

Abstract
Cancer therapies using genes and other macromolecules might realize their full clinical potential if they could be delivered to tumor tissue in optimal quantities. Unfortunately, the compromised circulation within tumors poses a formidable resistance to adequate and uniform penetration of these agents. Previously, we have proposed elevated interstitial fluid pressure (IFP) as a major physiological barrier to delivery of macromolecules. Here we postulate that modulation of tumor microvascular pressure (MVP) and associated changes in IFP would enhance macromolecular delivery into a solid tumor. To test our hypothesis, we altered tumor MVP by either periodic injection or continuous infusion of angiotensin II (AII) and measured the resulting changes in IFP and uptake of macromolecules. We used the nicotinyl hydrazine derivative of human polyclonal IgG (HYNIC-IgG) as a nonspecific macromolecule and CC49 antibody as a specific macromolecule. We found that both chronic and periodic modulation of tumor MVP enhances transvascular fluid filtration, leading to a 40% increase in total uptake of the specific antibody within 4 hr of its administration. Conversely, neither continuous nor periodic infusion of AII induced any increase in uptake of nonspecific antibodies. Strategies to improve delivery of macromolecules and limitations of this approach are identified.
The unprecedented advances in genetic engineering and hybridoma technology have led to the development and use of various high molecular weight agents for cancer detection and treatment. These agents include DNA and its fragments, growth factors, immunomodulators, and mAbs. However, inadequate delivery of these agents into tumor tissue represents the major obstacle for their use in cancer therapy (1–4). Therefore, novel strategies are needed to improve delivery of these agents (5). A mounting research effort today is focused on this topic (6–8). The main focus of our research in the past 20 years has been to identify and quantitatively assess the transport barriers that a blood-borne macromolecule has to overcome to reach its target in the tumor interstitium. Because the diffusion rate of macromolecular drugs across blood vessels is slow, their transport principally relies on convection (9, 10), which is driven by pressure gradients. Unfortunately, pathophysiological conditions in a tumor lead to a decrease in transvascular pressure gradients, which reduces the fluid filtration in solid tumors (11). Indeed, we have shown that (i) interstitial fluid pressure (IFP) is elevated and uniform throughout a solid tumor, with a sharp gradient only at its periphery (11–18), (ii) transvascular gradient is close to zero, because microvascular pressure (MVP) is very close to the IFP (17), (iii) oncotic pressure gradient across the tumor vascular wall is almost negligible,¶ and (iv) elevated IFP coupled with a high vascular permeability of the tumor vessels could reduce blood perfusion in solid tumors (19). As a result, the convective component of macromolecular transport across the vessel wall is hindered throughout most of the tumor. We recently have shown that changes in the microvascular pressure induce a rapid fluid redistribution within the tumor tissue through a transvascular fluid exchange (20). This finding has led us to postulate that an enhancement in fluid extravasation could improve the delivery of macromolecular drugs and diagnostic agents in tumors (20). We test this hypothesis here by enhancing the convection through an increase in the transmural pressure gradient in a solid tumor by both continuous and periodic infusion of angiotensin II (AII).
EXPERIMENTAL DESIGN AND METHODS
To test our hypothesis that an enhancement in fluid extravasation improves the delivery of macromolecular drugs in tumors, we pharmacologically modulated the tumor MVP. For this purpose, we used AII because it has been reported to increase the mean arterial blood pressure (MABP) and MVP as well as selectively increase tumor blood flow (21, 22). The latter is possible because newly formed tumor vessels are devoid of smooth muscle cells (23, 24). Moreover, AII is a nontoxic, vasoactive agent that leads to selective and reproducible blood pressure changes (22, 25, 26). Thus, a systemic injection of AII can be used to selectively modulate tumor MVP.
Tumor and Transplantation.
The human colon adenocarcinoma LS174T was grown s.c. in female, 8- to 10-week-old (22 ± 3 g body weight) severe combined immunodeficient mice bred in the Edwin L. Steele Laboratory (Massachusetts General Hospital, Boston). Before all surgical procedures were performed, the animals were anesthetized with a s.c. injection of ketamine/xylazine (100/10 mg/kg) in 0.9% NaCl solution. Institutional guidelines for animal welfare and experimental conduct were followed. The LS174T tumor line was chosen for this study because several of its physiological parameters have been characterized previously in our laboratory (27–30). Moreover, this tumor expresses TAG-72 antigen, which is a target of the mAb CC49 (31).
Blood Pressure Modulation and IFP Measurements.
