

Abstract
Doublecortin is a neuronal microtubule-stabilising protein, mutations of which cause mental retardation and epilepsy in humans. How doublecortin influences microtubule dynamics, and thereby brain development, is unclear. We show here by video microscopy that purified doublecortin has no effect on the growth rate of microtubules. However, it is a potent anti-catastrophe factor that stabilises microtubules by linking adjacent protofilaments and counteracting their outward bending in depolymerising microtubules. We show that doublecortin-stabilised microtubules are substrates for kinesin translocase motors and for depolymerase kinesins. In addition, doublecortin does not itself oligomerise and does not bind to tubulin heterodimers but does nucleate microtubules. In cells, doublecortin is enriched at the distal ends of neuronal processes and our data raise the possibility that the function of doublecortin in neurons is to drive assembly and stabilisation of non-centrosomal microtubules in these doublecortin-enriched distal zones. These distinct properties combine to give doublecortin a unique function in microtubule regulation, a role that cannot be compensated for by other microtubule-stabilising proteins and nucleating factors.
Keywords: doublecortin, lissencephaly, MAPs, microtubule, neuron
Introduction
Microtubules (MTs) are involved in many cellular processes including mitosis, motility and organelle transport (Feng and Walsh, 2001; Guzik and Goldstein, 2004). The building blocks of MTs are αβ-tubulin heterodimers, which associate laterally and longitudinally to form polar, cylindrical MTs (Desai and Mitchison, 1997). The ability of αβ-tubulin to polymerise is controlled by the guanine nucleotide bound to β-tubulin. GTP fuels MT growth because GTP-tubulin adopts a conformation that facilitates dimer–dimer interactions, thereby favouring polymerisation. GDP-tubulin, on the other hand, has a curved structure that does not fit well within the straight MT walls (Ravelli et al, 2004). When GTP-tubulin is incorporated into the MT lattice, hydrolysis is stimulated so that the MT is built mainly from GDP-tubulin, held in place by the inter-dimer contacts within the MT wall. As long as MT growth is favoured, GTP-tubulin will be added to the MT end, but when the rate of hydrolysis exceeds the rate of polymerisation—catastrophe—the constraints on the GDP-tubulin held within the lattice are released by depolymerisation. In solutions of pure tubulin, the switches between growth and shrinkage of individual MTs occur at random, a behaviour called dynamic instability. In vivo, MT-associated proteins (MAPs) are employed to control and harness stochastic MT dynamics (Cassimeris and Spittle, 2001), enabling them to be coupled to cellular processes, to act as tracks for molecular motors and to generate force (Inoué and Salmon, 1995; Grischchuk et al, 2005).
Doublecortin (DCX) is the best-described member of a recently discovered and expanding family of MAPs (des Portes et al, 1998; Gleeson et al, 1998, 1999; Francis et al, 1999; Gönczy et al, 2001; Edelman et al, 2005). These proteins are involved in cell division and/or cell migration, but have no sequence or structural homology with other MAPs such as MAP2/tau and XMAP215 (Spittle et al, 2000; Seitz et al, 2002; Kim et al, 2003). Mutations in DCX cause lissencephaly in humans (des Portes et al, 1998; Gleeson et al, 1998), a disease in which the layers of the cerebral cortex are disorganised and mental retardation and epilepsy result. DCX is a neuronal MAP expressed in migrating and differentiating neurons (Francis et al, 1999; Gleeson et al, 1999) and this pattern of expression, together with other data (Horesh et al, 1999; Sapir et al, 2000; Taylor et al, 2000), demonstrates that DCX is centrally involved in organising the MT cytoskeleton and that this function is essential for neuronal migration and differentiation (Kappeler et al, 2006; Koizumi et al, 2006a). DCX-like kinase (DCLK) is also important for neuronal development and has recently been shown to act in coordination with DCX to ensure both accurate cell division to generate neuronal precursors and efficient cell migration (Deuel et al, 2006; Koizumi et al, 2006b; Shu et al, 2006). Thus, a growing body of evidence supports the idea that this family of MAPs plays important roles in many aspects of cytoskeletal regulation and acts through a common mechanism of MT interaction.
DCX and its relatives bind to MTs through an MT-binding domain that is built of two DC (DCX-homology) domains. Lissencephaly-causing mutations cluster within these tandem DC domains, supporting the significance of MT binding for DCX function (Sapir et al, 2000; Taylor et al, 2000; Kim et al, 2003). DCX binds at an unusual site on the MT lattice, between the protofilaments (pfs) from which MTs are built (Moores et al, 2004). This confers specificity for MT architecture so that DCX preferentially stabilises 13pf MTs, its in vivo substrate. How does DCX affect MT organisation, dynamics and function? Answers to these questions are crucial for understanding what aspects of MT function are lost when DCX and its relatives are compromised. We have therefore analysed the properties of MTs in the presence of DCX, revealing novel insights into its essential role in neurons.
Results
DCX is an anti-catastrophe factor
MT growth is essential for cell migration and differentiation (Andersen, 2005). To probe the effects of DCX on MTs, we began by using light microscopy to observe directly the effect of DCX on MT growth and shrinkage. We examined the dynamics of MT plus-ends grown from axonemes at 5 μM tubulin (below the critical concentration) and measured MT growth and shrinkage parameters. Pure tubulin had an average growth rate of +0.66 μm/min (∼1–2 tubulin dimers/pf/s), a catastrophe frequency of 0.12 catastrophes/min and an average depolymerisation rate of 34.40 μm/min (Table I), values that are consistent with those previously observed for pure tubulin (e.g. Dreschel et al, 1992). When increasing concentrations of DCX (1.25, 2.5 and 5 μM) were present, no significant difference in MT growth rate was observed (Student's t-test, P>0.04; Figure 1B and Table I). However, with increasing [DCX], the catastrophe frequency was reduced until at 5 μM DCX, equivalent to a 1:1 ratio of DCX:tubulin, no catastrophes were observed (Figure 1B). At lower DCX concentrations when catastrophes did occasionally occur, the rate of depolymerisation was slowed (Table I). These observations show that DCX does not enhance MT growth rate but instead stabilises MTs by inhibiting depolymerisation.
Table 1.
Dynamic instability parameters of DCX-MTs
| [DCX] (μM) | Average growth rate (μm/min) (mean±s.d.) | No. of growth events | Average depol. rate (μm/min) (mean±s.d.) | No. of catastrophes | No. of rescues | % time growth | % time pause | % time depol. | Total time (min) | # MTs |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.66 (±0.18) | 38 | −34.40 (±6.36) | 25 | 0 | 95 | 3 | 2 | 208 | 25 |
| 1.25 | 0.64 (±0.22) | 23 | −9.36 (±3.40) | 10 | 0 | 89 | 3 | 8 | 122 | 22 |
| 2.5 | 0.58 (±0.12) | 31 | −9.17 (±7.40) | 10 | 4 | 85 | 12 | 4 | 207 | 32 |
| 5 | 0.70 ((±0.19) | 13 | N/A | 0 | N/A | 79 | 21 | 0 | 81 | 14 |
| [Tubulin]=5 μM. | ||||||||||
Figure 1.
