

Abstract
The AAA-ATPase (ATPase associated with various cellular activities) p97 has been implicated in the degradation of misfolded and unassembled proteins in the endoplasmic reticulum (ERAD). To better understand its role in this process, we used a reconstituted cell-free system to define the precise contribution of p97 in degrading immature forms of the polytopic, multi-domain protein CFTR (cystic fibrosis transmembrane conductance regulator). Although p97 augmented both the rate and the extent of CFTR degradation, it was not obligatorily required for ERAD. Only a 50% decrease in degradation was observed in the complete absence of p97. Moreover, p97 specifically stimulated the degradation of CFTR transmembrane (TM) domains but had no effect on isolated cytosolic domains. Consistent with this, p97-mediated extraction of intact TM domains was independent of proteolytic cleavage and influenced by TM segment hydrophobicity, indicating that the relative contribution of p97 is partially determined by substrate stability. Thus, we propose that p97 functions in ERAD as a nonessential but important ancillary component to the proteasome where it facilitates substrate presentation and increases the degradation rate and efficiency of stable (TM) domains.
Keywords: CFTR, ERAD, ER-associated degradation, polytopic protein, p97
Introduction
A fundamental obstacle in membrane protein degradation is the presentation of substrate to cytosolic and/or extracytosolic proteases. In this regard, two AAA-ATPases (ATPases associated with various cellular activities) (Ogura and Wilkinson, 2001 and references therein) have been proposed to facilitate retrotranslocation and membrane extraction of misfolded and unassembled proteins in the endoplasmic reticulum (ER) (Mayer et al, 1998; Plemper et al, 1998; Ye et al, 2001; McCracken and Brodsky, 2003; Pickart and Cohen, 2004). One ATPase is comprised of a ring of six subunits located at the base of the 19S proteasome regulatory complex (RC) (Glickman et al, 1998). The second is the homohexameric protein p97 (VCP/cdc48) that functions together with cofactors ufd1 and npl4 (Bays and Hampton, 2002). Both ATPases associate with the 20S proteasome, bind directly to polyubiquitinated proteins and facilitate degradation of both ER and cytosolic substrates (Dai et al, 1998; Bays and Hampton, 2002; Pickart and Cohen, 2004). The base of the 19S RC has been reported to interact directly with the Sec61 translocation machinery (Kalies et al, 2005). In contrast, p97 binds the ER via one or more large complexes that include several ubiquitin ligases (gp78, Doa10 and/or Hrd1), VIMP1, Derlin-1 and Ubx2 (Lilley and Ploegh, 2004; Ye et al, 2004; Zhong et al, 2004; Neuber et al, 2005; Schuberth and Buchberger, 2005).
Despite genetic, biochemical and functional evidence implicating the 19S RC and p97 in ER-associated degradation (ERAD), their precise roles remain unknown. AAA-ATPases (Rpt1–6) in the 19S RC collectively open the gate into the 20S core (Kohler et al, 2001), exhibit chaperone-like activity (Braun et al, 1999; Liu et al, 2002) and have been reported to facilitate 26S proteasome assembly and disassembly during the degradation cycle (Babbitt et al, 2005). Based on structural and functional homology to archaebacterial ATP-dependent proteases PAN (Navon and Goldberg, 2001) and ClpX (Ortega et al, 2000), the eukaryotic 19S RC is thought to unfold and actively translocate substrates into the 20S catalytic chamber. Thus an appealing, although largely untested possibility is that 19S unfolding activity might play a direct role in dislocating ERAD substrates from the ER lumen and/or bilayer. Consistent with this notion, functional proteasomes are required for coupling membrane extraction and proteolytic cleavage during ERAD (Mayer et al, 1998; Xiong et al, 1999), and purified 19S RC is sufficient for ATP-dependent dislocation of the lumenal substrate, pro-alpha factor (Lee et al, 2004).
On the other hand, accumulating evidence has suggested that p97 rather than the 19S RC is primarily responsible for ER dislocation. Like the 19S RC, p97 exhibits unfolding/segregating activity and is involved in presenting substrates to the proteasome (Verma et al, 2004; Richly et al, 2005). Indeed, inactivation of p97 or its cofactors, ufd1 or npl4, inhibits degradation of ubiquitinated membrane and luminal ERAD substrates including the ATP binding cassette (ABC) transporters PDR5, Ste6 and CFTR (cystic fibrosis transmembrane conductance regulator) (Gnann et al, 2004; Huyer et al, 2004), HmgCoA reductase (Bays et al, 2001; Rabinovich et al, 2002), ER-localized transcription factors, Spt23 and Mga2 (Hitchcock et al, 2001; Rape et al, 2001), and CPY* (Jarosch et al, 2002; Elkabetz et al, 2004). p97 inhibition or addition of dominant-negative p97 also decreases accumulation of cytosolic intermediates generated in the presence of proteasome inhibitors, reduces US11-dependent retrotranslocation and degradation of MHC I (Ye et al, 2001) and stabilizes a mutant form of CFTR (ΔF508) (Dalal et al, 2004). However, despite the growing perception that p97 is an essential and required component of the ERAD pathway, interference with p97 function results in an incremental decrease but does not completely abolish degradation of most ERAD substrates with the possible exception of MHC I. p97 is also not required for retrotranslocation of cholera toxin (Kothe et al, 2005). Thus, understanding the role of p97 in ERAD has broad and significant implications for the function and regulation of cellular degradation pathways. This has been a particularly challenging issue to address in vivo because p97 is involved in a wide range of cellular activities. p97/cdc48 null mutants are lethal in yeast (Giaever, 2002), and p97 (VCP) siRNA blocks cell cycle progression, arrests HeLa cells in prometaphase/metaphase and induces apoptosis (Wojcik et al, 2004).
To define the precise contribution of p97, we used an in vitro system that efficiently reconstitutes the ERAD pathway (Xiong et al, 1999; Gusarova et al, 2001) and examined the stability of a prototype substrate, CFTR. CFTR is an epithelial, ATP-gated chloride channel in the ABC transporter superfamily (Figure 1). It contains two transmembrane (TM) domains with six TM segments each, two large cytosolic nucleotide binding domains (NBDs) and a cytosolic regulatory (R) domain (Riordan et al, 1989). In most cell types studied to date, approximately 60–80% of wild-type CFTR and nearly 100% of common mutant variants are rapidly degraded via ERAD (Cheng et al, 1990; Jensen et al, 1995; Ward et al, 1995; Meacham et al, 2001). Importantly, this phenotype is recapitulated in vitro, where newly synthesized, membrane-integrated CFTR is efficiently degraded into trichloroacetic acid (TCA)-soluble fragments in an ATP- and proteasome-dependent manner (Xiong et al, 1999; Oberdorf et al, 2001, 2006).
Figure 1.