The systemic blood pressure was altered by infusion of AII (20 μg/min per kg body weight) via the tail vein. The infusion was carried out with a volumetric pump (Harvard Apparatus) at a flow rate of 1 μl/min. Arterial blood pressure was measured by a pressure transducer (model 23Gb; Gould, Cleveland) connected directly to the left carotid artery via a PE-10 catheter as described earlier (22). The IFP in the central region of a tumor was measured with the “wick-in-needle” technique (16).
Tracer Molecules.
We used antibodies as macromolecular probes: a nicotinyl hydrazine derivative of human polyclonal IgG (HYNIC-IgG) as a nonspecific antibody and CC49 as a specific antibody (kind gift of J. Schlom, National Cancer Institute, Bethesda, MD).
Nonspecific antibody.
The nicotinyl hydrazine derivative of human polyclonal IgG (HYNIC-IgG) was prepared as described previously (32). Briefly, a 4-fold molar excess of succinimidyl 6-hydrazinonicotinate hydrochloride (30 mM in dimethylformamide) was added dropwise to a stirred solution of IgG (1.0 g in 20 ml of 0.1 M phosphate buffer, pH 7.8), and the solution was stirred gently for 5 hr at room temperature. The mixture was dialyzed against 10 mM citrate (pH 5.2) at 4°C and filtered through a 0.2-μm filter. Protein concentration was determined by the Bradford method (33). The solution was diluted to 5 mg/ml with 100 mM NaCl/20 mM citrate/1% mannitol (pH 5.2), divided into 200-μl aliquots, and stored at −20°C. The number of hydrazino groups per IgG was determined by conversion to the corresponding hydrazone by reaction with p-nitrobenzaldehyde and measuring the optical density at 385 nm. Typically, ≈2.6 nicotinyl hydrazine groups were present per IgG molecule.
Specific antibody.
mAb CC49 was iodinated by using the Iodo-Gen method (34) with modification (15). Briefly, 40 μl of CC49 (2 mg/ml) and 80 μl 0.1 M sodium phosphate buffer, pH 7.2, was added to a polypropylene microfuge tube coated with 50 μg of Iodo-Gen. Na125I (10 μl, 1 mCi) then was added and the tube was vortexed for 30 sec and allowed to incubate for 2 min. To separate free 125I, the reaction mixture then was diluted with ≈2.5 ml of phosphate buffer and loaded onto a PD-10 column (Sephadex G-25M, catalog no. 17-0851-01; Pharmacia), which had been flushed previously with 25 ml of sterile 0.01 M sodium phosphate, pH 7.2. The column was eluted with 0.01 M sodium phosphate buffer using 1-ml aliquots, which were collected and assayed for radioactivity. The fractions were analyzed by using size-exclusion chromatography (HPLC column, SEC Zorbax GF250, UV: 280 nm, 0.1 M sodium phosphate + 0.05% azide, pH 6.85, 1 ml/min), and the fractions containing 125I-CC49 were pooled and used for injections. Specific activity was ≈12.5 μCi/μg.
Animal Groups.
Six experimental groups of five animals each were studied. The first two groups were control groups in which no alteration of blood pressure was attempted. In one of the control groups a nonspecific antibody, mAb, was used, and the other control group received the specific antibody CC49. The next two groups (a mAb group and a CC49 group) had a continuous infusion of AII for 40 min at a rate of 20 μg/min per kg body weight. In the last two groups (mAb and CC49 groups) AII was injected in a cyclical manner: a 3-min AII injection followed by a 5-min interval. The animals were randomly assigned to an experimental group.
Infusion Protocol.
The labeled antibody was injected systemically via the tail vein (0.1 ml of ≈20 μg/ml solution). The tail vein of anaesthetized mice was cannulated with a 5-cm-long PE-10 tube (0.28 mm i.d.; 0.97 mm o.d.) ending with a 26-gauge needle. After the antibody injection, the tubing was washed with saline (0.05–0.1 ml). For the AII-treated mice, the same tubing was used to infuse the AII.
Imaging Protocol.
The tissue uptake kinetics were measured up to 4 hr after injection by using a gamma camera (Sigma 410; Ohio Nuclear, Solon, OH). A 20% photopeak window was centered at 140 keV for technetium-99m and at 35 keV for 125I-labeled antibody. Sequential whole-body images of each animal were acquired by using a pinhole collimator at 20 min, 1, 2, 3, and 4 hr after the i.v. administration of the antibody. At 4 hr after injection the mice were sacrificed, and samples of tumor, kidney, liver, blood, muscle, lung, gall bladder, spleen, and tail were analyzed. The value of activity per g of tissue was determined by a gamma counter (CompuGamma CS; LKB).