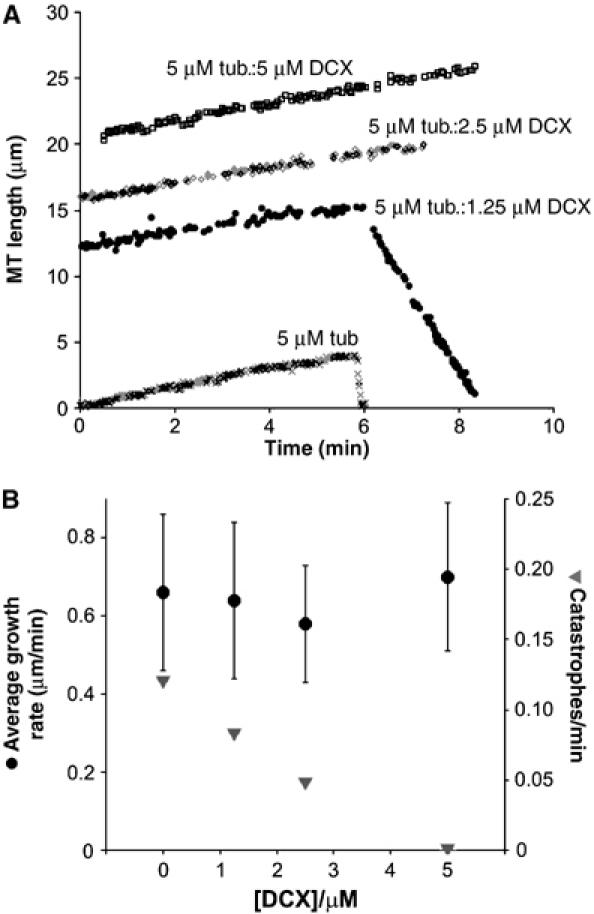
DCX is an anti-catastrophe factor but has no effect on MT growth rate. (A) Representative life histories of individual MT plus-ends grown from axonemes and 5 μM tubulin in the presence of 0, 1.25, 2.5 and 5 μM DCX. The parallel positive gradient of each MT trace illustrates that DCX has no effect on MT growth rate, whereas the difference in negative gradient between two of the traces illustrates that when catastrophes do occur in the presence of DCX, they are slower. The individual growth and shrinkage rates are as follows: 5 μM tubulin (x) +0.68 μm/min, −32.77 μm/min, respectively; 5 μM tubulin:1.25 μM DCX (•) +0.53 μm/min, −5.80 μm/min, respectively; 5 μM tubulin:2.5 μM DCX (◊) +0.58 μm/min; 5 μM tubulin:5 μM DCX (□) +0.64 μm/min. (B) DCX has no effect on the average MT plus-end growth rate (•) but reduces the frequency of catastrophes (▾).
Concentration dependence of MT binding and bundling by DCX
Catastrophe occurs when the GDP-tubulin dimers within the MT wall undergo a structural transition from a strained but polymerised conformation to a bent, depolymerisation-favouring conformation. It is driven by release of mechanical strain stored within the lattice (Caplow et al, 1994; Inoué and Salmon, 1995). As DCX inhibits catastrophe, the energy associated with DCX binding to the MT lattice is likely to balance this stored energy. We therefore wanted to know the affinity with which DCX binds to the MT lattice and we used a cosedimentation assay to estimate this value (Figure 2A).
Figure 2.

DCX MT lattice binding. (A) Cosedimentation assay of DCX with 13pf-enriched MTs. A constant concentration of MTs (2.9 μM polymerised tubulin) was titrated with a range of [DCX], the mixture was centrifuged and the supernatant (s) and pellet (p) fractions were visualised by SDS–PAGE. (B) Binding of DCX to 13pf-enriched MTs is bimodal, one mode of which corresponds to simple decoration with a 1:1 stoichiometry of DCX:tubulin dimer and an apparent affinity of 2 μM (left side of graph), and another in the presence of excess DCX where MT bundles are formed (right side). The curve was fitted to the equation [DCX]bound/[polymerised tubulin]=(stoichiometrymax*[DCX]total)/(Kd+[DCX]total)+[DCX]total/p as described previously (Ackmann et al, 2000). (C) MTs polymerised in the presence of substoichiometric DCX (0.8:1 DCX:tubulin dimer) form individual filaments (i), whereas in the presence of excess DCX (3:2 DCX:tubulin dimer), MT bundles form (ii) and exhibit aggregates on their surface (arrows in iii and iv). Bar=500 Å. (D) GDP-tubulin copolymerised with DCX for 30 min at 37°C forms MTs. Bar=2500 Å. (E) DCX favours polymerisation of 13pf MTs independent of the guanine nucleotide bound to β-tubulin.
We obtained a binding curve for DCX to MTs (Figure 2B), which shows two main features: (1) at lower [DCX], DCX binding appears to approach saturation at ∼1 DCX molecule for every tubulin dimer (≡6 μM DCX); (2) at higher [DCX], the ratio of bound DCX:tubulin rises to well above a 1:1 DCX:tubulin ratio. This type of binding curve was also reported for tau MT binding (Ackmann et al, 2000) and we modelled our binding data similarly, according to saturable, single site binding with an apparent Kd of 2 μM (±0.8 μM), which is masked at higher [DCX] by a linear increase in DCX binding to MTs defined by the overloading parameter (p)=10 μM (see Materials and methods). We also incubated DCX and MTs at a range of [DCX] corresponding to different parts of the binding curve and examined these samples by electron microscopy (EM). We found that MTs incubated with DCX at sub-stoichiometric concentrations (left side of the binding curve) were present as individual filaments (Figure 2Ci). MTs incubated with excess amounts of DCX formed large, electron-dense bundles of MTs, as described previously (Figure 2Cii; Gleeson et al, 1999; Horesh et al, 1999). This is also consistent with the observation that when DCX is overexpressed in cells, the MT cytoskeleton is distorted to form large arrays of MT bundles (Gleeson et al, 1999; Kim et al, 2003). We also saw protein aggregates along and between MTs at these higher [DCX] (Figure 2Ciii and iv), consistent with our modelling, and this is also seen for aggregates of tau (Ackmann et al, 2000). Taken together, these experiments reveal two modes of DCX interaction with MTs: one in which individual DCX molecules interact at well-defined binding sites within the MT lattice, and an additional one in which higher concentrations of DCX are incorporated into MT bundles.
DCX binding balances mechanical strain within the MT lattice
The anti-catastrophe activity of DCX occurs at stoichiometric and sub-stoichiometric ratios of DCX where little or no MT bundling was observed. Therefore, we focused on the left-hand side of the binding curve, corresponding to a 1:1 or less DCX:tubulin ratio with an apparent affinity of DCX for the MT lattice of 2.0 μM, in good agreement with a previous estimate (Francis et al, 1999). This apparent affinity can be estimated as corresponding to a free energy of binding of ∼−56 pN nm/molecule of DCX (ΔG°=−RT ln Kd). By comparing this value with the estimated strain energy stored in the MT lattice (−26 pN nm/tubulin dimer; Inoué and Salmon, 1995) it is apparent that DCX binding could act to oppose the mechanical strain within the lattice of a dynamic MT. In support of the hypothesis that DCX inhibits MT catastrophe by opposing the mechanical strain derived from GDP-tubulin dimers held within the MT wall, we could induce MT polymerisation of GDP-tubulin in the presence of stoichiometric amounts of DCX, when polymerisation would not otherwise occur (Figure 2D).
