alanine at position 126 (CapG-sev+A126) also caused a marked reduction in severing activity (Supplementary Figure S5). (B) Severing activity of the LDDYL CapG mutant before and after the deletion of histidine at position 126. The same assay described in Figure 1B was performed to compare the severing of these two mutant proteins. Note that a final concentration of 1000 nM LDDYL CapG mutant (open squares) failed to accelerate actin filament assembly as previously observed (Southwick, 1995). Following the deletion of H126 (LDDYL-H126 mutant), significant severing was observed at a concentration of 200–300 nM (closed circles and squares), and a final concentration of 500 nM of LDDYL-H126 (closed triangles) resulted in severing that was equivalent to 2000 nM of the LDDYL mutant (open inverted triangles).
Table 1.
Actin severing, capping and monomer binding of CapG and CapG mutant proteins
| Protein | Concentration causing significant severing (nM) | Capping (KI nM) | Actin monomer binding (KD) | CapG:monomer binding stoichiometrya | |||||
|---|---|---|---|---|---|---|---|---|---|
| CapG | No severing⩾4 μM | 0.5 nM | 25 nMb | 1:1 | |||||
| 1 μM | |||||||||
| CapG-sev | 30–50 nM | 0.5 nM | 0.1 nMb | 1:2 | |||||
| 220 nM | |||||||||
| CapG-sev+H124 | No severing⩾4 μM | 1–2 nM | 0.1 nMb | 1:2 | |||||
| 220 nM | |||||||||
| CapG-sev+A124 | No severing⩾4 μM | 1–2 nM | 0.1 nMb | 1:2 | |||||
| 220 nM | |||||||||
| Stoichiometry was determined by actual experimental data (Figure 2C and Supplementary Figure S4C). | Estimates of binding for sites 1 and 2 based on modeling of fluorescence intensity values after addition of CapG proteins to a subcritical concentration of NBD or pyrene conjugated monomeric actin (Figure 2A and B and data not shown). | ||||||||
Next, realizing that native CapG actually has an extra amino acid in this same region, His-126 (Figure 1A), we predicted that removal of this single amino acid should enhance severing. To test this possibility, we utilized a CapG mutant containing the 84LDDYLGG90 mutations in the S1 region of CapG-sev, and the WT CapG sequence in S1–S2 linker region, called the LDDYL mutant. Previous experiments have revealed that this S1 mutation is required for a gain of severing function; however, this mutation alone results in only weak severing activity (Southwick, 1995). As predicted, deletion of H-126 (called LDDYL-H126 mutant) enhanced severing, a final concentration of 500 nM demonstrating similar severing activity to a 2 μM final concentration of the original LDDYL mutant (Figure 5B).
Discussion
The present studies represent a detailed structural and functional analysis of CapG and gain-of-function mutants of CapG. These investigations provide new insights into how gelsolin family members are activated by calcium to sever and cap actin filaments. These advances were made possible because CapG, as compared to gelsolin, has a lower affinity for actin that is within the range of actin kinetic assays. This characteristic has allowed the detection of functional changes that could not be measured using gelsolin (Supplementary Figure S1). As a consequence, many recent investigators of gelsolin have relied on structural studies alone to infer structure–function relationships. Furthermore, as compared to CapG, gelsolin is a far more complex molecule containing 6, rather than 3 repeat subunits. To simplify their analysis many investigators have truncated gelsolin, creating artificial molecules that may poorly approximate the behavior of the full-length native molecule. For example, despite the existence of three calcium-binding sites, severing by gelsolin G1–G3 does not require calcium (Bryan and Hwo, 1986). In our studies, we have utilized full-length CapG mutants, and made discrete amino-acid changes that would not be expected to significantly alter the overall conformation of the molecule. In order to maximize severing activity, one relatively long stretch of 15 amino acids known to link S1–S2 was converted from the sequence of CapG to the sequence of gelsolin.