At the end of the study, images were transferred into a PowerMacintosh 8100/80 workstation and analyzed off-line to determine the antibody uptake rate in the tumor, liver, kidney, and lung. This was accomplished by a region-of-interest technique. All the measurements were corrected for the physical half-life (6 hr for technicium-99m; 60 days for 125I) of the radioisotope used.
RESULTS
Enhancement of Transcapillary Fluid Filtration.
Changes in MABP were selectively transmitted to tumor IFP, indicating an enhancement of fluid filtration across blood vessels (Fig. 1). Fig. 1 shows typical changes in tumor IFP induced by a cyclic change in MABP. The IFP, measured in the center of the tumor, closely follows variation of MABP as a result of transvascular fluid exchange. The average variation of MABP was 64.5 ± 11 mmHg (from 97 ± 12 to 161 ± 13 mmHg), leading to a 5 ± 0.94 mmHg change in IFP in the tumor center. Because there is no lymphatic fluid drainage in these tumors, IFP approximates MVP (11), and the variation in IFP closely corresponds to that of MVP.
Figure 1.

Arterial pressure modulation in a s.c. tumor LS174T by a periodic infusion of AII at a rate of 20 μg/min per kg body weight for 3 min followed by a 5-min interval. The IFP measured with a wick-in-needle in the center of the tumor closely follows the arterial pressure changes, indicating a transvascular fluid exchange. The vascular–interstitial fluid exchange occurs mainly during the initial phase of the MVP change; when IFP reaches the plateau, the fluid filtration is minimal. The amount of fluid that extravasates at each cycle is very low because the change of IFP is not higher than 5 mmHg and the time lag between the change of arterial pressure and IFP is approximately 5 sec.
The accuracy of the wick-in-needle measurements for the IFP transients was tested by measuring the IFP changes with a micropipette connected to a servo-null controller (11, 35). No differences in IFP readings were obtained when comparing the wick-in-needle with the micropipette technique. The micropipette has a shorter time response and a smaller measurement orifice (2–4 μm) than the wick-in-needle and, hence, reflects more accurate measurements of the transient redistribution of the IFP (11).
Effect of MABP Modulation on Macromolecular Uptake.
Fig. 2 shows the biodistribution of the nonspecific antibody (HYNIC-IgG) and the specific antibody (CC49) 4 hr after injection for three different cases: (i) baseline MABP (control, no administration of AII); (ii) chronic increase of MABP (continuous administration of AII); and (iii) cyclic changes in MABP (periodic infusion of AII). Modulation of MABP did not induce any significant difference in the tumor uptake of the nonspecific antibody as compared with the control group (Fig. 2A). This was true for the normal tissues as well. Conversely, a significant increase in specific antibody CC49 tumor uptake was observed with both cyclic changes (P < 0.04, ANOVA) and chronic increases in MABP (P < 0.03, ANOVA) (Fig. 2B). Furthermore, continuous AII administration induced an increase of accumulation of the CC49 antibody in the lung and in the kidney compared with the control, whereas periodic administration of AII led to an altered accumulation only in the lung. Besides the compartments shown in Figs. 2, we also evaluated the concentration of antibody in the tail, spleen, and gall bladder; no differences between MABP modulation groups and controls were found in those tissues. From our data it is not possible to establish the mass balance because it would require knowledge of mAb accumulation in all body compartments as well as its clearance. The increased drug concentration in blood induced by the continuous infusion of AII could be a result of a prolonged clearance time of drug induced by interactions between the specific antibody and circulating antigen. Indeed, HPLC runs on blood samples of tumor-bearing mice and mice without a tumor xenograft indicated that there was an increase of about 50% in molecular weight of the circulating radiolabeled macromolecules in the tumor-bearing mice group compared with the group of mice without a tumor xenograft (data not shown). The complex mAb–antigen may have a higher circulating time than the mAb, and this may lead to a high blood level. This effect can be more evident in the group treated with a continuous infusion of AII because of the increased tumor blood flow induced by chronic infusion of AII (36).
Figure 2.

Biodistribution of a nonspecific (A) and a specific (B) mAb in LS174T tumor-bearing mice at 4 hr postinjection. The data, expressed as a percentage of injected dose per g of tissue, are averaged over five animals and represent the sum of interstitial and vascular uptake (bars = SEM). There is no significant effect of MABP modulation on nonspecific mAb uptake (A). A net increase in total tumor uptake of CC49 specific antibody resulted from both continuous (P < 0.03, ANOVA) and cyclic (P < 0.04, ANOVA) infusion of AGII (B). Modulation of MABP also induced significant increases in lung and kidney uptake.