These GDP-MTs exhibited the preferred 13pf architecture typical of DCX-nucleated MTs (Figure 2E), demonstrating that DCX acts energetically to favour MT polymerisation and spatially to ensure that 13pf MTs are formed. The dominance of this architecture was confirmed when 13pf MTs were found to be the prevalent species when MTs were polymerised in the presence of DCX and the non-hydrolysable GTP analogue, GMPCPP (Figure 2E). Polymerisation of GMPCPP-tubulin shows a strong preference for a 14pf MT architecture (Hyman et al, 1992) presumably through effects on lateral interactions within the lattice. However, DCX overcame this architectural preference, demonstrating that DCX will always stabilise growth of 13pf MTs.
DCX-MTs support kinesin motility
MTs act as structural supports within neurons and are used in force generation during migration; however, they are also the principal tracks for long-distance cellular transport. In particular, the growth cone of a migrating neuron requires a steady stream of cellular components to be channelled to its tip and such transport is performed by plus-end--directed kinesin motors (Guzik and Goldstein, 2004). It is therefore critical to determine whether kinesins can move along DCX-MTs. Although the DCX binding site is in between MT pfs, only one-third of the DCX molecule has been visualised in EM maps (Moores et al, 2004). It therefore seems probable that the rest of the DCX molecule (∼25 kDa) protrudes from the inter pf groove and might be expected to impede kinesin movement. To investigate the effect of DCX-MTs on kinesin transport, we used an ensemble gliding assay to evaluate motility of MTs stabilised by paclitaxel, GMPCPP and DCX using mammalian neuronal kinesin (Figure 3A; Supplementary Movies 1 and 2; Crevel et al, 1997). To our surprise, DCX-MTs display motility in this assay, demonstrating that DCX stabilisation does not block kinesin-dependent transport in the neuron. Recent studies have shown that in DCX/DCLK knockout mice, intracellular transport in neurons is severely compromised (Deuel et al, 2006). This suggests that DCX and its relatives not only support neuronal motility but are also important for providing stable tracks for kinesins in vivo.
Figure 3.

DCX-MTs are substrates for transport and depolymerisation by kinesin motors. (A) Series of frames, separated by 2 s, from movies of in vitro ensemble gliding assays showing mammalian neuronal kinesin-1 moving paclitaxel-stabilised (left frames) or DCX-stabilised (right frames) MTs (see also Supplementary Movies). The trailing end of one MT is marked by an arrow at 0 s and tracked by a dot as it moves between consecutive frames. Bar=5 μm. (B) (i) Graph showing the increase of tubulin released from stabilised MTs as a result of increasing concentrations of the ATP-dependent depolymerising motor pkin13; n=4. (ii) Characteristic pkin13-induced tubulin rings (arrows) were observed by negative stain EM at the ends of DCX-MTs in the presence of the non-hydrolysable analogue AMPPNP. This supports the idea that individual DCX-MTs are substrates for depolymerising kinesins. Bar=500 Å.
Further analysis of the exact speed at which DCX-MTs were moved compared to paclitaxel- and GMPCPP-stabilised MTs revealed that DCX-MTs moved slightly but significantly slower than both MT populations: paclitaxel-MTs=0.64 μm/s±0.071 (mean±s.d.), n=403 MTs; GMPCPP-MTs=0.65 μm/s±0.062, n=417 MTs; DCX-MTs=0.55 μm/s±0.057, n=385 MTs (16% slower, Student's t-test, P<0.0001). This suggests that while DCX does not block the passage of the motor, it hinders kinesin slightly at some point in its ATP-dependent step. The significance of this observation is unclear, but we are undertaking the calculation of a three-dimensional reconstruction of DCX on 13pf MTs that will provide structural insight into this result.
DCX-MTs are substrates for depolymerising kinesins
Kinesin motors are also important in reorganising the MT cytoskeleton and in particular, the MT depolymerising Kif2A (kinesin class 13) is essential during brain development (Homma et al, 2003). We therefore wanted to know the effect DCX stabilisation had on the activity of these depolymerising kinesins. Kinesin-13s in vivo are dimeric but the depolymerisation mechanism of the dimer is unknown; so we decided to use the well-understood (but catalytically less efficient) kinesin-13 motor core (pkin13) to dissect the properties of DCX-MTs (Moores et al, 2002). MTs were incubated with pkin13 and ATP, and the kinesin-13-catalysed release of tubulin from the MTs was evaluated (Figure 3Bi and Supplementary Figure 1). ATP-dependent MT depolymerisation was catalysed by pkin13 from paclitaxel-stabilised MTs as expected. Similarly, pkin13-catalysed depolymerisation was observed in DCX-MTs that were stabilised with 0.5:1 DCX:tubulin. In fact, these MTs appear to be slightly better substrates for the pkin13 depolymerase than paclitaxel-stabilised MTs, but this is probably because paclitaxel-MTs are more protected against stochastic catastrophe than the 0.5:1 DCX-MTs. On the other hand, stoichiometrically stabilised DCX-MTs (i.e. 1:1 DCX:tubulin dimer) were well protected from depolymerisation; however, examination by EM suggested that this protection was afforded by MT bundling under these conditions. In support of the idea that non-bundled DCX-MTs could be depolymerised by pkin13, the tubulin ring structures that are characteristic of pkin13 activity (Moores et al, 2002) were observed at the end of individual DCX-MTs in the presence of pkin13 and the non-hydrolysable ATP analogue AMPPNP (Figure 3Bii and Supplementary Figure 2). This shows that while DCX stabilises neuronal MTs against stochastic catastrophe, the activities of kinesin motors are largely unimpeded, either in their transport or depolymerising activities.
MT nucleation by DCX
Cells employ a number of mechanisms to regulate the temporal and spatial organisation of the MT cytoskeleton in response to environmental cues. These include extension of existing polymers (Brown et al, 1992), transport of pre-existing MTs into a new location (Dent et al, 1999) and growth of new MTs (Vorobjev et al, 1997). The necessity of adopting these different strategies is particularly obvious for neurons because their most distal processes are distant from their MT organising centre (MTOC). At the MTOC, MT nucleation is mediated by large (2.2 MDa), multi-component γ-tubulin ring complexes (γ-TuRCs; Moritz and Agard, 2001). Mounting evidence suggests that γ-TuRCs nucleate MTs by providing a complementary surface from which cellular MTs can grow (Aldaz et al, 2005), with the geometry of the ring complex imposing a 13pf architecture on the nucleated MTs.
With the essential role that MTs play in growth cone extension and directionality, control of new MT growth is of primary concern for migrating neurons (Buck and Zheng, 2002; Andersen, 2005). γ-TuRCs, however, are not present in the extended processes of neurons (Baas and Joshi, 1992); so other mechanisms for MT nucleation must be in operation here. The mechanism of MT nucleation in the absence of a γ-TuRC template is not well understood but must involve two main steps: formation of some kind of nucleation structure followed by elongation (Job et al, 2003).