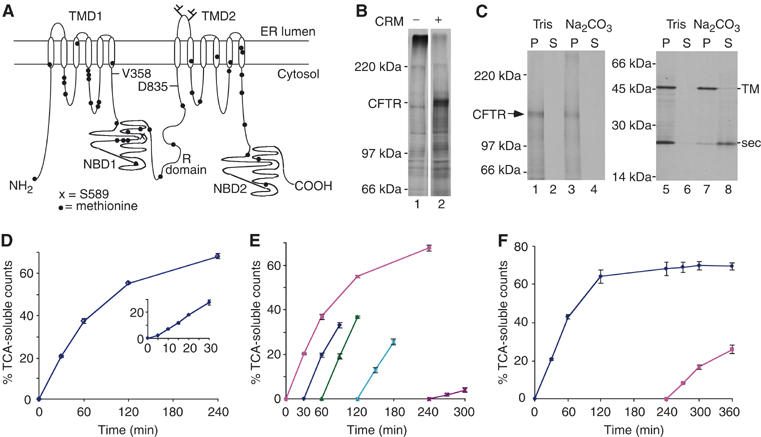
CFTR is degraded into TCA-soluble peptides. (A) Schematic representation of CFTR showing relative location of methionine residues and topology of TM domains (TMDs), NBDs and regulatory (R) domain. Residues V358, S589 and D835 are also indicated. (B) CFTR translated in the presence of CRMs (lane 2) yields the expected full-length ∼150–160 kDa glycosylated protein. (C) Carbonate extraction of CFTR (lanes 1–4) and control TM and secretory (sec) proteins (lanes 5–8) (Skach and Lingappa, 1993). (D) In vitro CFTR degradation showing the percent of [35S]methionine-labeled protein that is converted into radiolabeled TCA-soluble fragments at each time point. The inset shows brief delay and then linear degradation during the first 30 min. (E) RRL was preincubated at 37oC for 30, 60, 120 and 240 min before addition of substrate. Degradation activity remains intact for 2 h but is severely decreased after 4 h. (F) Degradation was carried out as in panel D except that at 240 min, membranes containing residual CFTR protein were isolated and added to a fresh degradation assay and reactions were continued for an additional 2 h. All values are reported as mean of three or more experiments ±s.e.m.
We now show that although p97 specifically augments CFTR degradation, it is not obligatorily required for ERAD. Rather, both the rate and efficiency of CFTR degradation were decreased by only 50% in the absence of p97. Interestingly, p97 had no stimulatory effect on the degradation of isolated CFTR cytosolic domains but specifically increased both the extraction efficiency and degradation rate of CFTR TM domains. p97-mediated domain extraction occurred independently of proteolytic cleavage and was specifically influenced by TM segment hydrophobicity. Together, these data demonstrate that p97 plays a non-essential but important ancillary role in ERAD by increasing the degradation rate (and efficiency) of selected and possibly more thermodynamically stable protein domains.
Results
Reconstitution of in vitro CFTR degradation
CFTR was expressed in vitro in the presence of [35S]methionine and ER microsomal membranes to generate a 160 kDa core-glycosylated protein that was cotranslationally integrated into the ER membrane (Figure 1B and C). Microsomes were then collected and added to fresh rabbit reticulocyte lysate (RRL) lacking exogenous hemin, and degradation was monitored directly by substrate conversion into TCA-soluble peptides. In this system, degradation is specific for misfolded proteins (Supplementary Figure 1), entirely ATP-dependent and sensitive to proteasome inhibitors (Xiong et al, 1999; Oberdorf et al, 2001). After a brief 5-min lag, the rate of TCA-soluble fragment production was linear for 30–60 min, and 65–70% of CFTR was converted into TCA-soluble fragments within 4 h (Figure 1D). At later time points, a gradual decrease in degradation was observed (Figure 1E). This was primarily due to run-down of degradation activity because addition of ATP or fresh RRL failed to restore degradation (data not shown), whereas residual membrane-bound CFTR was readily degraded when microsomes were repelleted and added to fresh RRL (Figure 1F). Because proteasome-mediated CFTR degradation is a multistep process, the rate depends on ubiquitination, dislocation, translocation into the catalytic core and, lastly, peptide cleavage. We have previously shown that for fully functional proteasomes, peptide cleavage exceeds the rate of CFTR unfolding and extraction (Oberdorf et al, 2006). Thus, the initial rate of TCA-soluble fragment production (expressed as % CFTR converted into TCA-soluble fragments/minute) provides an approximate measure of the rate-limiting steps that include membrane extraction, unfolding and delivery of substrate to the 20S catalytic core.
p97 stimulates CFTR degradation
To determine the precise contribution of p97 to CFTR degradation, RRL was depleted of p97 and/or its cofactors by affinity chromatography using two Ni-immobilized recombinant His-tagged proteins (Figure 2A). The first was a nonhydrolyzable ubiquitin fusion protein in which ubcH5a fused to the C-terminus of ubiquitinG76V (uu5). The second was p47, an independent p97 binding protein implicated in membrane fusion (Kondo et al, 1997). Adsorption of RRL to uu5-coated beads removed ∼50% of p97 (Figure 2B, lanes 1–4), and no further reduction was observed upon serial adsorption (data not shown). p97 quantitation was based on immunoblotting standard curves of purified recombinant protein (Supplementary Figure 2). Interestingly, uu5 adsorption depleted ufd1 to below the limits of detection (Figure 2B, lane 4), consistent with reports that ubiquitin fusion proteins bind p97 via the specific adapters ufd1/npl4 (Johnson et al, 1995). In contrast, adsorption using p47-coated beads depleted ∼99% of p97 from RRL but had no effect on ufd1 levels (Figure 2B, lane 6). Sequential adsorption with uu5 followed by p47 depleted both ufd1 and p97 (Figure 2B, lane 5). Importantly, p47 did not appreciably deplete other RRL AAA-ATPases (19S RC) or proteasomes (Figure 2C). Microsome preparations also contain similar amounts of p97 to that found in RRL (0.250 and 0.3 μM, respectively; Supplementary Figure 2). However, p97 affinity is relatively weak, and >90% of p97 was released from membranes by a 50-fold dilution, which reduced the total concentration to 8 nM (Figure 2D). Membrane binding was reversible and could be restored by incubation in intact RRL or purified p97 (Figure 2E).
Figure 2.
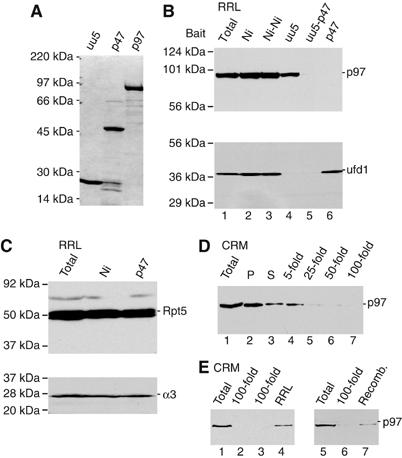
RRL depletion of p97 and p97 complexes. (A) Coomassie-stained gel of recombinant His-tagged proteins used as bait for RRL affinity depletion. (B) Immunoblots for p97 (top) and ufd1 (bottom) of intact RRL (lane 1), RRL following a single adsorption with Ni-NTA beads (lane 2), uu5-coated beads (lane 4) or p47-coated beads (lane 6), and two sequential adsorptions with Ni-NTA beads (lane 3) or uu5 followed by p47 (lane 5). (C) Immunoblots for 19S RC subunit Rpt5 (top) and 20S α3 subunit (bottom) using intact, mock- and p47-adsorbed RRL. (D) Immunoblot of membrane-associated p97. Data show p97 recovered in pellet (P) and supernatant (S) after pelleting microsomes (lanes 2 and 3) and the amount of p97 from the same starting material that remained associated with membranes after dilution indicated (lanes 4–7). (E) CRMs (lanes 1 and 5) and p97-depleted membranes (100-fold dilution) were reincubated with RRL (lane 4) or recombinant p97 (lane 7), pelleted and analyzed by immunoblotting.