Based on comparisons of CapG to the other members of the gelsolin family that all sever actin filaments, we were able to generate a gain-of-function mutation that required 5–10 times the concentration of gelsolin to achieve the same rate of severing (Figure 1B). Functional comparisons of our mutant severing protein, called CapG-sev, to native CapG that lacks severing activity have allowed us to measure the functional changes associated with actin filament severing. First, we have found the acquisition of severing activity had no detectable effect on actin filament capping (Figure 1C). This observation suggests that the structural requirements for efficient capping are less stringent than for actin filament severing. Secondly, we found that a gain in severing was accompanied by an increase in actin monomer binding affinity (estimated KD 1–2 orders of magnitude lower than native CapG) and a doubling of actin monomer binding capacity (Figure 2A–C and Supplementary Figure S2). Our experiments suggest that gelsolin family members, in particular CapG, may possess additional actin monomer binding sites that are usually masked. Our findings also raise the possibility that efficient severing may require two actin monomer bindings sites. We propose that one site binds the actin monomer directly above the point of severing, altering the actin monomer conformation and weakening its interaction with the adjacent monomer. This weakening of monomer–monomer interaction within the filament would allow the second actin monomer-binding domain, containing the LDDYL loop, to more easily dissociate these two monomers within the filament. The changes in fluorescence observed when CapG-sev binds NBD and pyrene labeled-actin are consistent with this mechanism.
Our ability to solve the structure of CapG-sev has allowed us to make specific functional predictions concerning the calcium activation of CapG and guided further structure–function studies of actin filament severing. Our crystallographic studies resulted in the unexpected formation a domain-swapped dimer (Figure 3B) that has allowed us to predict the inactive conformation of CapG-sev and by extension WT CapG. The proposed structure consists of a tight interaction between S1 and S2 with S3 being loosely associated (Figure 6A and Supplementary Figure S3B). This arrangement differs significantly from the folded conformation of gelsolin G1–G3 in which a tight interface is formed between G1 and G3, as well as G1 and G2 (Burtnick et al, 1997, 2004; Figure 6A; Supplementary Figure S3A). The folded structure of the first three segments of gelsolin has been termed the G1–G3 latch and differs from CapG, which in our model forms a shorter S1–S2 latch (Figure 6A).
Figure 6.
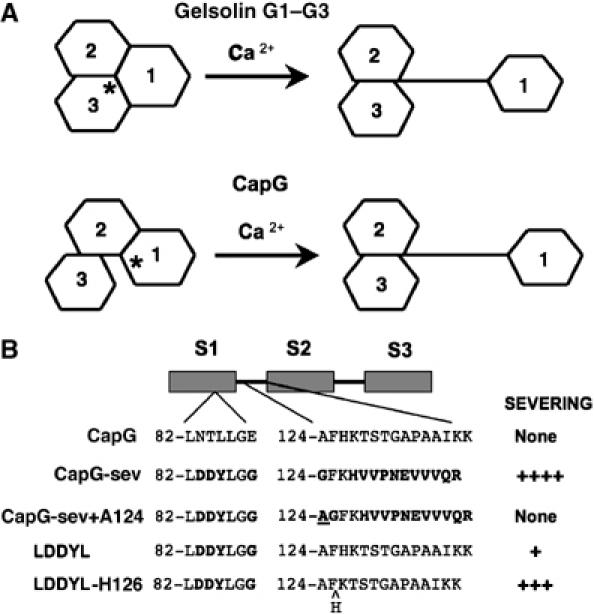
Models for calcium activation of gelsolin and CapG, and a summary of CapG severing mutants. (A) Cartoon comparing the proposed models for gelsolin G1–G3 and CapG Ca2+ activation. In the inactive conformation, the area of interface between S1 and S3 in CapG is smaller, as compared to the gelsolin; while the S1–S2 and G1–G2 areas of interface are similar. In functional terms, CapG has an S1–S2 latch while gelsolin has a G1–G3 latch. The asterisk denotes the site where Ca2+ first binds to initiate unfolding (S1 for CapG and G3 for gelsolin). Note that in the active conformation the arm linking S1 to S2 in CapG is longer than for gelsolin, and this greater length impairs actin filament severing. The gelsolin model is based on Burtnick et al (2004). (B) Schematic depictions of the recombinant mutant CapG proteins summarizing their relative actin filament severing activity. Mutations were only made in S1 and in the S1–S2 linker. The specific amino acids modified are depicted for each mutant. Bold letters indicate the amino acids that were changed. The alanine added to the S1–S2 linker of CapG-sev is underlined, and the histidine deleted in the LDDYL mutant S1–S2 linker is indicated by a ^. Severing activity was roughly quantified as none or by plus signs. None=no severing up to a final concentration of ⩾4 μM, +=significant severing at 1–2 μM, +++=significant severing at 100–300 nM and ++++=significant severing at 30–50 nM.