DCX nucleates 13pf MTs in vitro and in vivo (Horesh et al, 1999; Taylor et al, 2000; Moores et al, 2004) and this property is likely to be an important aspect of the role of DCX in migrating and differentiating neurons, particularly because of its specific enrichment at their extremities (Supplementary Figure 3; Francis et al, 1999; Friocourt et al, 2003; Schaar et al, 2004). The potent nucleation activity of DCX is illustrated in Figure 4A. When 10 μM tubulin alone was polymerised at 37°C, a few very long MTs were observed against a high background of unpolymerised tubulin (Figure 4A, left). In contrast, when stoichiometric amounts of DCX were coincubated with 10 μM tubulin, many MTs were observed (Figure 4A, right). Nucleation was also seen at 5 μM DCX versus 10 μM tubulin, but the background of unpolymerised tubulin was higher (data not shown).
Figure 4.
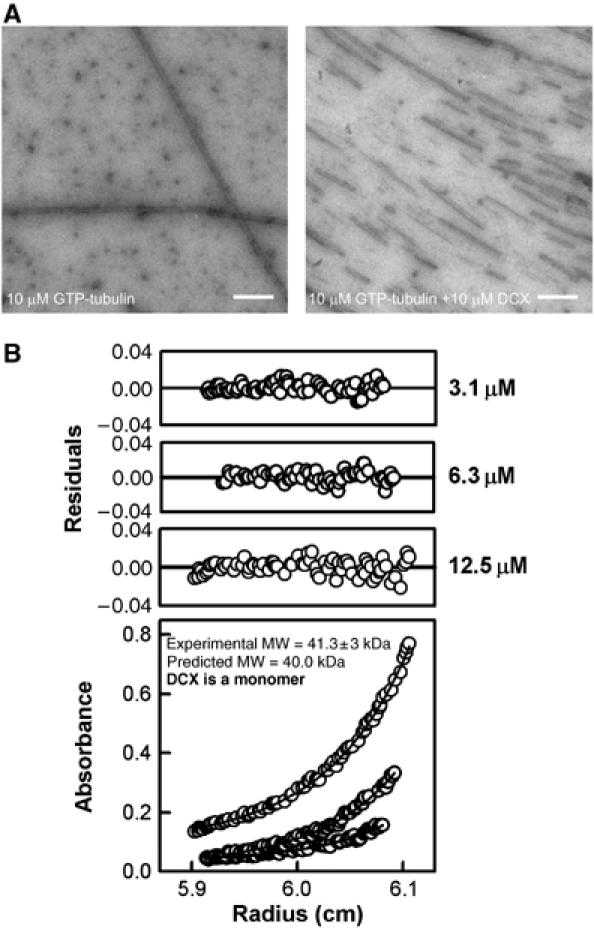
MT nucleation by monomeric DCX. (A) Electron micrographs of 10 μM tubulin polymerised in the absence (left) and presence (right) of DCX, imaged 5 min after polymerisation was initiated. Bar=5000 Å. (B) Absorbance data, curve fits (bottom panel) and residual of the fits (top panels) from the analytical ultracentrifugation experiment with 3.1, 6.3 and 12.5 μM DCX centrifuged at 20 000 r.p.m. The curve fits show that DCX is predominantly monomeric.
By analogy with the proposed mechanism for MT nucleation by γ-TuRCs, DCX, a 40 kDa protein, could form a large oligomeric structure that acts as a template for 13pf MTs. We used equilibrium analytical ultracentrifugation to examine the oligomeric state of DCX at concentrations and in buffers where MT nucleation is promoted. We observed that DCX behaved predominantly as a monomer at three different concentrations (3.1, 6.3 and 12.5 μM; Figure 4B), suggesting that DCX, unlike the γ-TuRC, does not nucleate MTs by itself oligomerising to form a template. Instead, a DCX monomer must favour nucleation through its interaction with multiple tubulin dimers (Taylor et al, 2000; Kim et al, 2003). Thus, DCX's mechanism of MT nucleation is distinct from that of γ-TuRCs.
To dissect the mechanism of DCX MT nucleation further, we examined the interaction of DCX with dimeric tubulin using size-exclusion chromatography. Individually, DCX and dimeric tubulin migrated as discrete species (Figure 5A; dimeric kinesin-1 is included for comparison). A mixture of DCX and tubulin showed no change in elution volume, suggesting that a co-complex between DCX and dimeric tubulin (which would be faster eluting) does not form (Figure 5B). To confirm this, we pre-incubated tubulin with dimeric kinesin-1 and AMPPNP to form a larger complex (Figure 5B). As our motility assay demonstrated that DCX does not significantly interfere with the kinesin–tubulin interaction, DCX should co-migrate with the kinesin–tubulin complex if it can bind individual tubulin dimers. However, DCX eluted in later fractions than the kinesin–tubulin complex (Figure 5B), confirming that DCX does not bind individual tubulin dimers. One possible explanation for our results is that the conditions of our gel filtration experiment (low temperature, presence of kinesin-1) prevented us from observing the interaction between tubulin heterodimers and DCX. However, given that (1) DCX did not block kinesin-1 motility (Figure 3A) and (2) the protein concentrations used were twice that at which substantial MT nucleation is observed (Figure 5C), we believe that these gel filtration experiments provide evidence that DCX cannot bind single tubulin dimers.
Figure 5.

DCX does not bind dimeric tubulin but nucleates MTs. (A) Size-exclusion chromatography trace showing the migration of dimeric tubulin, DCX and dimeric kinesin-1, which were run separately down a Superose-12 column. SDS–PAGE of the relevant fractions is shown above the trace. (B) Size-exclusion chromatography trace and SDS–PAGE analysis of co-complexes of DCX-tubulin, tubulin+kinesin and tubulin+kinesin+DCX, demonstrating that whereas kinesin binds dimeric tubulin and shifts its migration profile, DCX and tubulin do not interact, either as a bi-complex or as a tri-complex in the presence of kinesin. (C) Electron micrograph of 5 μM tubulin polymerised in the presence of 5 μM DCX imaged 5 min after polymerisation was initiated. The asterisk and arrows indicate a fully polymerised MT and small DCX-induced tubulin sheets, respectively. Bar=1000 Å.
If DCX does not interact with individual tubulin dimers and yet strongly nucleates MTs, it must act by stabilising early nucleation structures. We examined the early products of MT polymerisation of 5 μM tubulin/5 μM DCX (below the tubulin critical concentration under these experimental conditions). As shown in Figure 5C, this mixture contained fully formed MTs (labelled *) and also many small sheets of pfs (arrows). These sheets display variable dimensions, with the shortest visible structure being just a few tubulin dimers in length and the longest being more than 20 dimers long. A range of pf number is also observed in these structures but the average pf spacing is always 50–55 Å, which is the spacing seen in MTs (Desai and Mitchison, 1997). Following longer incubation times, MTs and not sheets are observed (data not shown), suggesting that they are the building blocks for the MTs that ultimately form. Given that DCX binds between the pfs from which MTs are built (Moores et al, 2004) and that this binding site is only created when tubulin dimers form lateral contacts, it is extremely likely that DCX favours MT nucleation principally by stabilising lateral contacts in the incipient MT wall. We studied the architecture of DCX-nucleated MTs by EM every 5 min for 30 min and observed that the proportion of 13pf MTs did not change over time (>98%; results of four experiments, data not shown). This observation is consistent with our hypothesis that DCX directs the nucleation of 13pf MTs as opposed to preferentially stabilising them. The imposition of the 13pf MT architecture by DCX supports the dominance of lateral stabilisation in this nucleation mechanism.