The understanding of the mechanism by which AII leads to an increase of MVP is important to explain our experimental results. AII induces vasoconstriction, leading to an increase of systemic blood pressure. This leads to an increase of tumor MVP because tumor vessels are devoid of smooth muscle cells (23, 37). This mechanism also explains the increase of tumor blood flow and vascular volume induced by AII (36).
The accumulation of antibodies reported in Fig. 2 represents the total vascular and interstitial uptake measured in tissue samples (tumor tissue, kidney, liver, blood, muscle, and lung) harvested at 4 hr after injection and measured with a gamma counter. To gather information about the vascular space and interstitial uptake rate we also acquired gamma images of the whole mouse over the 4-hr period of treatment. Fig. 3 shows a typical curve of tumor uptake kinetics obtained by measuring the intensity of the tumor region. The data are normalized to the blood counts and presented as a percentage. Because the labeled antibody reaches a uniform plasma distribution within a couple of minutes after injection (38), an estimate of the vascular space can be obtained by the intercept of the line with the y axis. The interstitial uptake rate, on the other hand, can be evaluated by the slope. The mean values of the slopes and the intercepts obtained for the three animal groups for nonspecific and specific antibody are reported in Table 1. The data are reported as average values of five animals ± SD. The higher values of SD obtained for the specific antibodies is a result of the different radioisotope used for labeling the specific and nonspecific antibodies. The accuracy of the gamma-camera measurements is higher with 99 m-Tc than with 125I.
Figure 3.

Typical uptake kinetics curve obtained by a gamma camera. The data are reported as a percentage of the ratio between the tumor and the blood counts versus time. The slope is representative of the interstitial uptake of the antibody, and the intercept is representative of the tumor vascular space.
Table 1.
Statistical analysis of the slopes and intercepts of the uptake kinetics of the nonspecific (HYNIC) and the specific CC49 antibodies
| Group | Slope × 104 (mean ± SD)
|
Intercept × 102 (mean ± SD)
|
||
|---|---|---|---|---|
| Nonspecific mAb | Specific mAb | Nonspecific mAb | Specific mAb | |
| Control | 3.3 ± 0.2 | 10 ± 1 | 4.0 ± 0.7 | 6.7 ± 3.9 |
| Continuous | 2.8 ± 0.4 | 10 ± 1 | 5.5 ± 0.9 | 8.2 ± 9.9 |
| Cycling | 2.4 ± 0.6 | 20 ± 10 | 3.5 ± 0.3 | 6.7 ± 3.5 |
The data, obtained by a linear fit of the gamma camera counts vs. time as shown in Fig. 3, are averaged over five animals and represent the interstitial uptake rate (slope) and vascular space (intercept). There is no significant effect of chronic increase or periodic modulation of MABP on the nonspecific mAb interstitial uptake rate. The data indicate that periodic modulation of MABP induced a 2-fold increase of the average value of specific antibody interstitial uptake rate compared with the control.
The good agreement between the data obtained by this method with those published in the literature supports the validity of our measurements. The vascular volume of 4% evaluated by the intercept of the uptake kinetics for the nonspecific mAbs (Table 1) is in good agreement with the data reported in the literature for the same tumor type (30). Furthermore, similar to Hori and coworkers (21, 39), we found that chronic infusions of AII induced a 30% increase of tumor vascular area, although the difference was not significant. Chronic infusion of AII can increase the tumor vascular area by a recruitment of redundant or collapsed blood vessels (21, 39). By comparing the intercept values obtained for the nonspecific and specific antibodies, an apparent increase in measured vascular area for the specific antibody results. This may be explained by a possible binding of the specific antibody within the tumor vessel, suggesting the presence of cancer cells within the endothelial layer lining, the so-called “mosaic vessels” (23). Therefore, the intercept of the uptake kinetics for the specific antibody would represent not only the vascular volume but also the number of specific sites for the CC49 antibody binding that are within the tumor endothelium.
The effect of periodic infusion of AII on vascular volume has never been reported in the literature. We found that continuous infusion of AII did not induce any variation in the vascular space compared with the control (Table 1).
For the nonspecific antibody, neither periodic modulation nor constant increase of MABP influences significantly the interstitial uptake rate (Table 1). A similar result also was found for the uptake rate of the specific antibody CC49. However, although the difference was not significant, periodic modulation of blood pressure increased the interstitial uptake rate of the specific antibody by 2-fold compared with the control group. No difference in the mean value of the slope was found between the continuous and control groups.
Comparing the slopes for the nonspecific and specific antibodies, it is possible to appreciate the important effect that the specificity of the antibody has on the interstitial uptake. There is a 3- to 4-fold increase in the interstitial uptake rate when using a specific compared with a nonspecific antibody (Table 1).