Both CapG and gelsolin are activated by calcium. Modeling suggests that in gelsolin G1–G3 calcium binding to G3 initiates unfolding (Burtnick et al, 2004). In the case of CapG, we predict that binding to the S1 calcium-binding site initiates activation. This site is relatively open as compared to its S3 calcium-binding site, and would be expected to alter the conformation of S1, causing S1 to dissociate from S2 and extend away from S2, uncovering the S1 actin-binding site. As S1 moves away from S2, the S1–S2 linker extends, forming a linear arm (Figures 3A, 4B, and 6A). Because CapG-sev and by extension CapG has a loose association between S1 and S3, the proposed resting conformation for CapG-sev is predicted to be less thermodynamically favorable than the resting state of gelsolin, and would, therefore, be expected to more readily open in response to calcium binding. The extended disposition of domain 1, which leads to the formation of the dimer in the crystalline state, demonstrates that CapG-sev has a natural propensity to form an extended conformation. This same tendency was also observed for native CapG in our analytical ultracentrifugation analysis (Supplementary Figure S2B), and is likely to explain why in the absence of calcium, but in the presence of the high concentrations of NH4-formate, crystallized CapG-sev maintained an open conformation. As compared to both the G1 and G3 calcium-binding sites of gelsolin, the S1 binding site of the inactive conformation of CapG is more readily accessible. This difference may explain why CapG requires lower concentrations of Ca2+ to initiate activation (Yu et al, 1990). The smaller S1–S2 latch also renders activation of CapG more thermodynamically favorable. These structural arrangements may explain why CapG is able to quickly bind and dissociate from actin filaments in response to changes in calcium concentration during cell membrane ruffling (Southwick and DiNubile, 1986; Witke et al, 2001).
Finally, our modeling of CapG-sev with F-actin based on previous structural analysis of gelsolin (Figure 4B) indicates that the WH2 containing S1–S2 linker region is tightly stretched across the longitudinal axis of one actin monomer in the filament. We predicted that the length of this segment would be critical for proper positioning of the LDDYLGG loop for severing. To test this prediction we performed two sets of experiments (Figure 6B). First we added a single amino acid to the S1–S2 linker region of our CapG-sev mutant that contained the identical G1–G2 linker sequence of gelsolin. Lengthening of the linker by one amino acid markedly impaired severing (Figures 5A and 6B). We also observed that CapG has an extra amino acid in its S1–S2 linker region. Therefore, we also performed the reverse experiment utilizing a mutant CapG containing the S1 mutations of CapG-sev known to be critical for severing, and the WT CapG S1–S2 linker sequence (LDDYL mutant). The reduction of the CapG linker region sequence by one amino acid enhanced actin filament severing (Figures 5B and 6B). These observations indicate the exact length of the WH2-containing region linking S1 to S2 is critical for actin filament severing. However, these changes had no significant effect on actin filament capping or actin monomer binding suggesting that length of this region is not as critical for these actin-regulatory functions.
In conclusion, crystallographic analysis combined with functional assays has allowed us to gain greater insight into the structural and functional determinants of calcium-sensitive actin filament capping and severing. Our model suggests that CapG has a different inactive conformation as compared to gelsolin, consisting of a shorter S1–S2 latch rather than the longer G1–G3 latch. Furthermore, our structural studies reveal that the S1 calcium-binding site is more exposed in CapG as compared to the gelsolin calcium-binding sites. The rapid reversibility of CapG-actin binding in response to fluctuations in calcium concentration and the activation of CapG by lower calcium concentrations than gelsolin may be attributed to the shorter S1–S2 latch and a more exposed calcium-binding site. Our functional studies of CapG and CapG-sev demonstrate that severing is accompanied by an increase in the affinity for monomeric actin, and the gain of a second actin monomer-binding site. Finally, our understanding of the active CapG-sev conformation has led us to experimentally prove that the addition of a single amino-acid residue to the WH2 containing S1–S2 linking arm markedly impairs severing, but not capping activity. These findings highlight the functional and structural differences between CapG and gelsolin, and emphasize the importance of S1–S2 linker arm length for efficient severing.
Materials and methods
Protein preparations
Actin was prepared from rabbit muscle acetone powder as described previously (Spudich and Watt, 1971) and gel-filtered through a HiLoad 16/60 Superdex-200 column (Pharmacia Biotech, Piscataway, NJ). Purified actin was maintained in G-buffer (5 mM Tris–HCl, 0.1 mM calcium chloride, 0.2 mM dithiothreitol, 0.2 mM ATP, 0.02% sodium azide, pH 7.9) at 4°C. Pyrene actin and NBD-actin (7-chloro-4-nitrobenzeno-2-oxa-1,3-diazole actin) were made using standard protocols (Detmers et al, 1981; Kouyama and Mihashi, 1981) and gel-filtered.