Efficient removal of p97 from RRL and microsomal membranes enabled us to carry out CFTR synthesis and degradation entirely in a p97-depleted system. Although control adsorption with Ni-NTA beads slightly decreased the initial degradation rate (0.53±0.02/min versus 0.64±0.03/min) owing to dilution of RRL (Figure 1E versus Figure 3B), p97 depletion had no specific effect on CFTR translation, glycosylation, or membrane integration (data not shown). In contrast, p97 depletion resulted in an ∼50% reduction in the initial rate and overall extent of CFTR degradation that was partially restored by the addition of purified recombinant His-p97 (Figure 3A and B). Depletion of both p97 and ufd1 resulted in a similar (∼50%) decrease in degradation, but in this case activity was not restored upon addition of wild-type p97 (Figure 3C and D). These results provide biochemical evidence that p97 directly contributes to the degradation of membrane-bound CFTR and supports previous studies that its function in ERAD requires ufd1. However, CFTR continued to be degraded at a reduced rate even after highly effective p97 (and ufd1) depletion. This suggested either that CFTR degradation can occur independently of p97 or that small amounts of residual p97 might account for the remaining degradation activity. Indeed, depletion of more than 80% of RRL p97 was required before a significant effect on degradation rate was observed (Supplementary Figure 3).
Figure 3.
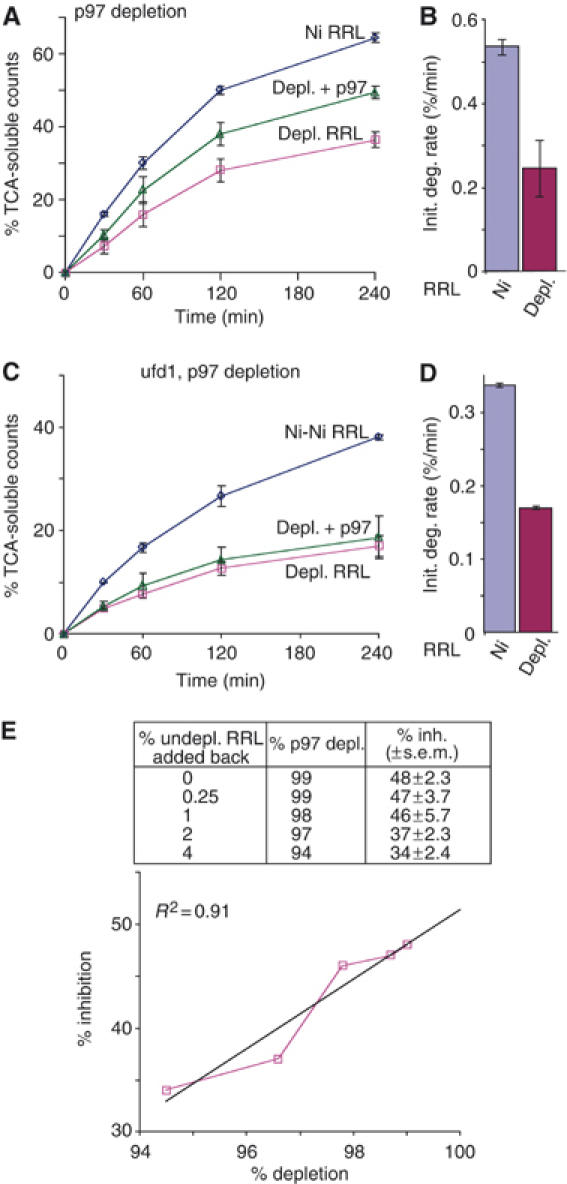
p97 effect on CFTR degradation. (A) Degradation was carried out as in Figure 1, using mock-depleted RRL (Ni RRL), RRL depleted of p97 using p47 (depl. RRL) or p97-depleted RRL plus recombinant p97 (depl.+p97). (B) Initial degradation rates were calculated from panel A based on the percent of intact [35S]methionine-labeled protein converted into radiolabeled TCA-soluble fragments (% CFTR converted/min) during the first 30 min. (C) CFTR degradation in sequential mock-depleted RRL (Ni-Ni), ufd1 and p97-depleted RRL (depl. RRL) and depleted RRL with the addition of recombinant p97 (depl.+p97). (D) Degradation rates calculated from panel C. The slight decrease in the baseline degradation was caused by dilution of RRL with beads used during affinity adsorption. (E) RRL was serially depleted (× 2) with p47 Ni-NTA beads and degradation reactions were supplemented with the indicated amounts of undepleted RRL (inset) to quantitate the % inhibition of CFTR degradation at very low p97 concentrations. Values represent mean of three or more experiments ±s.e.m.
To rule out the possibility that trace residual amounts of p97 were sufficient for CFTR degradation, RRL was subjected to two serial depletions with p47 to remove >99% of p97 and used in conjunction with depleted microsomes. Because translation reactions contain 40% RRL but only 4% microsomes (by volume), depleted microsomes contain only 1% of the total p97 present in undepleted RRL. In addition, microsomes are pelleted twice through buffer after translation to remove unlabeled [35S]methionine. Thus, the residual amount of p97 contributed by membranes represents significantly less than 1% of the total present in undepleted degradation reactions. Consistent with the results of Figure 2, this extent of p97 depletion decreased the rate of CFTR degradation by 48±2.3% (Figure 3E, table). Small amounts of undepleted RRL were then added back to titrate the effects of very low p97 levels. When the amount of residual p97 was titrated between <1 and 5%, a roughly linear relationship was obtained between the initial degradation rate and the p97 concentration (Figure 3E). Although the concentration dependence of p97 is likely complex, linear regression analysis (R2=0.91) predicted that at 100% p97 depletion, the rate of CFTR degradation would be decreased by only 51% (Figure 3E). These results provide strong evidence that CFTR degradation can be carried out in the complete absence of p97, and that the major effect of p97 is to increase the degradation rate by approximately two-fold.
p97 selectively stimulates degradation of CFTR TM domains
p97 exhibits the unique ability to discriminate among polyubiquitinated substrates and selectively present components of protein complexes to the proteasome (Dai et al, 1998; Hitchcock et al, 2001; Rape et al, 2001). However, it is unknown whether p97 exerts different effects on different domains within the same protein. CFTR provides an ideal ERAD substrate to examine this question because it contains multiple well-defined domains that reside either in the cytosol or within the ER membrane (Figure 1). We therefore compared p97 requirements for the degradation of isolated CFTR cytosolic and TM domains. His-tagged constructs encoding portions of cytosolic NBD1 (residues V358–S588) or NBD1-R (residues V358–D835) domains were translated in p97-depleted RRL, isolated by Ni-affinity purification and added directly to degradation reactions. Both constructs were converted into TCA-soluble fragments in an ATP- and proteasome-dependent manner (Figure 4 and Supplementary Figure 4). The initial degradation rate of NBD1 was ∼4-fold faster than that of full-length CFTR, and nearly 90% of the polypeptide was converted into TCA-soluble fragments (Figure 4A and B). Remarkably, p97 depletion had no effect on NBD1 degradation and only a minor effect on NBD1-R turnover (15% decrease in initial rate). Control experiments further showed that NBD1 degradation was not significantly affected by the presence or absence of microsomes in the degradation reaction (Supplementary Figure 5). Thus, in contrast to full-length CFTR, degradation of cytosolic domains was nearly independent of p97.