DISCUSSION
We have suggested previously that fluid and macromolecular extravasation in solid tumors is reduced because of pathophysiological conditions that lead to an equilibrium between MVP and IFP. Here we show that alterations of MABP induced by systemic injection of AII lead to an enhancement of fluid and macromolecular exchange between the vascular and interstitial space in tumors.
Mechanism of Enhanced Delivery.
Injection of AII leads to selective enhancement of fluid filtration across the wall of tumor vessels. Plasma extravasates and accumulates in the interstitium, leading to an increase in tumor IFP (Fig. 1). Analogously, when the MVP is lowered, the interstitial fluid is reabsorbed by the capillaries and IFP decreases (Fig. 1). Thus, periodic modulations of MABP induce cycles of fluid exchange between the vascular and interstitial space. However, the net fluid extravasation at each cycle is relatively small. Indeed, the increase of transmural pressure induced by the 50-mmHg increase of MABP is only 5 mmHg (Fig. 1), and the time scale for fluid redistribution is very fast (about 5 sec). This time scale is within the same order of magnitude as for tumors perfused ex vivo (20).
Transvascular transport of molecules occurs both by diffusion and convection, according to the equation
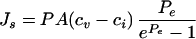 |
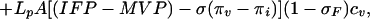 |
where Js is the flux of molecules; P and Lp are the permeability and the hydraulic conductivity of the endothelial wall; A is the vascular area; cv and ci are the drug concentrations in plasma and in the interstitium; σ and σF are the oncotic and plasma-drag reflection coefficients; πv and πi are the oncotic pressures in plasma and interstitial fluid; and Pe is the Peclet number (40). The first and second terms in the right-hand side of the equation represent the diffusive and convective component of the transport, respectively (41). In normal tissues, extravasation of low molecular weight drugs is dominated by diffusion, whereas for high molecular weight agents convection represents the principal mechanism of transport (9, 10). If the same notion held also for tumor tissues, one should conclude that macromolecular extravasation would depend mainly on hydrostatic and oncotic pressure gradients. Furthermore, because the oncotic gradients in tumor tissues are almost zero,¶ macromolecular extravasation should rely mostly on the difference between MVP and IFP. Based on these arguments, we have suggested previously that elevated IFP may represent a major physiological transport barrier to macromolecular uptake in solid tumors (2, 14). However, the relevance of elevated IFP to macromolecular uptake in tumors has not been established experimentally. To test the influence of elevated IFP on transport of macromolecules, we estimated the change in tumor macromolecular uptake induced by modulation of MABP. If convection were the dominant mechanism of transport, it is reasonable to expect that modulation of MABP would produce macromolecular extravasation along with the fluid extravasation. Our results on nonspecific antibody distribution apparently contradict this hypothesis. Indeed, neither continuous infusion nor periodic injection of AII led to a net increase of nonspecific antibody (Fig. 2A). However, the absence of a net increase in the total antibody uptake does not necessarily imply that macromolecules do not extravasate in tumors. In normal tissues, macromolecules extravasate from the capillaries and are transported into the lymphatics, whereas in tumors they may cross the vascular wall in both directions. Therefore, nonspecific antibodies may extravasate and be reabsorbed within a blood pressure cycle, and this would not lead to a net accumulation of macromolecules in the interstitium.
Nonspecific antibodies can cross the vascular wall in both directions because there is no specific binding in the extravascular space. In the case of high-affinity antibodies, on the other hand, the macromolecules that extravasate would remain within the extracellular matrix anchored to the cancer cell surface or the interstitial matrix. In this case, transvascular transport of macromolecules would be nearly unidirectional and a net increase in antibody accumulation should be expected as a result of MABP modulation. Indeed, our results show that the enhancement of capillary fluid filtration induced by periodic injections of AII led to a significant increase in tumor uptake. Twenty MABP cycles led to a 40% increase in total tumor uptake of the specific antibody at 4 hr after injection when compared with the control group (Fig. 2B). A similar increase also was produced by a continuous infusion of AII. By comparing Fig. 2 A with B it can be concluded that alterations of blood pressure lead to an improvement of delivery of specific antibody probably because of a combination of an enhanced transcapillary fluid filtration and specific binding with cancer cells. These two effects must be combined because the enhancement of fluid filtration alone does not lead to any improvement of delivery (Fig. 2A).