Discussion
MTs are essential for neuronal function, as are the cellular regulators of their dynamics (Feng and Walsh, 2001; Guzik and Goldstein, 2004). DCX cannot be compensated for by other MT-stabilising proteins such as tau and MAP2 (Teng et al, 2001) and we set out to understand the unique features of DCX that are essential for neuronal migration and differentiation.
Regulation of MT dynamics
Our findings show that DCX has no effect on MT growth rate and saturates MTs with ∼1:1 stoichiometry and an apparent affinity of ∼2 μM (Figures 1 and 2). In contrast, MAP1, MAP2 and their relatives enhance MT growth rate (Dreschel et al, 1992) and have a lower binding stoichiometry on, and tighter binding affinity for, MTs (tau Kd=0.075 μM, Ackmann et al, 2000; MAP2 Kd=0.3 μM, Roger et al, 2004). Elongated MAPs such as MAP2 have multiple points of contact with the MT lattice and this allows them to both stabilise MTs at lower stoichiometries and to capture free tubulin dimers from solution to enhance MT growth rates. A similar mechanism of growth rate enhancement has also been postulated for DCX (Kim et al, 2003), but our data demonstrate that DCX simply stabilises the MT lattice (Figure 6). The inability of DCX to bind dimeric tubulin is consistent with this observation (Figure 5A), supporting the idea that DCX reduces the off-rate of tubulin at MT ends while having no effect on the on-rate.
Figure 6.

A model for the effects of DCX on MT function. DCX does not bind individual tubulin dimers but stabilises the tubulin–tubulin lateral contacts in tubulin oligomers, thereby mediating nucleation in the early stages of MT polymerisation (left). DCX stabilises the fully polymerised MT lattice (viewed from the end) against spontaneous catastrophe by counteracting the outward-directed forces of MT depolymerisation (right) that disrupt inter-pf lateral contacts. It does not, however, greatly inhibit kinesin motors involved in either cargo movement or MT depolymerisation.
At super-stoichiometric ratios, DCX induces formation of large MT bundles (Gleeson et al, 1999; Horesh et al, 1999; Kim et al, 2003). As our data also show that DCX is predominantly monomeric (Figure 4B), MT bundling presumably occurs as a result of weak interactions at the MT surface when excess DCX is present (Figure 2C). However, none of the effects that we observed on MT polymerisation required excess DCX, suggesting that MT bundling by DCX is unlikely to occur naturally in neurons. Indeed, MT bundling by tau is associated with formation of pathological aggregates (Ackmann et al, 2000), implying that the balance between under- and overexpression of regulators of the neuronal MT cytoskeleton is very finely tuned.
DCX-MTs are tracks for molecular motors
As well as regulating the dynamics of cellular MTs, DCX-MTs also support motility of the plus-end-directed kinesins that are functionally critical in neurons (Guzik and Goldstein, 2004; Stokin et al, 2005). Removal of the MT-stabilising properties of DCX and its relative DCLK results in severe disruption of these transport processes in neurons (Deuel et al, 2006), reinforcing the idea that the properties imposed on MT tracks by DCX are an essential part of their function in neurons (Figure 6). Kinesin motility was slowed by 16% on DCX-MTs compared to undecorated MTs, presumably because part of DCX interferes slightly with the kinesin step. This is probably the C-terminus of DCX available for interaction with other cellular binding partners (Kizhatil et al, 2002; Friocourt et al, 2005).
Among neuronal MAPs, the different MT binding sites of DCX compared with tau/MAP2 on MTs predict that these stabilising proteins would have different effects on kinesin-mediated transport (Al-Bassam et al, 2002; Moores et al, 2004). This is exactly what we observed, because while kinesin motility on DCX-MTs was slightly slower than on paclitaxel-MTs, tau and MAP2 demonstrate a range of effects on both kinesin speed and the dynamics of motor attachment (Heins et al, 1991; Trinczek et al, 1999; Seitz et al, 2002).
Depolymerising kinesins and MT remodelling
In addition to acting as tracks for transport, the MT cytoskeleton must also be altered in response to extracellular cues. The role of Kif2 (kinesin-13) motors during brain development was recently demonstrated (Homma et al, 2003). Individual DCX-MTs appear to be substrates for the depolymerisation activity of these motors, but in contrast, tau has been shown to counteract kinesin-13 MT depolymerisation in cell extracts (Noetzel et al, 2005), supporting the idea that these two MAPs interact with their substrate in very different ways. Because of the central role that MTs play during neuronal migration and development, regulation of the balance of MT polymerisation and depolymerisation is essential. DCX allows MT growth and stabilisation without interfering with other critical cytoskeletal reorganisations (Figure 6).
MAPs and MT nucleation
The best-understood mechanism of MT nucleation is by γ-TuRCs (Moritz and Agard, 2001) that act as a template to a population of cellular MTs with a uniform 13pf architecture. However, cells may need very localised new MT growth at sites where γ-TuRCs are not found, for example in response to specific local cues during cell migration. DCX nucleates MTs not by binding individual tubulin dimers but by stabilising small, oligomeric tubulin nuclei, a mechanism that is consistent with current models of tubulin nucleation (Buey et al, 2006; Figure 6, left). Previous work described the ability of DCX to bind dimeric tubulin (Taylor et al, 2000; Kim et al, 2003) but the techniques used in these experiments could not accurately discriminate between single tubulin dimers and small clusters, as our size-exclusion chromatography experiments could.
Although other MAPs also have MT nucleation activity, none appears to direct specific formation of 13pf MTs (Arnal et al, 2004; Santarella et al, 2004). Both longitudinal and lateral tubulin contacts are essential for MT polymerisation, but the observation that stabilisation of longitudinal contacts by certain MAPs does not guarantee MT formation (Krebs et al, 2004; Santarella et al, 2004), together with the precision with which DCX defines MT architecture independent of the guanine nucleotide bound to the tubulin (Figure 2E), supports the idea that stabilisation of lateral contacts is the dominant mechanism in MT nucleation by DCX (Figure 6).
Tubulin has been shown to polymerise in vitro via the formation of long, wide tubulin sheets that close to form complete cylindrical MTs (Chretien et al, 1995). Sheet closure was suggested to be the limiting step in tubulin polymerisation. In contrast, only small tubulin sheets are seen in the presence of DCX, quickly followed by the formation of 13pf MTs. This latter MT polymerisation mechanism would be advantageous for the neuron and would bypass the rate-limiting step of large sheet closure seen with pure tubulin. It also removes the considerable steric problem of rolling up the elongated tubulin sheets (up to ∼2 μm in length) within the confined space of the neuronal process. DCX-induced polymerisation thus allows ready formation of complete MTs with the appropriate architecture.
Could DCX-rich zones be factories for stable MTs? As has previously been shown (Friocourt et al, 2003; Schaar et al, 2004; Supplementary Figure 3), the localisation of DCX in developing neurons is very tightly controlled and enrichment of DCX is seen in distal processes where γ-TuRCs are not found (Baas and Joshi, 1992). An intriguing possibility, therefore, is that these regions of DCX enrichment may be specialised sites for nucleation and stabilisation of 13pf, centrosome-independent MTs in neurons, acting in response to external guidance signals (Gdalyuhu et al, 2004; Schaar et al, 2004; Tanaka et al, 2004). Further work will be necessary to explore this possibility.