Figure 4.
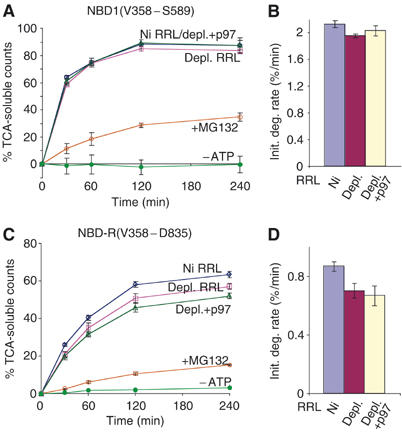
Degradation of CFTR cytosolic domains is unaffected by p97. Purified NBD1 (A) or NBD1-R (C) was added directly to degradation reactions containing mock-depleted RRL or p97-depleted RRL with or without added recombinant p97 as indicated. Also shown are degradation rates in ATP-depleted RRL (−ATP) and in the presence of MG132. (B, D) Initial degradation rates were calculated from panels A and C. Location of domain in the CFTR polypeptide is indicated in Figure 1A.
We next examined degradation requirements for the first CFTR TM domain (TMD1) (residues M1–V393), which contains the first six TM segments and connecting loops (Figure 5A). Previous in vitro and in vivo topological analysis of TMD1 by us and others has confirmed the six-spanning topology shown (Lu et al, 1998; Zhang et al, 1998; Carveth et al, 2002). Consistent with this, carbonate extraction studies confirmed that in vitro-translated TMD1 was fully integrated into the ER membrane (Figure 5A). Because 12 of the 13 TMD1 methionines (92%) are located within 27 residues of the membrane, and the linear distance from the 19S ATPase ring to proteolytic sites in the 20S core is ∼11 nm (Baumeister et al, 1998), only the N-terminal methionine should be accessible to proteolytic cleavage in the intact domain (Lee et al, 2001). TMD1 was also subject to ERAD, as demonstrated by ATP-dependent conversion into TCA-soluble fragments and MG132 sensitivity (Figure 5B). Approximately 45% of TMD1 methionines were converted into TCA-soluble fragments, confirming that TMD1 was also extracted from the ER membrane. The initial rate of degradation (0.44±0.04%/min) was similar to that of full-length CFTR (Figure 5C). In addition, p97 depletion resulted in an ∼50% reduction in both the rate and extent of TMD1 degradation, which was almost entirely restored by the addition of recombinant p97 (Figure 5B and C). These data indicate that CFTR TMD1 is degraded two- to five-fold slower than isolated cytosolic NBD1 and NBD1-R domains in intact RRL and three- to nine-fold slower in p97-depleted RRL. Proteasome inhibition also resulted in accumulation of ubiquitinated cytosolic (NBD1) and TM (TMD1) substrates, demonstrating that domains are degraded by the ubiquitin–proteasome pathway regardless of their location (Supplementary Figure 4). Thus, p97 selectively enhances the rate of TMD1 degradation over that of CFTR cytosolic domains.
Figure 5.

Degradation of CFTR TMD1. (A) TMD1 topology showing location of methionines and length of cytosolic and lumenal loops. Carbonate extraction of in vitro-expressed TMD1. (B) TMD1 degradation was assayed in the presence and absence of p97 as in Figure 4. Also shown are effects of MG132 and ATP depletion. (C) Initial rates of TMD1 degradation in mock RRL, p97-depleted RRL and after p97 supplementation.
p97 contribution is dependent on TM domain hydrophobicity
The inverse correlation between degradation rate and p97 dependence suggested that p97 might selectively facilitate the unfolding of more thermodynamically stable peptide regions. If this were true, then TMs that were less stable in the lipid bilayer might also show less p97 dependence. We therefore examined a shorter construct containing the CFTR N-terminus and first two TM segments (Figure 6A). This construct (TM1–2) efficiently acquires its correct two-spanning topology in vitro (Lu et al, 1998) and contains five methionines, four of which are located within 18 residues of the membrane. Like TMD1, in vitro-synthesized TM1–2 was membrane-integrated (Figure 6B) and was degraded by the proteasome slightly faster (0.73±0.08%/min) than full-length CFTR (Figure 6C and D). In contrast to TMD1, however, the rate of TM1–2 degradation was only modestly affected (∼20% reduction) by p97 depletion.
Figure 6.
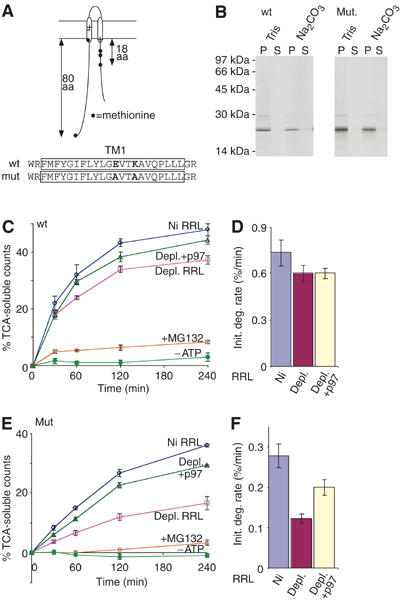
p97 effects are influenced by TM segment composition. (A) Topology and methionine distribution of TM1–2 constructs. Location of residues Glu92 and Lys95 is shown within the TM1 sequence. (B) Carbonate extraction of wild-type and E92A/K95A polypeptides. (C, E) Degradation assays performed in mock-depleted RRL and p97-depleted RRL with or without added recombinant p97. Effects of MG132 and ATP depletion are also shown. (D, F) Initial degradation rates calculated from panels C and E.
CFTR is somewhat unusual in that both TM1 and TM2 contain potentially charged residues (Riordan et al, 1989). E92 and K95 reside near the center of TM1 (Figure 6A), and R134 resides near the C-terminus of TM2. E92 and K95 were therefore replaced with alanine residues to generate a more stable TM structure. The TM1–2 E92A/K95A mutations had a striking effect, decreasing the initial degradation rate by ∼2.5-fold (0.28±0.03/min) in the presence of p97 (Figure 6E and F), and further decreasing the degradation rate by an additional 2.3-fold following p97 depletion (0.12±0.01/min). Again, degradation was proteasome dependent (Figure 6E and Supplementary Figure 4) and substantially restored by the addition of recombinant p97.
p97 augments TM domain extraction independently of proteolytic cleavage
Finally, to determine whether p97 facilitates extraction of CFTR TM domains directly and independently of proteasome-mediated peptide cleavage, we examined specific requirements for TMD1 release from the membrane. For these experiments, TMD1 degradation products were separated into membrane-bound and cytosolic fractions before analysis. Consistent with previous studies (Oberdorf et al, 2006), all cytosolic fragments were TCA soluble under control conditions (Figure 7A). In the presence and absence of p97, MG132 decreased the rate of TCA-soluble fragment production by 75 and 74%, respectively, but in both cases CFTR continued to be released into the cytosol as large TCA-insoluble polypeptides (Figure 7A and B, vertical arrows). These cytosolic fragments were visualized by SDS–PAGE and found to be predominantly comprised of full-length TMD1, which had been integrated into the membrane during translation and then extracted en block during the degradation reaction (Figure 7C, lanes 1–10). p97 depletion decreased the rate of fragment release as expected but, by itself, had no effect on the TCA solubility of cytosolic fragments (Figure 7B). However, even when degradation was inhibited by MG132, p97 depletion decreased both the rate of total TMD1 release from the ER membrane (65% decrease; Figure 7D and E) and the cytosolic accumulation of the full-length domain (Figure 7C, lanes 1–5 versus lanes 6–10). As expected, no cytosolic fragments were visualized following ATP depletion or in the absence of MG132 (Figure 7C, lanes 11–20). These results strongly suggest that p97 can facilitate extraction of intact CFTR TMDs in vitro, and that the stimulatory effect of p97 on CFTR degradation occurs independently and upstream of proteolytic cleavage.