The general procedures for producing CapG mutant were previously described in detail (Southwick, 1995). These recombinant proteins were purified according to Dabiri et al (1992) with the following modifications: bacterial pellets were diluted 1:1 with S1 buffer (10 mM imidazole HCl, 0.25 mM EGTA, 0.1 mM MgCl2, 1 mM DTT) and loaded on a fast protein liquid chromatography DEAE ion exchange column followed by a PD-10 column to desalt and separate away contaminating nucleotides. The PD-10 column step was then repeated to decontaminate nucleotides completely, and followed by gel filtration using a Superdex S-200 column (Pharmacia).
Crystallization and X-ray diffraction
The peak fractions of the gel filtration were concentrated to 8–10 mg/ml, and dialyzed against S1 buffer. Diffraction quality crystals of the CapG-sev were grown by hanging drop vapor diffusion at 20°C by mixing equal volumes of protein solution and reservoir solution containing 3.0–4.0 M NH4-formate. Diffraction quality crystals of the WT CapG could not be obtained.
All diffraction data were collected at 100 K using an ADSC Q-4 CCD detector on the X9B beam line at NSLS (BNL), using 10% glycerol in 5 M Na-formate as a cryo-protector. Data were indexed, integrated and scaled using the programs DENZO and SCALEPACK (Borek et al, 2003). The structure was solved by multiple isomorphous replacement using four derivatives: gold cyanide (Au), ethyl mercury phosphate, lead chloride, and di-μ-iodobisethylenediamine-diplatinum. Initial phases to 4.3 Å resolution were calculated with SOLVE (Terwilliger, 2004), overall figure-of-merit 0.6 and improved with DM (Cowtan and Main, 1998), implemented as part of the CCP4 suite (1994). Although the electron density was of reasonable quality, the limited resolution precluded manual chain tracing. The electron density for the three domains of CapG-sev was individually fitted using our web-based exhaustive phased translation function (http://russel.bioc.aecom.yu.edu/server/NYSGRC.html). The search models used for these calculations were the first domain of human gelsolin (PDB entry 1EQY, sequence identity 26%) and the second and third domains of horse plasma gelsolin (PDB entry 1D0N, sequence identity 54%). The model was corrected with O (Jones et al, 1991) and refined with CNS/X-PLOR (Brunger et al, 1998) against 2.5 Å resolution native data. Buried surface areas generated by intramolecular contacts were calculated using the AREAIMOL program from CCP4. To localize the putative Ca2+-binding sites, CapG-sev crystals were transferred into 4.0 M Na-formate and subsequently soaked in a solution of 10 mM EuCl3/4.0 M Na-formate for 15 min prior to freezing (soaking in CaCl2-containing solutions completely destroyed the crystals). Atomic coordinates of CapG-sev (accession code 1J72) and the CapG-sev–Eu3+ complex (accession code 1JHW) have been deposited in the PDB.
Actin kinetic and binding assays
The actin filament severing assay was performed as previously described (Southwick, 1995), S2 buffer (10 mM Imidizole HCl, 0.1 mM. CaCl2, 1 mM MgCl2, 0.25 mM ATP, 0.1 mM KCl and 1 mM DTT, pH 7.5) being used for these assays. Actin monomer fluorescence assays were performed by adding increasing concentrations of CapG or CapG mutants to a final concentration of 100 nM pyrene actin or NBD-conjugated actin in modified polymerization buffer (contained 0.4 mM MgCl2, rather than 1 mM MgCl2, to assure a relatively high actin critical concentration). After the addition of CapG or mutant CapG proteins, a fluorescence intensity increase was observed within 5 s and remained stable for over 30 min (Southwick and DiNubile, 1986). To assess actin monomer sequestration, elongation from gelsolin–actin seeds (final concentrations 2 nM gelsolin, 40 nM actin) in a final concentration of 1.5 μM actin (50% pyrene labeled) was measured in the presence of increasing concentrations of CapG and mutant CapG molecules as previously described (Young et al, 1994).