Fig. 2B shows that both chronic increase and cycling modulation of blood pressure induce an augmentation of specific mAb accumulation in tumor tissues. The question that arises here is whether or not the mechanisms by which chronic increase or cyclic changes of blood pressure induce an improvement of specific mAb accumulation are the same. An answer to this important question can be obtained by analyzing the data of Table 1. The cycling group shows a 2-fold increase of interstitial uptake rate compared with the control. Although this difference is not statistically significant, it suggests that the augmentation induced by the cycling results from a net increase of macromolecular extravasation whereas the increased antibody uptake induced by the continuous infusion presumably is a result of an increase in vascular area. This effect influences the total uptake in two ways. (i) It increases the amount of drug within the vascular space and (ii) enhances the diffusive component of transport.
The advantageous effect of cycling blood pressure on interstitial uptake is evident only for highly specific antibodies because of the unidirectionality of transvascular transport. Indeed, even without any blood pressure manipulation, the uptake rate increases by almost one order of magnitude when using a specific versus a nonspecific antibody (Table 1). This indicates that the transvascular transport of specific antibody occurs prevalently from the vascular to the interstitial space and not vice versa.
Taken together these data confirm that extravasation of large molecules is reduced in solid tumors by an equilibration between MVP and IFP, as previously suggested (14, 20), and suggest that an enhancement of macromolecular uptake can be achieved by combining an increase of transvascular convection with a high specificity of the drugs against tumor cells.
Clinical Implications and Limitations.
There are several reports in the literature showing that a chronic increase in arterial pressure leads to an enhancement of uptake of chemotherapeutic agents in solid tumors (42). Most of these investigations, however, have focused on low molecular mass chemotherapeutic agents (<10,000 molecular weight) that rely on tumor blood flow and diffusive extravasation, which are not affected by IFP. A chronic increase in arterial pressure may enhance the diffusive component of the macromolecular transport via an increase of tumor blood flow (36) and recruitment of blood vessels (21), but it should not have a significant effect on the convective component and thus on the tumor uptake of macromolecules. Indeed, a chronic increase of MABP did not induce any appreciable increase in total nonspecific antibody uptake (Fig. 2A) or any appreciable increase in interstitial uptake rate of specific antibody (Fig. 3B).
The effect of AII-induced chronic hypertension on macromolecular uptake in tumors is controversial. In concert with our results, Elizondo and Sung (43) reported that chronic increase of MABP did not induce any effect on specific or nonspecific immunotoxin uptake in rhabdomyosarcoma xenografts. In contrast to our results, others have reported an enhancement of uptake of nonspecific (42, 44) and tumor-specific (45, 46) high molecular weight agents with a chronic increase of MABP. However, none of these latter studies reported the values of tumor IFP; therefore, it is difficult to compare their results with our study. Indeed, if tumor IFP is low, a chronic increase of MABP may increase fluid filtration and, therefore, macromolecular uptake. Furthermore, these studies report the effect at long times (24, 48, and 72 hr) whereas our study was more focused at a short time effect. At long times, other effects, such as an AII-induced increase in permeability (47, 48), may play a role in determining macromolecular accumulation.
Our results show that it is possible to enhance tumor macromolecular uptake by combining alteration of blood pressure and specific binding. Furthermore, there is an indication, although not supported by statistical significance, that cyclic modulation of blood pressure led to an increase of interstitial uptake whereas chronic increase of blood pressure led to an increase of vascular uptake. Further investigations are needed to prove this last point. These data support the principle that elevated IFP is a strong barrier to delivery of macromolecules to tumors and suggest that strategies to enhance macromolecular delivery to solid tumors should be targeted at increasing the transmural pressure gradient.
The periodic injection of AII may not be directly applicable clinically because of potential side effects, although AII has been utilized successfully, especially in Japan (36, 39, 49), to improve tumor blood flow and accumulation of low molecular weight drugs. Furthermore, periodic modulation of MABP also induces an undesirable increase in specific mAb accumulation in the lung (Fig. 2B). The reason for this increase in lung uptake is not clear. It might be a result of an increase in lung fluid filtration induced by AII as reported previously (47, 48). An alternative and more clinically feasible approach to increasing transmural pressure is to reduce tumor IFP, which can be achieved by modulation of the tumor extracellular matrix. For instance, it has been shown that by blocking the integrin links between interstitial matrix and cells, it is possible to reduce IFP and to increase tissue fluid content (50). Irradiation also can reduce the IFP in tumors. There is a significant decrease in tumor IFP after a single dose of radiation, which probably is triggered by remodeling of the extracellular matrix that is induced by radiation (51).
In summary, by combining the effect of tumor MVP modulation with high-binding affinity therapeutic or diagnostic agents, we show that it is possible to increase macromolecular delivery to solid tumors.
Acknowledgments
We thank Ms. Sylve Roberge for her technical assistance, Drs. Jeffry Schlom and Diane Milenic for providing the CC49 antibody and the protocols to label it, and Dr. Larry Baxter for his helpful comments. This work was supported by an Outstanding Investigator Grant from the National Cancer Institute (R35-CA-56591) to R.K.J. P.A.N. was supported, in part, by the Italian National Research Council (Consiglio Nazionale delle Ricerche).