Figure 7.
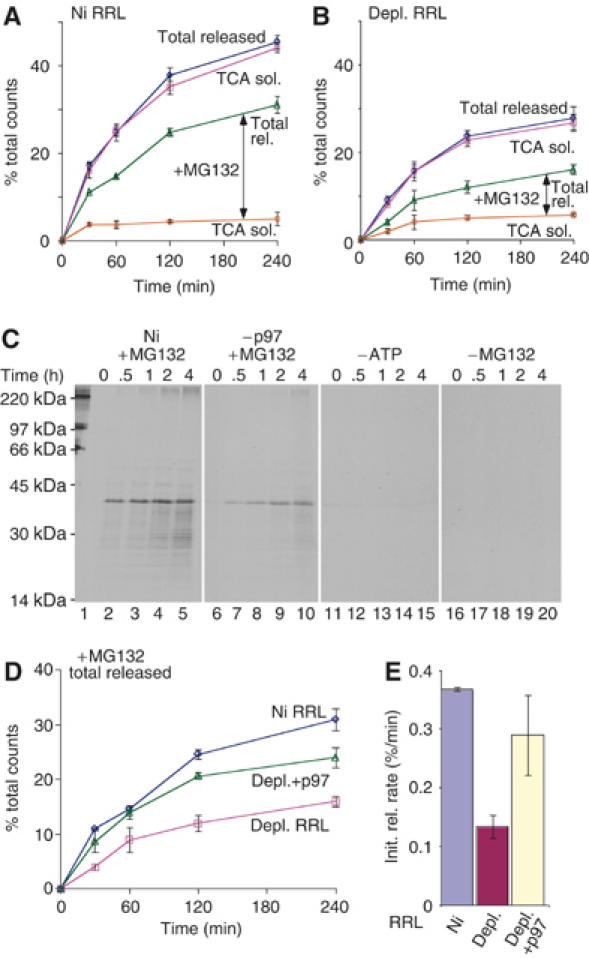
p97 facilitates membrane extraction independently of cleavage. TMD1 degradation reactions were carried out in (A) mock- or (B) p97-depleted RRL, and cytosolic supernatants were analyzed for CFTR fragments before (total released) or after (TCA sol) TCA precipitation. Vertical arrow indicates TCA-insoluble peptide fragments that accumulate in the supernatant. (C) Autoradiograms showing dislocated full-length TMD1 degradation products generated in the presence of MG132 (lanes 1–10), in mock-depleted (lanes 1–5) or p97-depleted (–p97, lanes 6–10) RRL. No fragments were visualized by SDS–PAGE in the absence of either ATP (lanes 11–16) or MG132 (lanes 16–20). The extent (D) and initial rate (E) of total TMD1 released in the presence of MG132 in mock-depleted (Ni), p97-depleted (depl.) or depleted RRL supplemented with recombinant p97 (depl.+p97).
Discussion
Two AAA-ATPase complexes, p97–ufd1–npl4 and the proteasome 19S RC, are currently implicated in the dislocation and degradation of misfolded proteins from the ER membrane. Using an in vitro system that reconstitutes the ERAD pathway, we now determine the relative contribution of p97 in facilitating degradation of a prototypical ERAD substrate, CFTR. This biochemical approach enabled us to quantitate both the degradation rate and extent of p97 depletion in a way not possible for most in vivo systems. We find that in the absence of p97, the rate of CFTR degradation was decreased by ∼50% as determined by proteasome-dependent conversion of substrate into TCA-soluble fragments. Detailed depletion and add-back experiments confirmed that residual traces of p97 did not account for the substantial residual degradation activity observed. These results are in general agreement with several recent studies that have also observed a partial degradation defect for CFTR after p97 depletion or inactivation in cells (Kobayashi et al, 2002; Dalal et al, 2004; Gnann et al, 2004; Vij et al, 2006). Thus, although p97 clearly facilitates ERAD, it is not obligatorily required for extraction, unfolding or proteolysis of a large, membrane-integrated polytopic protein.
Remarkably, the stimulatory effect of p97 on CFTR degradation was restricted to TM domains, whereas degradation of cytosolic domains was nearly entirely p97 independent. The relative contribution of p97 was also markedly influenced by changes in the hydrophobicity of individual TM segments. These findings raise the possibility that p97 primarily increases the degradation efficiency of specific domains that are more stable and/or more slowly degraded. Indeed, we found a strong inverse correlation between the baseline degradation rate of different CFTR domains and the stimulatory contribution of p97 (Figure 8). This also held true in the absence of p97 where the degradation rate of isolated NBD1 and NBD1-R domains was significantly faster than TMD1 and TM1–2(E92A/K95A). Although additional studies are clearly needed before this effect can be generalized, our analysis suggests that p97 stimulatory effects are most pronounced when the degradation rate is slow (<0.5%/min in our study). p97 also directly stimulated the extraction and membrane release of intact TM domains independently of proteolytic cleavage. Taken together, these results indicate that p97 functions as a non-essential, yet important ancillary component of the ERAD pathway that stimulates presentation of thermodynamically stable substrates or domains to the 26S proteasome.
Figure 8.
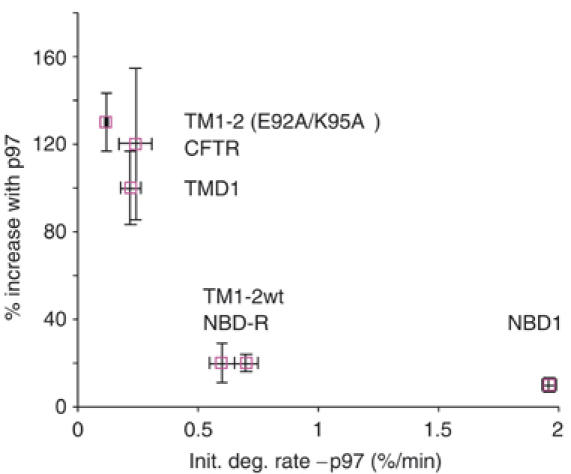
P97 effect is inversely related to the rate of degradation. The stimulatory effect (% increase) of p97 on the degradation rate was plotted as a function of the initial degradation rate in the absence of p97.