Analytical ultracentrifugation experiments
The sedimentation coefficients and the solution molecular mass for CapG and CapG-sev alone in the presence or absence of Ca2+ were determined by sedimentation velocity and equilibrium, respectively, using a Beckman Optima XL-A analytical ultracentrifuge (Bubb et al, 1994). Final concentrations of 12 μM CapG and CapG-sev in S1 buffer (for CaCl2-containing solutions, EGTA was omitted) were used and data were analyzed using DCDT+ and Svedberg software. To examine CapG and CapG-sev binding to actin, pyrene-conjugated actin was used for analytical centrifugation as previously described (Bubb et al, 1991). All samples with actin included pyrene actin (78% labeled) in 0.1 mM ATP, 0.5 mM DTT, 0.1 mM CaCl2 and 10 mM imidazole, pH 7.5. For samples containing the CapG-sev, the absorbance at any radius was assumed to be the sum of free pyrene actin, and CapG-sev bound to either one or two pyrene actin subunits. The interaction between the first actin subunit and CapG or CapG-sev was assumed to go to completion (Kd≈0), and the KD for the interaction of CapG-sev with a second subunit was a fitted parameter. Molecular weights were based on sequence and the only other fitting parameters were the concentration of the free species at the base of the centrifuge cell and a baseline error term, which was negligible in all cases. Partial specific volume was assumed to be 0.73 for all species.
Supplementary Material
Supplementary Figure Legends
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
supplementary Tables
Acknowledgments
We would like to thank Dr Vladimir Malashkevich his assistance in constructing the CapG-sev and gelsolin models. This work was funded by NIH Grant RO1AI-23262 (to FSS); the Medical Research Service of the Department of Veterans Affairs and the NSF Grant NSF-0316015 (to MRB), NIH Grant 5K25AR048918 (to EGY).
References
- Borek D, Minor W, Otwinowski Z (2003) Measurement errors and their consequences in protein crystallography. Acta Crystallogr D 59: 2031–2038 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54 (Part 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Bryan J, Hwo S (1986) Definition of an N-terminal actin-binding domain and a C-terminal Ca2+ regulatory domain in human brevin. J Cell Biol 102: 1439–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan J, Kurth MC (1984) Actin–gelsolin interactions. Evidence for two actin-binding sites. J Biol Chem 259: 7480–7487 [PubMed] [Google Scholar]
- Bubb MR, Knutson JR, Porter DK, Korn ED (1994) Actobindin induces the accumulation of actin dimers that neither nucleate polymerization nor self-associate. J Biol Chem 269: 25592–25597 [PubMed] [Google Scholar]
- Bubb MR, Lewis MS, Korn ED (1991) The interaction of monomeric actin with two binding sites on Acanthamoeba actobindin. J Biol Chem 266: 3820–3826 [PubMed] [Google Scholar]
- Burtnick LD, Koepf EK, Grimes J, Jones EY, Stuart DI, McLaughlin PJ, Robinson RC (1997) The crystal structure of plasma gelsolin: implications for actin severing, capping, and nucleation. Cell 90: 661–670 [DOI] [PubMed] [Google Scholar]
- Burtnick LD, Urosev D, Irobi E, Narayan K, Robinson RC (2004) Structure of the N-terminal half of gelsolin bound to actin: roles in severing, apoptosis and FAF. EMBO J 23: 2713–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 suite (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Cowtan K, Main P (1998) Miscellaneous algorithms for density modification. Acta Crystallogr D 54 (Part 4): 487–493 [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Stossel TP, Kwiatkowski DJ (1991) Enhanced motility in NIH 3T3 fibroblasts that overexpress gelsolin. Science 251: 1233–1236 [DOI] [PubMed] [Google Scholar]
- Dabiri GA, Young CL, Rosenbloom J, Southwick FS (1992) Molecular cloning of human macrophage capping protein cDNA. A unique member of the gelsolin/villin family expressed primarily in macrophages. J Biol Chem 267: 16545–16552 [PubMed] [Google Scholar]
- Detmers P, Weber A, Elzinga M, Stephens RE (1981) 7-Chloro-4-nitrobenzeno-2-oxa-1,3-diazole actin as a probe for actin polymerization. J Biol Chem 256: 99–105 [PubMed] [Google Scholar]
- dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ (2003) Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev 83: 433–473 [DOI] [PubMed] [Google Scholar]