The cell employs myriad cellular factors to modulate the intrinsic dynamic properties of the MT cytoskeleton according to cellular circumstance. Our characterisation of the properties of DCX provides insight into the regulatory mechanisms that neurons employ. Very precise and subtle tuning of expression, location and function are essential and it is clear that incorrect regulation can be devastating for human health.
Materials and methods
DCX preparation and MT polymerisation
Full-length DCX was expressed and purified as described previously (Moores et al, 2004). Before use, DCX was dialysed against 80 mM Pipes, pH 6.8, 2 mM MgCl2, 1 mM EGTA (BrB80)+1 mM TCEP and clarified by centrifugation. Bovine brain tubulin and human MCAK motor domain (kinesin-13) were purchased from Cytoskeleton Inc. DCX-MTS were polymerised at 37°C in 80 mM Pipes, pH 6.8, 1 mM MgCl2, 8% DMSO and 2.5 mM GTP, GDP or GMPCPP (Jena Bioscience GmbH, Jena, Germany). Paclitaxel-stabilised MTs were polymerised in the same buffer at 5 mg/ml tubulin for 30 min, after which 1 mM paclitaxel (Cal-Biochem) dissolved in DMSO was added. To prepare GMPCPP-stabilised MTs, GMPCPP was added instead of GTP and no paclitaxel was required.
MT dynamics assays
MT dynamics assays were performed essentially as described previously (Walker et al, 1988). Coverslips and slides were cleaned using the method described at www.technicalvideo.com/Products/CCP.html for both MT dynamics assays and motility assays (below). Purified fragments of sea urchin sperm axonemes in BrB80, 1 mM GTP and 1 mM DTT were bound to the coverslip of an ∼12 μl flow cell and were incubated at room temperature for 5 min to ensure binding to the coverslip surface. The flow cell was rinsed with BrB80/GTP/DTT and 5 μM tubulin, 1 mM acetyl phosphate+1 U/μl acetate kinase (to ensure constant [GTP]) in BrB80/GTP/DTT and a range of [DCX] was added to the flow cell, which was then sealed. MT plus-end dynamics were observed using video-enhanced differential interference contrast microscopy on microscope stage warmed to 37°C. Images were digitally background subtracted and contrast enhanced and two-frame averaged in real time using a Hammamatsu Argus 10 and were imported into the freeware package NIH Image (http://rsb.info.nih.gov/nih-image/Default.html). A custom-written macro was used to digitise the position of the MT end (Drummond and Cross, 2000; http://mc11.mcri.ac.uk/retrac.html).
Cosedimentation assay to determine affinity of DCX for MTs
DCX preferentially binds 13pf MTs (Moores et al, 2004); so to determine the affinity of DCX for this MT architecture, MTs were polymerised in the presence of sea urchin sperm axonemes in MES buffer (100 mM MES, 1 mM MgCl2, 1 mM EGTA, pH 6.5) to favour growth of 13pf MTs (Ray et al, 1993). 8 μM tubulin was polymerised with 1.6 μg/ml axonemes under these conditions at 37°C for 1 h, stabilised by addition of 1 mM paclitaxel and left at room temperature overnight. These MTs were then pelleted and resuspended in BrB80 ready for use in the cosedimentation assay. The extent of 13pf enrichment was evaluated by EM: ∼70% of the MTs had 13pf architecture. The cosedimentation assay was performed as described previously (Moores et al, 2003), such that a range of [DCX] was titrated against a fixed [MT] (2.9 μM polymerised tubulin). These mixtures were incubated in BrB80 at room temperature for 20 min, the supernatant (free protein) and pellet (MTs and bound protein) fractions were separated by centrifugation at 400 000 g for 15 min, were analysed by SDS–PAGE and the protein bands in each fraction were visualised by Coomassie blue. The intensity of the gel bands was quantitated and the proportions of DCX and tubulin in each fraction were calculated. Several controls were incorporated into these calculations including background corrections, the amount of DCX that sediments into the pellet in the absence of MTs (10%) and the amount of protein that sediments owing to nonspecific trapping by MTs (13% calculated using BSA). We checked if the low concentration of axonemal protein alone could cause DCX to sediment in the assay, but found that the proportion of DCX found in the pellet in the presence of axonemes was the same as that seen for DCX alone (data not shown). This allowed us to conclude that our calculated binding curve is a consequence of the DCX–MT interaction alone. The previously described biphasic analysis (Ackmann et al, 2000) was used to fit the binding curve using the equation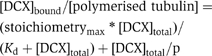 where n is the binding stoichiometry, Kd is the apparent dissociation constant and p is the so-called overloading parameter.
where n is the binding stoichiometry, Kd is the apparent dissociation constant and p is the so-called overloading parameter.
The cosedimentation assay was also used to evaluate MT depolymerisation by pkin13 (a kind gift from Roman Sakowicz, Cytokinetics, South San Francisco, prepared as described previously; Moores et al, 2002). A 4 μM portion of paclitaxel-stabilised MTs or DCX-MTs was incubated with a range of pkin13 (25 nM–0.4 μM) with 0.5 mM MgATP at room temperature for 30 min. The incubations were subjected to centrifugation and the supernatant and pellet fractions were analysed using SDS–PAGE, as described above. To observe pkin13-AMPPNP-induced tubulin rings at the ends, DCX-MTs were incubated with 1 μM pkin13 and 1 mM AMPPNP for 10 min and the incubation mixture was placed on an EM grid.
Electron microscopy
MTs were adsorbed to carbon-coated grids and were negatively stained with 1% uranyl acetate. Micrographs were taken on an FEI Tecnai T10 EM at 100 kV or on an FEI Tecnai F20 at 200 kV. Preparation of cryo-EM samples and performance of cryo-EM for evaluating the pf number of MTs were performed as described previously (Moores et al, 2004).
Motility assays
Motility assays were performed as described (Crevel et al, 1997) using a dimeric rat kinesin-1 construct (residues 1–430) with a C-terminal GST tag. 0.8 μM kinesin motor was bound to the coverslip of an ∼12 μl flow cell in BrB80, 5 mM DTT and 0.1 mg/ml casein (Sigma) and was incubated at room temperature for 5 min to ensure saturation of the coverslip surface by the motor. The flow cell was rinsed with BrB80/DTT. Stabilised MTs diluted to ∼1 μM polymerised tubulin in BrB80/DTT, 2 mM ATP, 10 mM phosphocreatine and 50 μg/ml creatine phosphokinase (added to ensure that [ATP] was not limiting) were added to the flow cell. MT movement was observed using video-enhanced differential interference contrast microscopy and digitally processed as above. The freeware package RETRAC (by Nick Carter, http://mc11.mcri.ac.uk/Retrac) was used to measure the rates of MT sliding by tracking the trailing end of the MTs. The data presented are averages of five different recordings from at least five separate slides with at least 10 MTs analysed per recording.