Cryo-EM studies have suggested that unfolding by AAA-ATPases occurs via conformational changes caused by ATP binding/hydrolysis (Ogura and Wilkinson, 2001). For example, unfolding by the prokaryotic PAN AAA-ATPase takes place on the surface of the hexameric ring and leads directly to peptide translocation through the central pore (Navon and Goldberg, 2001). In the case of the 26S proteasome, substrate must translocate through the 19S AAA-ATPases to reach the 20S cylinder. Although it is unclear whether translocation occurs through the p97 ring, there may be an analogous unfolding on the surface of the hexamer. However, our findings clearly demonstrate that such an interaction with p97 is not obligatory even for substrates where p97 can and normally does facilitate degradation. This raises a significant mechanistic question. How and when is p97 recruited to substrate? One possibility is that the proteasome provides a baseline unfolding activity that (processively) degrades substrate until it encounters a stable region that it is unable to unfold (Lee et al, 2001). This stalled complex could then recruit p97 (or possibly other as yet unknown ATPases) to assist in extraction/unfolding and thus stimulate the degradation rate. In a second, and perhaps more plausible model, both the 26S proteasome and p97 could processively unfold substrates together. However, p97 would not contribute substantially to the rate of unfolding until a specific region of protein was encountered in which its ATPase activity would increase the basal rate provided by the proteasome. In support of the former possibility, purified 26S proteasomes are clearly capable of degrading substrates in vitro in the absence of p97 (Lee et al, 2004).
A third explanation for our findings is that CFTR could use multiple pathways for ubiquitination and presentation to the proteasome (Verma et al, 2004). Consistent with this, we recently reported that during proteasome-mediated degradation, CFTR N-terminal, C-terminal and R-domain epitopes were lost at similar rates, suggesting that degradation could be initiated at multiple sites within the polypeptide (Oberdorf et al, 2006). Moreover, CFTR ubiquitination and degradation can be initiated by a cytosolic complex involving the U-box protein CHIP, Hsc70 and ubcH5 (Meacham et al, 2001) as well as membrane-bound E2/E3 proteins Hrd1/Hrd3, Doa10 and Ubc6 (Gnann et al, 2004). Thus, different recognition machinery may operate on different CFTR domains depending on their cellular compartment and the nature of the folding defect. This model is further supported by recent studies indicating that a large protein complex at the ER membrane involving Der1, Rma1 and Ubc6 is specifically involved in recognizing mutations within TMD1 (D Cyr, personal communication), whereas the CHIP–Hsc70–UbcH5 complex primarily recognizes cytosolic (NBD) folding defects (Younger et al, 2004). Intriguingly, the Hrd1/3 and Doa10 pathways in yeast, and the Rma1 pathway in mammalian cells, all converge at p97/cdc48. If p97 functions to present substrates to the proteasome as predicted, this may also explain why CFTR TMDs preferentially display augmented degradation in the presence of p97. Recently, it was reported that ubx2 is a central component of this complex in yeast and through its UBA and UBX domains links substrates with E3 ligases, Der-1 and p97 (Neuber et al, 2005; Schuberth and Buchberger, 2005). It will therefore be most interesting to determine whether Ubx2 and additional components are also required for p97 augmentation of TMD degradation.
To our knowledge, this is the first study demonstrating that p97 contributes differentially to the degradation of different domains derived from the same protein. Although recruitment of degradation machinery likely plays a role in this process, it is also important to consider that the inefficient maturation of CFTR observed in most cell types is caused by failure to acquire proper tertiary and/or quaternary structure. Although the timing of NBD folding during synthesis is somewhat controversial (Du et al, 2005; Kleizen et al, 2005), NBDs are thought to fold relatively inefficiently in cells, and this kinetic defect is amplified by deletion of F508 in NBD1, a mutation found in nearly 90% of CF patients (Qu et al, 1997; Du et al, 2005; Thibodeau et al, 2005). Thus, the relatively unfolded state of isolated NBD1 (and NBD-R) in RRL may contribute to the rapid baseline degradation rate and the lack of requirement for p97. By contrast, TM helices are degraded much more slowly, and demonstrate a clear augmentation by p97. These findings parallel observations for the cytosolic substrate, titin, which can also exhibit two degradation rates, a slow rate that requires domain unfolding and fast rate when the substrate is presented in a pre-denatured state (Kenniston et al, 2003). Each pathway is characterized by marked differences in energy consumption as a result of the inefficient, iterative engagement of folded substrates with the AAA-ATPase ClpXP, which is cooperatively promoted during denaturation at the surface of the ring (Kenniston et al, 2003). This process provides a mechanism for partitioning stable from misfolded or denatured substrates based on their affinity for the AAA-ATPase (Kenniston et al, 2005). By analogy the eukaryotic 19S RC is predicted to have a similar cooperative unfolding behavior such that an unfolded or partially unfolded substrate would be energetically less costly and more efficiently degraded.
Importantly, the unfolding process is highly cooperative and is influenced by local structure (helices and surface loops) at the site of initial unfolding (Lee et al, 2001). In the case of membrane proteins, extraction of TMDs may represent a significant energy barrier, particularly where short lumenal loops (e.g. CFTR TM3–4, TM5–6) necessitate pairwise extraction of TM segments. Indeed, TMD1–6 was degraded at a much slower rate than cytosolic domains, suggesting that membrane extraction may be generally rate-limiting in polytopic protein degradation. In the TM1–2 construct, polar/charged residues could provide a locally unstable environment in the lipid bilayer in which p97 is not required for extraction. Interestingly, replacing TM1 charged residues with alanine resulted in the most stable of the constructs examined and slowed degradation by nearly five-fold (in the absence of p97) when compared to wild-type TM1–2 and nearly 20-fold when compared to the isolated NBD1. Thus, p97 may play a key role in stimulating TM1–2 E92A/K95A degradation by providing a second step for substrate partitioning, thereby generating a locally unfolded domain that preferentially engages the AAA-ATPase ring of the 19S RC. The net result of two serial partitioning steps would be to dramatically increase the efficiency of engagement by both AAA-ATPases and the ultimate delivery of substrate into the proteasome catalytic core.
Materials and methods
Constructs/recombinant proteins
Details for plasmid cloning and synthesis of recombinant proteins are described in Supplementary data.
Affinity depletion of RRL and CRM
RRL and canine pancreatic rough microsomes (CRMs) were affinity depleted as described (Carlson et al, 2005). Briefly, Ni-NTA beads containing near-saturating amounts of His-tagged recombinant proteins were added to RRL at a ratio of 5:1 (RRL:Ni-NTA) and incubated at least for 4 h at 4°C with continuous mixing. The supernatants were removed and frozen in liquid nitrogen. Mock depletions were performed using equal volumes of uncoupled Ni-NTA beads. CRMs were depleted of p97 by 50-fold dilution in Buffer B (0.25 M sucrose, 1 mM DTT and 50 mM triethanolamine acetate, pH 7.5) at 24°C for 15 min. Membranes were pelleted at 180 000 g for 20 min, and resuspended in their original volume of Buffer B. Membranes were repopulated with p97 by incubating in an equal volume of RRL or 5 μg/ml recombinant p97 before pelleting through Buffer B. Immunoblotting was performed using standard protocols described in Supplementary data.