- Hartwig JH, Kwiatkowski DJ (1991) Actin-binding proteins. Curr Opin Cell Biol 3: 87–97 [DOI] [PubMed] [Google Scholar]
- Holmes KC, Popp D, Gebhard W, Kabsch W (1990) Atomic model of the actin filament. Nature 347: 44–49 [DOI] [PubMed] [Google Scholar]
- Irobi E, Burtnick LD, Urosev D, Narayan K, Robinson RC (2003) From the first to the second domain of gelsolin: a common path on the surface of actin? FEBS Lett 552: 86–90 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Kouyama T, Mihashi K (1981) Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur J Biochem 114: 33–38 [PubMed] [Google Scholar]
- Kurth MC, Wang LL, Dingus J, Bryan J (1983) Purification and characterization of a gelsolin–actin complex from human platelets. Evidence for Ca2+-insensitive functions. J Biol Chem 258: 10895–10903 [PubMed] [Google Scholar]
- Kwiatkowski DJ (1999) Functions of gelsolin: motility, signaling, apoptosis, cancer. Curr Opin Cell Biol 11: 103–108 [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Janmey PA, Yin HL (1989) Identification of critical functional and regulatory domains in gelsolin. J Cell Biol 108: 1717–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGough AM, Staiger CJ, Min JK, Simonetti KD (2003) The gelsolin family of actin regulatory proteins: modular structures, versatile functions. FEBS Lett 552: 75–81 [DOI] [PubMed] [Google Scholar]
- McLaughlin PJ, Gooch JT, Mannherz HG, Weeds AG (1993) Structure of gelsolin segment 1–zactin complex and the mechanism of filament severing. Nature 364: 685–692 [DOI] [PubMed] [Google Scholar]
- Parikh SS, Litherland SA, Clare-Salzler MJ, Li W, Gulig PA, Southwick FS (2003) CapG(−/−) mice have specific host defense defects that render them more susceptible than CapG(+/+) mice to Listeria monocytogenes infection but not to Salmonella enterica serovar Typhimurium infection. Infect Immun 71: 6582–6590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast GC, Ziff EB (1991) Mbh 1: a novel gelsolin/severin-related protein which binds actin in vitro and exhibits nuclear localization in vivo. EMBO J 10: 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puius YA, Fedorov EV, Eichinger L, Schleicher M, Almo SC (2000) Mapping the functional surface of domain 2 in the gelsolin superfamily. Biochemistry 39: 5322–5331 [DOI] [PubMed] [Google Scholar]
- Southwick FS (1995) Gain-of-function mutations conferring actin-severing activity to human macrophage cap G. J Biol Chem 270: 45–48 [DOI] [PubMed] [Google Scholar]
- Southwick FS, DiNubile MJ (1986) Rabbit alveolar macrophages contain a Ca2+-sensitive, 41,000-dalton protein which reversibly blocks the ‘barbed' ends of actin filaments but does not sever them. J Biol Chem 261: 14191–14195 [PubMed] [Google Scholar]
- Spudich JA, Watt S (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin–troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem 246: 4866–4871 [PubMed] [Google Scholar]
- Stossel TP (1993) On the crawling of animal cells. Science 260: 1086–1094 [DOI] [PubMed] [Google Scholar]
- Stossel TP, Hartwig JH, Janmey PA, Kwiatkowski DJ (1999) Cell crawling two decades after Abercrombie. Biochem Soc Symp 65: 267–280 [PubMed] [Google Scholar]
- Terwilliger T (2004) SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchrotron Radiat 11: 49–52 [DOI] [PubMed] [Google Scholar]
- Witke W, Li W, Kwiatkowski DJ, Southwick FS (2001) Comparisons of CapG and gelsolin-null macrophages: demonstration of a unique role for CapG in receptor-mediated ruffling, phagocytosis, and vesicle rocketing. J Cell Biol 154: 775–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witke W, Sharpe AH, Hartwig JH, Azuma T, Stossel TP, Kwiatkowski DJ (1995) Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell 81: 41–51 [DOI] [PubMed] [Google Scholar]
- Young CL, Feierstein A, Southwick FS (1994) Calcium regulation of actin filament capping and monomer binding by macrophage capping protein. J Biol Chem 269: 13997–14002 [PubMed] [Google Scholar]
- Yu FX, Johnston PA, Sudhof TC, Yin HL (1990) gCap39, a calcium ion- and polyphosphoinositide-regulated actin capping protein. Science 250: 1413–1415 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure Legends
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
supplementary Tables