ABBREVIATIONS
- MVP
microvascular pressure
- IFP
interstitial fluid pressure
- AII
angiotensin II
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Stohrer, M., Boucher, Y., Stangassinger, M. & Jain, R. K., 43rd Annual Meeting of the Radiation Research Society, April 1–6, 1995, San Jose, CA, p. 187.
References
- 1.Jain R K. Microcirculation. 1997;4:1–23. doi: 10.3109/10739689709148314. [DOI] [PubMed] [Google Scholar]
- 2.Jain R K. Sci Am. 1994;271:58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 3.Sands H. J Nucl Med. 1992;33:29–32. [PubMed] [Google Scholar]
- 4.Hall S S. Science. 1995;270:915–916. doi: 10.1126/science.270.5238.915. [DOI] [PubMed] [Google Scholar]
- 5.Jain R K. Science. 1996;271:1079–1080. doi: 10.1126/science.271.5252.1079. [DOI] [PubMed] [Google Scholar]
- 6.Bobo H R, Laske D W, Akbasac A, Morrison P F, Dedrick R L, Oldfield E H. Proc Natl Acad Sci USA. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langer R. Nature (London) 1998;392:5–10. [PubMed] [Google Scholar]
- 8.Jain R K. Nat Med. 1998;4:655–657. doi: 10.1038/nm0698-655. [DOI] [PubMed] [Google Scholar]
- 9.Rippe B, Haraldsson B. Phys Rev. 1994;74:163–218. doi: 10.1152/physrev.1994.74.1.163. [DOI] [PubMed] [Google Scholar]
- 10.Flessner M F, Dedrick R L. Cancer Res. 1994;54:4376–4384. [PubMed] [Google Scholar]
- 11.Boucher Y, Baxter L T, Jain R K. Cancer Res. 1990;50:4478–4484. [PubMed] [Google Scholar]
- 12.Young J S, Lumsden C E, Stalker A L. J Pathol Bacteriol. 1950;62:313–333. doi: 10.1002/path.1700620303. [DOI] [PubMed] [Google Scholar]
- 13.Gullino P M, Clark S H, Grantham F H. Cancer Res. 1964;24:780–797. [PubMed] [Google Scholar]
- 14.Jain R K, Baxter L T. Cancer Res. 1988;48:7022–7032. [PubMed] [Google Scholar]
- 15.Bakir M A, Eccles S A, Babich J W, Aftab N, Styles J M, Dean C J, Ott R J. J Nucl Med. 1992;33:2154–2160. [PubMed] [Google Scholar]
- 16.Boucher Y, Kirkwood J, Opacic D, DeSantis M, Jain R K. Cancer Res. 1991;51:6691–6694. [PubMed] [Google Scholar]
- 17.Boucher Y, Jain R K. Cancer Res. 1992;52:5110–5114. [PubMed] [Google Scholar]
- 18.Less R J, Posner M C, Boucher Y, Borochovitz D, Wolmark N, Jain R K. Cancer Res. 1992;52:6371–6374. [PubMed] [Google Scholar]
- 19.Netti P A, Roberge S, Boucher Y, Baxter L T, Jain R K. Microvascular Res. 1996;52:27–46. doi: 10.1006/mvre.1996.0041. [DOI] [PubMed] [Google Scholar]
- 20.Netti P A, Baxter L T, Boucher Y, Skalak R, Jain R K. Cancer Res. 1995;55:5451–5458. [PubMed] [Google Scholar]
- 21.Hori K, Suzuki M, Abe I, Saito S, Sato H. J Natl Cancer Inst. 1985;74:453–459. [PubMed] [Google Scholar]
- 22.Zlotecki R A, Boucher Y, Lee I, Baxter L T, Jain R K. Cancer Res. 1993;53:2466–2468. [PubMed] [Google Scholar]
- 23.Jain R K. Cancer Res. 1988;48:2641–2658. [PubMed] [Google Scholar]
- 24.Krylova N V. Bibl Anat. 1969;10:301–303. [PubMed] [Google Scholar]
- 25.Zlotecki R A, Baxter L T, Boucher Y, Jain R K. Microvasc Res. 1995;50:429–443. doi: 10.1006/mvre.1995.1069. [DOI] [PubMed] [Google Scholar]
- 26.Hori K, Suzuki M, Saito S, Tanda S, Zhang Q-H, Li H-C. Microvasc Res. 1994;48:246–256. doi: 10.1006/mvre.1994.1052. [DOI] [PubMed] [Google Scholar]
- 27.Kristjansen P E G, Roberge S, Lee I, Jain R K. Microvasc Res. 1994;48:389–402. doi: 10.1006/mvre.1994.1063. [DOI] [PubMed] [Google Scholar]