In vitro transcription/translation
mRNA was transcribed with SP6 RNA polymerase for 1 h at 40°C and translated in a transcription-linked reaction for 1–2 h at 24°C as described in detail elsewhere (Carlson et al, 2005). Water was substituted for DNA in mock transcription reactions. Canine pancreatic rough microsomal membranes (A280=4) were added at the start of all translations, except the cytosolic NBD and NBD-R constructs. Aurin tricarboxylic acid (25–50 μM) was added after 15–20 min to synchronize translation. Following translation, CRMs containing in vitro-synthesized protein were isolated twice by centrifugation, 180 000 g for 10 min, through a cushion containing 0.5 M sucrose in Buffer C (100 mM KCl, 5 mM MgCl2, 1 mM DTT, 50 mM HEPES, pH 7.5) and resuspended in 1/2 original volume in 0.1 M sucrose in Buffer C. Before pelleting, TM1–2 E92A/K95A was released from ribosomes by addition of 1 mM puromycin. Soluble NBD and NBD-R proteins were isolated by diluting translation reactions with three volumes of Buffer A and mixing with 10 μl Ni-NTA at 4°C for 30 min. Beads were then washed in 10 × 1 ml 10 mM imidazole in Buffer A, and proteins were eluted with 250 mM imidazole in Buffer A.
Degradation assay
Isolated CRMs from translation reactions were added to RRL supplemented with 1 mM ATP, 12 mM creatine phosphate, 5 mM MgCl2, 3 mM DTT, 10 mM Tris–HCl (pH 7.5) and 4 μg creatine kinase/50 μl reaction (Carlson et al, 2005) and incubated at 37°C. Then, 100 μM MG132 or 20 mM 2-deoxyglucose and 20 U hexokinase were added to the reaction mixture as indicated to inhibit proteasomes or deplete ATP, respectively. Recombinant p97 (200 ng/μl RRL) was also added to p97-depleted RRL where indicated. Samples were removed (Tn), precipitated in 20% TCA and centrifuged at 16 000 g for 10 min. TCA supernatants (TCA Sol) were then counted in ScintiSafe (Fisher Chemicals) using a Beckmann LS6500 scintillation counter. Total incorporated [35S] in each sample was determined by directly counting an aliquot of the degradation reaction. Mock reactions were used as a control to correct for small amounts of nonspecific, ATP-independent association of [35S] with the membranes (Carlson et al, 2005). The percent of protein degraded at each time point was determined using the following formula:
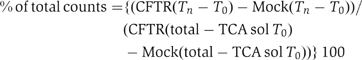
where Tn=TCA-soluble counts at T=n and T0=TCA-soluble counts at T=0 for indicated CFTR and mock translation reactions. All values are reported as mean of three or more experiments ±s.e.m.
Membrane release assay
At each time point, microsomes from degradation reactions were first pelleted at 180 000 g for 10 min through 0.5 M sucrose in Buffer B. The cytosolic supernatant was then either counted directly to measure total CFTR released from the membrane, precipitated in 20% TCA to determine TCA-soluble counts released or analyzed by SDS–PAGE and autoradiography. The percentage of membrane-bound CFTR substrate released into the supernatant and %TCA-soluble counts released at each time point were calculated by the same formula as used for the degradation assay.
Carbonate extraction
CRMs containing translation products were added to 1 ml of 0.25 M sucrose, 1 μg BSA and 0.1 M Tris–HCl (pH 7.5) or 1 ml of 0.1 M Na2CO3 (pH 11.5) and incubated on ice for 30 min, followed by centrifugation at 180 000 g for 30 min. Pellets were solubilized in 30 μl of SDS–PAGE loading buffer. Supernatants were precipitated in 20% TCA, and the resulting pellets were solubilized in 30 μl SDS–PAGE loading buffer. Samples were analyzed by SDS–PAGE and EN3HANCE (Perkin Elmer, Boston, MA) fluorography and imaged on Kodak film.
Supplementary Material
Supplemental Information
Acknowledgments
We thank Dr H Meyer for the gifts of p47 and p97 cDNAs, Dr M Hochstrasser for human ubiquitin, cDNA, Dr P Howley for ubcH5a cDNA and Dr K Frueh for c9 antisera. Also, we thank Drs C Enns, K Frueh, L Musil and S Lutsenko for helpful comments on the manuscript.
References
- Babbitt SE, Kiss A, Deffenbaugh AE, Chang YH, Bailly E, Erdjument-Bromage H, Tempst PTB, Sklar LA, Baumler J, Gogol E, Skowyra D (2005) ATP hydrolysis-dependent disassembly of the 26S proteasome is part of the catalytic cycle. Cell 121: 553–565 [DOI] [PubMed] [Google Scholar]
- Baumeister W, Walz J, Zuehl F, Seemueller E (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92: 367–380 [DOI] [PubMed] [Google Scholar]
- Bays N, Hampton R (2002) Cdc48–Ufd1–Npl4: stuck in the middle with Ub. Curr Biol 12: R366–R371 [DOI] [PubMed] [Google Scholar]
- Bays N, Wilhovsky S, Goradia A, Hodgkill-Harlow K, Hampton R (2001) HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell 12: 4114–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun B, Glickman M, Kraft R, Dahlmann B, Kloetzel P-M, Finley D, Schmidt M (1999) The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat Cell Biol 1: 221–226 [DOI] [PubMed] [Google Scholar]
- Carlson E, Bays N, David L, Skach W (2005) Reticulocyte lysate as a model system to study endoplasmic reticulum membrane protein degradation. In Ubiquitin–Proteasome Protocols, Patterson C, Cyr D (eds). Totowa, NJ: Humana Press Inc. [DOI] [PubMed] [Google Scholar]
- Carveth K, Buck T, Anthony V, Skach W (2002) Cooperativity and flexibility of cystic fibrosis transmembrane conductance regulator transmembrane segments participate in membrane localization of a charged residue. J Biol Chem 277: 39507–39514 [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834 [DOI] [PubMed] [Google Scholar]
- Dai RM, Chen E, Longo DL, Gorbea CM, Li CH (1998) Involvement of valosin-containing protein, an ATPase co-purified with IκBα and 26S proteasome, in ubiquitin–proteasome-mediated degradation of IκBα. J Biol Chem 273: 3562–3573 [DOI] [PubMed] [Google Scholar]
- Dalal S, Rosser MF, Cyr DM, Hanson PI (2004) Distinct roles for the AAA-ATPases NSF and p97 in the secretory pathway. Mol Biol Cell 15: 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Sharma M, Lukacs GL (2005) The ΔF508 cystic fibrosis mutation impairs domain–domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol 12: 17–25 [DOI] [PubMed] [Google Scholar]
- Elkabetz Y, Shapira I, Rabinovich E, Bar-Nun S (2004) Distinct steps in dislocation of luminal endoplasmic reticulum-associated substrates. J Biol Chem 279: 3980–3989 [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian K-D, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hernpel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathem JN, Valle G, Voet M, Volckaert G, Wang C-y, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391 [DOI] [PubMed] [Google Scholar]
- Glickman MH, Rubin DM, Fried VA, Finley D (1998) The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol Cell Biol 18: 3149–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnann A, Riordan JR, Wolf DH (2004) Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the cdc48 protein complex in yeast. Mol Biol Cell 15: 4125–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarova V, Caplan AJ, Brodsky JL, Fisher EA (2001) Apoprotein B degradation is promoted by the molecular chaperones hsp90 and hsp70. J Biol Chem 276: 24891–24900 [DOI] [PubMed] [Google Scholar]
- Hitchcock AL, Krebber H, Frietze S, Lin A, Latterich M, Silver PA (2001) The conserved npl4 protein complex mediates proteasome-dependent membrane-bound transcription factor activation. Mol Biol Cell 12: 3226–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S (2004) Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal substrate. J Biol Chem 279: 38369–38378 [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf D, Sommer T (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol 4: 134–139 [DOI] [PubMed] [Google Scholar]