Analytical ultracentrifugation
Ultracentrifugation runs were performed at 20°C at 14 000, 20 000 and 25 000 r.p.m. in a Beckman Optima XL-I analytical ultracentrifuge equipped with both interference and absorbance optics. The starting sample concentrations were monitored using their absorbance at 280 nm. Three concentrations of DCX were used (3.1, 6.3 and 12.5 μM); sample volumes of 0.1 ml and buffer volumes of 0.11 ml were loaded into six-sector cells and the equilibrium run lasted 90 h. The sedimentation curves were fitted by performing analyses of individual curves at equilibrium using the self-association mathematical model based on a single species, using Beckman software provided as an add-on in ORIGIN V4.1 (MicroCal Software Inc.). The partial specific volume was calculated from the sequence to be 0.7308 ml/g and the buffer density was calculated as 1.00067 g/ml using the programme SEDNTERP (Laue et al, 1992).
Size-exclusion chromatography
Combinations of 10 μM dimeric GTP-tubulin, 10 μM DCX and 10 μM dimeric kinesin-1 (rat kinesin-1, residues 1–430) with 2 mM AMPPNP were incubated for 10 min at 4°C before loading onto a Superose-12 size-exclusion column (Amersham Biosciences) pre-equilibrated with BrB80 at 4°C, and were eluted in 0.3 ml fractions. Fractions were evaluated by SDS–PAGE.
Supplementary Material
Supplementary Data, Movies 1
Supplementary Data, Movies 2
Supplementary Material
Acknowledgments
CAM is a BBSRC David Phillips Fellow and gratefully acknowledges a grant from the University of London Central Research Fund and insightful discussions with Ronald Milligan (The Scripps Research Institute). This work was also supported by grants from the European Commission (QLG3-CT-2000-00158), INSERM, CNRS, the Fédération pour la Recherche sur le Cerveau (AH and FF), the Fondation pour la Recherche Médicale (20000293/3-INE) and the Association pour la Recherche sur le Cancer (ARC 5505) (AH). SJP thanks the Wellcome Trust and BBSRC for analytical ultracentrifugation equipment and Jayesh Gor for his assistance. CK and FF thank Pierre Bourdoncle for discussions concerning fluorescence intensity quantifications. We would like to thank Maria Alonso for technical assistance and other members of the Cross lab for useful discussions.
References
- Ackmann M, Wiech H, Mandelkow E (2000) Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament-like conformation. J Biol Chem 275: 30335–30343 [DOI] [PubMed] [Google Scholar]
- Al-Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA (2002) MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol 157: 1187–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldaz H, Rice LM, Stearns T, Agard DA (2005) Insights into microtubule nucleation from the crystal structure of human γ-tubulin. Nature 435: 523–527 [DOI] [PubMed] [Google Scholar]
- Andersen SSL (2005) The search and prime hypothesis for growth cone turning. BioEssays 27: 86–90 [DOI] [PubMed] [Google Scholar]
- Arnal I, Heichette C, Diamantopoulos GS, Chretien D (2004) CLIP-170/tubulin-curved oligomers coassemble at microtubule ends and promote rescues. Curr Biol 14: 2086–2095 [DOI] [PubMed] [Google Scholar]
- Baas PW, Joshi HC (1992) γ-tubulin distribution in the neuron: Implications for the origins of neurite microtubules. J Cell Biol 119: 171–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Slaughter T, Black MM (1992) Newly assembled microtubules are concentrated in the proximal and distal regions of growing axons. J Cell Biol 119: 867–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck KB, Zheng JQ (2002) Growth cone turning induced by direct local modification of microtubule dynamics. J Neurosci 22: 9358–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buey RM, Diaz JF, Andreu JM (2006) The nucleotide switch of tubulin and microtubule assembly: a polymerization-driven structural change. Biochemistry 45: 5933–5938 [DOI] [PubMed] [Google Scholar]
- Caplow M, Ruhlen RL, Shanks J (1994) The free energy for hydrolysis of a microtubule-bound nucleotide triphosphate is near zero: all of the free energy for hydrolysis is stored in the microtubule lattice. J Cell Biol 127: 779–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassimeris L, Spittle C (2001) Regulation of microtubule-associated proteins. Int Rev Cytol 210: 163–226 [DOI] [PubMed] [Google Scholar]
- Chretien D, Fuller SD, Karsenti E (1995) Structure of growing microtubule ends: two-dimensional sheets close into tubes at variable rates. J Cell Biol 129: 1311–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevel IM-TC, Lockhart A, Cross RA (1997) Kinetic evidence for low chemical processivity in ncd and Eg5. J Mol Biol 273: 160–170 [DOI] [PubMed] [Google Scholar]
- Dent EW, Callaway JL, Szebenyi G, Baas PW, Kalil K (1999) Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J Neurosci 19: 8894–8908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrié A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J (1998) A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92: 51–61 [DOI] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ (1997) Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13: 83–117 [DOI] [PubMed] [Google Scholar]
- Deuel TAS, Liu JS, Corbo JC, Yoo S-Y, Rorke-Adams LB, Walsh CA (2006) Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron 49: 41–53 [DOI] [PubMed] [Google Scholar]
- Dreschel DN, Hyman AA, Cobb MH, Kirschner MW (1992) Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 3: 1141–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DR, Cross RA (2000) Dynamics of interphase microtubules in Schizosaccharomyces pombe. Curr Biol 10: 766–775 [DOI] [PubMed] [Google Scholar]
- Edelman AM, Kim W-Y, Higgins D, Goldstein EG, Oberdoerster M, Sigurdson W (2005) Doublecortin kinase-2, a novel doublecortin-related protein kinase associated with terminal segments of axons and dendrites. J Biol Chem 280: 8531–8543 [DOI] [PubMed] [Google Scholar]
- Feng Y, Walsh CA (2001) Protein–protein interactions, cytoskeletal regulation and neuronal migration. Nat Rev Neurosci 2: 408–416 [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet M-C, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell S, Berwald-Netter Y, Denoulet P, Chelly J (1999) Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23: 247–256 [DOI] [PubMed] [Google Scholar]
- Friocourt G, Kappeler C, Sailloui Y, Fauchereau F, Rodriguez MS, Bahi N, Vinet MC, Chafey P, Poirier K, Taya S, Wood SA, Dargemont C, Francis F, Chelly J (2005) Doublecortin interacts with the ubiquitin protease DFFRX, which associated with microtubules in neuronal processes. Mol Cell Neurosci 28: 153–164 [DOI] [PubMed] [Google Scholar]
- Friocourt G, Koulakoff A, Chafey P, Boucher D, Fauchereau F, Chelly J, Francis F (2003) Doublecortin functions at the extremeties of growing neuronal processes. Cereb Cortex 13: 620–626 [DOI] [PubMed] [Google Scholar]
- Gdalyuhu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, Reiner O (2004) DCX, a new mediator of the JNK pathway. EMBO J 23: 823–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Copper EC, Dobyns WB, Minnerath SR, Ross EM, Walsh CA (1998) doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92: 63–72 [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh C (1999) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23: 257–271 [DOI] [PubMed] [Google Scholar]