- 28.Leunig M, Yuan F, Menger D M, Boucher Y, Goetz A F, Messmer K, Jain R K. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 29.Yuan F, Dellian M, Fukumura D, Leunig M, Berk D A, Torchilin V P, Jain R K. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 30.Yuan F, Leunig M, Berk D A, Jain R K. Microvasc Res. 1993;45:269–289. doi: 10.1006/mvre.1993.1024. [DOI] [PubMed] [Google Scholar]
- 31.Milenic D E, Yokota T, Filpula D R, Finkelman M A J, Dodd S W, Wood J F, Whitlow M, Snoy P, Schlom J. Cancer Res. 1991;51:6363–6371. [PubMed] [Google Scholar]
- 32.Abrams M J, Juweid M, tenKate C I, Schwartz D A, Hauser M M, Gaul F E, Fuccello A J, Rubin R H, Strauss H W, Fischman A J. J Nucl Med. 1990;31:2022–2028. [PubMed] [Google Scholar]
- 33.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 34.Fraker P J, Speck J C. Biochem Biophys Res Commun. 1978;80:849–853. doi: 10.1016/0006-291x(78)91322-0. [DOI] [PubMed] [Google Scholar]
- 35.Intaglietta M, Pawula R F, Tompkins W R. Microvasc Res. 1970;2:212–220. doi: 10.1016/0026-2862(70)90009-9. [DOI] [PubMed] [Google Scholar]
- 36.Hori K, Zhang Q, Saito S, Tanda S, Li H, Suzuki M. Cancer Res. 1993;53:5528–5534. [PubMed] [Google Scholar]
- 37.Less J R, Skalak T C, Sevick E M, Jain R K. Cancer Res. 1991;51:265–273. [PubMed] [Google Scholar]
- 38.Baxter L T, Zhu H, Mackensen D G, Jain R K. Cancer Res. 1994;54:1517–1528. [PubMed] [Google Scholar]
- 39.Hori K, Suzuki M, Tanda S, Saito S, Zhang Q H. Int J Oncol. 1993;2:289–296. doi: 10.3892/ijo.2.2.289. [DOI] [PubMed] [Google Scholar]
- 40.Jain R K. Cancer Metastasis Rev. 1987;6:559–593. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- 41.Fung Y C. Biomechanics: Motion, Flow, Stress and Growth. New York: Springer; 1990. [Google Scholar]
- 42.Abe I, Hori K, Saito S, Tanda S, Li Y, Suzuki M. Jpn J Cancer Res. 1988;79:874–879. doi: 10.1111/j.1349-7006.1988.tb00050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elizondo F G J, Sung C. Cancer Chemother Pharmacol. 1996;39:113–121. doi: 10.1007/s002800050546. [DOI] [PubMed] [Google Scholar]
- 44.Li C J, Miyamoto Y, Kojima Y, Maeda H. Br J Cancer. 1993;67:975–980. doi: 10.1038/bjc.1993.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noguchi A, Takahashi T, Yamaguchi T, Kitamura K, Noguchi A, Tsumuri H, Takashina K, Maeda H. Jpn J Cancer Res. 1992;83:240–243. doi: 10.1111/j.1349-7006.1992.tb00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda A, Kikuki T, Ozaki M, Ariga T, Nagashima T, Isono K, Miyoski T. Nippon Ega Gakkai Zasshi. 1991;92:475. [PubMed] [Google Scholar]
- 47.Hansen T N, Le Blanc A L, Gest A L. J Appl Physiol. 1985;58:812–818. doi: 10.1152/jappl.1985.58.3.812. [DOI] [PubMed] [Google Scholar]
- 48.Roy B J, Pitts V H, Townsley M I. Am J Physiol. 1996;271:H222–H227. doi: 10.1152/ajpheart.1996.271.1.H222. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki M, Hori K, Abe I, Saito S, Sato H. J Natl Canc Inst. 1981;67:663–669. [PubMed] [Google Scholar]
- 50.Reed R K, Rubin K, Wiig H, Rodt S A. Circulation Res. 1992;71:978–983. doi: 10.1161/01.res.71.4.978. [DOI] [PubMed] [Google Scholar]
- 51.Znati C A, Rosenstein M, Boucher Y, Epperly M W, Bloomer W D, Jain R K. Cancer Res. 1996;56:964–968. [PubMed] [Google Scholar]