- Jensen T, Loo M, Pind S, Williams D, Goldberg A, Riordan J (1995) Multiple proteolytic systems, including the proteosome, contribute to CFTR processing. Cell 83: 129–136 [DOI] [PubMed] [Google Scholar]
- Johnson ES, Ma PC, Ota IM, Varshavsky A (1995) A proteolytic pathway that recognizes ubiquitin as a degradation signal. J Biol Chem 270: 17442–17456 [DOI] [PubMed] [Google Scholar]
- Kalies K-U, Allan S, Sergeyenko T, Kroger H, Romisch K (2005) The protein translocation channel binds proteasomes to the endoplasmic reticulum. EMBO J 24: 2284–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Sauer RT (2005) Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA 102: 1390–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Fernandez JM, Sauer RT (2003) Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell 114: 511–520 [DOI] [PubMed] [Google Scholar]
- Kleizen B, van Vlijmen T, de Jonge H, Braakman I (2005) Folding of CFTR is predominately cotranslational. Mol Cell 20: 277–287 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Tanaka K, Inoue K, Kakizuka A (2002) Functional ATPase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 Cells. J Biol Chem 277: 47358–47365 [DOI] [PubMed] [Google Scholar]
- Kohler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D (2001) The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol Cell 7: 1143–1152 [DOI] [PubMed] [Google Scholar]
- Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G (1997) p47 is a cofactor for p97-mediated membrane fusion. Nature 388: 75–78 [DOI] [PubMed] [Google Scholar]
- Kothe M, Yihong T, Wagner J, De Luca H, Kern E, Rapoport TA, Lencer W (2005) Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J Biol Chem 280: 28127–28132 [DOI] [PubMed] [Google Scholar]
- Lee C, Schwartz M, Prakash S, Iwakura M, Matouschek A (2001) ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell 7: 627–637 [DOI] [PubMed] [Google Scholar]
- Lee RJ, Liu C, Harty C, McCracken AA, Latterich M, Romisch K, DeMartino GN, Thomas PJ, Brodsky JL (2004) Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J 23: 2206–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL (2004) A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429: 834–840 [DOI] [PubMed] [Google Scholar]
- Liu C, Millen L, Roman TB, Xiong H, Gilbert H, Noiva R, DeMartino GN, Thomas PJ (2002) Conformational remodeling of proteasome substrates by PA700, the 19S regulatory complex of the 26S proteasome. J Biol Chem 277: 26815–26820 [DOI] [PubMed] [Google Scholar]
- Lu Y, Xiong X, Helm A, Kimani K, Bragin A, Skach W (1998) Co- and posttranslational mechanisms direct CFTR N-terminus transmembrane assembly. J Biol Chem 273: 568–576 [DOI] [PubMed] [Google Scholar]
- Mayer T, Braun T, Jentsch S (1998) Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO J 17: 3251–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken A, Brodsky J (2003) Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD). BioEssays 25: 868–877 [DOI] [PubMed] [Google Scholar]
- Meacham G, Patterson C, Zhang W, Younger J, Cyr D (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 3: 100–105 [DOI] [PubMed] [Google Scholar]
- Navon A, Goldberg A (2001) Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol Cell 8: 1339–1349 [DOI] [PubMed] [Google Scholar]
- Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T (2005) Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol 7: 993–998 [DOI] [PubMed] [Google Scholar]
- Oberdorf J, Carlson E, Skach W (2001) Redundancy of proteasome ß subunit function during endoplasmic reticulum associated degradation. Biochemistry 40: 13397–13305 [DOI] [PubMed] [Google Scholar]
- Oberdorf J, Carlson E, Skach W (2006) Uncoupling proteasome peptidase and ATPase activities results in cytosolic release of an ER polytopic protein. J Cell Sci 119: 303–313 [DOI] [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure-diverse function. Genes Cells 6: 575–597 [DOI] [PubMed] [Google Scholar]
- Ortega J, Singh SK, Ishikawa T, Maurizi MR, Steven AC (2000) Visualization of substrate binding and translocation by the ATP-dependent protease, ClpX. Mol Cell 6: 1515–1521 [DOI] [PubMed] [Google Scholar]
- Pickart CM, Cohen RE (2004) Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol 5: 177–187 [DOI] [PubMed] [Google Scholar]
- Plemper R, Egner R, Kuchler K, Wolf D (1998) Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a consorted action of Sec61 and the proteasome. J Biol Chem 273: 32848–32856 [DOI] [PubMed] [Google Scholar]
- Qu B, Strickland EH, Thomas PJ (1997) Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J Biol Chem 272: 15739–15744 [DOI] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frölich K-U, Diamant N, Bar-Nun S (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 22: 626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S (2001) Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(Ufd1/Npl4), a ubiquitin-selective chaperone. Cell 107: 667–677 [DOI] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S (2005) A series of ubiquitin binding factors connects cdc48/p97 to substrate multiubiquitination and proteasomal targeting. Cell 120: 73–84 [DOI] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Collins FS, Tsui L-C (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1072 [DOI] [PubMed] [Google Scholar]
- Schuberth C, Buchberger A (2005) Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated degradation. Nat Cell Biol 7: 999–1006 [DOI] [PubMed] [Google Scholar]
- Skach W, Lingappa V (1993) Amino terminus assembly of human P-glycoprotein at the endoplasmic reticulum is directed by cooperative actions of two internal sequences. J Biol Chem 268: 23552–23561 [PubMed] [Google Scholar]
- Thibodeau PH, Brautigam CA, Machius M, Thomas PJ (2005) Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol 12: 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Oania R, Graumann J, Deshaies RJ (2004) Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin–proteasome system. Cell 118: 99–110 [DOI] [PubMed] [Google Scholar]
- Vij N, Fang S, Zeitlin P (2006) Selective inhibition of ERAD rescues ΔF508-CFTR and suppresses IL8 levels: therapeutic implications. J Biol Chem 281: 17369–17378 [DOI] [PubMed] [Google Scholar]
- Ward C, Omura C, Kopito R (1995) Degradation of CFTR by the ubiquitin–proteosome pathway. Cell 83: 121–128 [DOI] [PubMed] [Google Scholar]
- Wojcik C, Yano M, Demartino GN (2004) RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J Cell Sci 117: 281–292 [DOI] [PubMed] [Google Scholar]
- Xiong X, Chong E, Skach W (1999) Evidence that endoplasmic reticulum (ER)-associated degradation of cystic fibrosis transmembrane conductance regulator is linked to retrograde translocation from the ER membrane. J Biol Chem 274: 2616–2624 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer H, Rapoport T (2001) The AAA-ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414: 652–656 [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA (2004) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429: 841–847 [DOI] [PubMed] [Google Scholar]
- Younger JM, Ren H-Y, Chen L, Fan C-Y, Fields A, Patterson C, Cyr DM (2004) A foldable CFTR ΔF508 biogenic intermediate accumulates upon inhibition of the Hsc70-CHIP E3 ubiquitin ligase. J Cell Biol 167: 1075–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JT, Chen M, Han E, Wang C (1998) Dissection of de novo membrane insertion activities of internal transmembrane segments of ATP-binding-cassette transporters: toward understanding topological rules for membrane assembly of polytopic membrane proteins. Mol Biol Cell 9: 853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S (2004) AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem 279: 45676–45684 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information