- Gönczy P, Bellanger JM, Kirkham M, Pozniakowski A, Baumer K, Phillips JB, Hyman AA (2001) zyg-8, a gene required for spindle positioning in C. elegans, encodes a doublecortin-related kinase that promotes microtubule assembly. Dev Cell 1: 363–375 [DOI] [PubMed] [Google Scholar]
- Grischchuk EL, Molodtsov MI, Ataullakhanov FI, McIntosh JR (2005) Force production by disassembling microtubules. Nature 438: 384–388 [DOI] [PubMed] [Google Scholar]
- Guzik BW, Goldstein LS (2004) Microtubule-dependent transport in neurons: steps towards an understanding of regulation, function and dysfunction. Curr Opin Cell Biol 16: 443–450 [DOI] [PubMed] [Google Scholar]
- Heins S, Song YH, Wille H, Mandelkow E, Mandelkow EM (1991) Effect of MAP2, MAP2c and tau on kinesin-dependent microtubule motility. J Cell Sci Suppl 14: 121–124 [DOI] [PubMed] [Google Scholar]
- Homma N, Takei Y, Tanaka Y, Nakata T, Tereda S, Kikkawa M, Noda Y, Hirokawa N (2003) Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell 114: 229–239 [DOI] [PubMed] [Google Scholar]
- Horesh D, Sapir T, Francis F, Wolf SG, Caspi M, Elbaum M, Chelly J, Reiner O (1999) Doublecortin, a stabilizer of microtubules. Hum Mol Genet 8: 1599–1610 [DOI] [PubMed] [Google Scholar]
- Hyman AA, Salser S, Drechsel DN, Unwin N, Mitchison TJ (1992) Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolysable analogue, GMPCPP. Mol Biol Cell 3: 1155–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoué S, Salmon ED (1995) Force generation by microtubule assembly/disassembly in mitosis and related movements. Mol Biol Cell 6: 1619–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job D, Valiron O, Oakley B (2003) Microtubule nucleation. Curr Opin Cell Biol 15: 111–117 [DOI] [PubMed] [Google Scholar]
- Kappeler C, Saillour Y, Baudoin J-P, Tuy FPD, Alvarez C, Houbron C, Gaspar P, Hamard G, Chelly J, Métin C, Francis F (2006) Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum Mol Genet 15: 1387–1400 [DOI] [PubMed] [Google Scholar]
- Kim MH, Cierpicki T, Derewenda U, Krowarsch D, Feng Y, Devedjiev Y, Dauter Z, Walsh CA, Otlewski J, Bushweller JH, Derewenda ZS (2003) The DCX-domain tandem of doublecortin and doublecortin-like kinase. Nat Struct Biol 10: 324–333 [DOI] [PubMed] [Google Scholar]
- Kizhatil K, Wu Y-X, Sen A, Bennett V (2002) A new activity of doublecortin in recognition of the phospho-FIGQY tyrosine in the cytoplasmic domain of neurofascin. J Neurosci 22: 7948–7958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi H, Higginbotham H, Poon T, Tanaka T, Brinkman BC, Gleeson JG (2006a) Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat Neurosci 9: 779–786 [DOI] [PubMed] [Google Scholar]
- Koizumi H, Tanaka T, Glesson JG (2006b) doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron 49: 55–66 [DOI] [PubMed] [Google Scholar]
- Krebs A, Goldie KN, Hoenger A (2004) Complex formation with kinesin motor domains affects the structure of microtubules. J Mol Biol 335: 139–153 [DOI] [PubMed] [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM, Pelletier SL (1992) Computer-aided interpretation of analytical sedimentation data for proteins. In Harding SE, Rowe AJ, Horton JC (eds) pp 90–125. Cambridge: The Royal Society of Chemistry
- Moores CA, Hekmat-Nejad M, Sakowicz R, Milligan RA (2003) Regulation of KinI kinesin ATPase activity by binding to the microtubule lattice. J Cell Biol 163: 963–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moores CA, Perderiset M, Francis F, Chelly J, Houdusse A, Milligan RA (2004) Mechanism of microtubule stabilisation by doublecortin. Mol Cell 18: 833–839 [DOI] [PubMed] [Google Scholar]
- Moores CA, Yu M, Guo J, Beraud C, Sakowicz R, Milligan RA (2002) A mechanism for microtubule depolymerisation by KinI kinesins. Mol Cell 9: 903–909 [DOI] [PubMed] [Google Scholar]
- Moritz M, Agard DA (2001) Gamma-tubulin complexes and microtubule nucleation. Curr Opin Struct Biol 11: 174–181 [DOI] [PubMed] [Google Scholar]
- Noetzel TL, Dreschel DN, Hyman AA, Kinoshita K (2005) A comparison of the ability of XMAP215 and tau to inhibit the microtubule destabilizing activity of XKCM1. Philos Trans R Soc 360: 591–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli RBG, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M (2004) Insight into tubulin regulation from a complex with colchicines and a stathmin-like domain. Nature 428: 198–202 [DOI] [PubMed] [Google Scholar]
- Ray S, Meyhofer E, Milligan RA, Howard J (1993) Kinesin follows the microtubule's protofilament axis. J Cell Biol 121: 1083–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger B, Al-Bassam J, Dehmalt L, Milligan RA, Halpain S (2004) MAP2c, but not tau, binds and bundles F-actin via its microtubule binding domain. Curr Biol 14: 363–371 [DOI] [PubMed] [Google Scholar]
- Santarella RA, Skiniotis G, Goldie KN, Tittmann P, Gross H, Mandelkow E-M, Mandelkow E, Hoenger A (2004) Surface-decoration of microtubules by human tau. J Mol Biol 339: 539–553 [DOI] [PubMed] [Google Scholar]
- Sapir T, Horesh D, Caspi M, Atlas R, Burgess HA, Wolf SG, Francis F, Chelly J, Elbaum M, Pietrokovski S, Reiner O (2000) Doublecortin mutations cluster in evolutionarily conserved functional domains. Hum Mol Genet 9: 703–712 [DOI] [PubMed] [Google Scholar]
- Schaar BT, Kinoshita K, McConnell S (2004) Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron 41: 203–213 [DOI] [PubMed] [Google Scholar]
- Seitz A, Kojima H, Oiwa K, Mandelkow E-M, Song Y-H, Mandelkow E (2002) Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J 21: 4896–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Tseng H-C, Sapir T, Stern P, Zhou Y, Sanada K, Fischer A, Coquelin FM, Reiner O, Tsai L-H (2006) Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron 49: 25–39 [DOI] [PubMed] [Google Scholar]
- Spittle C, Charrasse S, Larroque C, Cassimeris L (2000) The interaction of TOGp with microtubules and tubulin. J Biol Chem 275: 20748–20753 [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LSB (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307: 1282–1288 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Tseng H-C, Kulkarni AB, Tsai L-H, Gleeson JG (2004) Cdk5 phosphorylation of doublecortin ser297 regulates its effect on neuronal migration. Neuron 41: 215–227 [DOI] [PubMed] [Google Scholar]
- Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG (2000) Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem 275: 34442–34450 [DOI] [PubMed] [Google Scholar]
- Teng J, Takei Y, Harada A, Nakata T, Chen J, Hirokawa N (2001) Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J Cell Biol 155: 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E (1999) Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles. J Cell Sci 112: 2355–2367 [DOI] [PubMed] [Google Scholar]
- Vorobjev IA, Svitkina TM, Borisy GG (1997) Cytoplasmic assembly of microtubules in cultured cells. J Cell Sci 110: 2635–3645 [DOI] [PubMed] [Google Scholar]
- Walker RA, O'Brien ET, Pryer NK, Soboeiro MF, Voter WA, Erickson HP, Salmon ED (1988) Dynamic instability of individual microtubules analysed by video light microscopy: rate constants and transition frequencies. J Cell Biol 107: 1437–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data, Movies 1
Supplementary Data, Movies 2
Supplementary Material