

Abstract
The derivation of a quantitative model of phenylalanine metabolism in humans is described. The model is based on the kinetic properties of pure recombinant human phenylalanine hydroxylase and on estimates of the in vivo rates of phenylalanine transamination and protein degradation. Calculated values for the steady-state concentration of blood phenylalanine, rate of clearance of phenylalanine from the blood after an oral load of the amino acid, and dietary tolerance of phenylalanine all agree well with data from normal as well as from phenylketonuric patients and obligate heterozygotes. These calculated values may help in the decision about the degree of restriction of phenylalanine intake that is necessary to achieve a satisfactory clinical outcome in classical patients and in those with milder forms of the disease.
The initial and rate-limiting step in the complete catabolism of phenylalanine to CO2 and water is its hydroxylation to tyrosine, a reaction catalyzed by the phenylalanine hydroxylating system. The system is complex, consisting of phenylalanine hydroxylase (PAH), the pterin coenzyme tetrahydrobiopterin (BH4), and several enzymes that serve to regenerate BH4, i.e., dihydropteridine reductase and pterin 4α-carbinolamine dehydratase (1, 2).
Although the benzene ring of phenylalanine cannot be ruptured without first being hydroxylated in the para position, the alanine side-chain of the amino acid can be metabolized even in the absence of the ring-hydroxylation step. This alternate pathway is initiated by transamination of phenylalanine to phenylpyruvate followed by conversion of the latter compound to metabolites such as phenyllactate, phenylacetate, and o-hydroxyphenylacetate. Products of the transaminase pathway are excreted in the urine. The steps in these alternate pathways of phenylalanine metabolism are outlined in Fig. 1.
Figure 1.

Phenylalanine metabolism. Reaction 1 is catalyzed by phenylalanine hydroxylase. Reaction 2 is catalyzed by phenylalanine transaminase.
Mutations in PAH that decrease the activity of the enzyme cause hyperphenylalaninemia (HPA). When the HPA is severe (blood phenylalanine > 1.2 mM, 20 times above normal levels of 0.055–0.060 mM), it leads to the disease known as classical phenylketonuria (PKU), which, if not treated by a low-phenylalanine diet from early infancy, leads to postnatal brain damage and mental retardation (3).
The consensus view is that all classical PKU patients should be treated with a low-phenylalanine diet. The decision to treat milder forms of the disease (blood phenylalanine levels in the range of 0.4–0.7 mM), which are associated with lower risk of brain damage (4), however, is less clear cut. Fortunately, results from a variety of procedures can provide useful guidelines about which patients require treatment. In addition to the severity of the HPA itself, which, as indicated above, is a useful treatment criterion, methods range from semiquantitative measurements of the patient’s tolerance to dietary phenylalanine, i.e., the amount of phenylalanine that can be consumed without elevating the blood phenylalanine concentration to unacceptable levels, to quantitative measurements of residual PAH activity in vivo by the use of one of several different methods involving the infusion of phenylalanine labeled with heavy isotopes (5–7). But even the availability of these methods does not resolve all of the problems in this area: the dietary criterion is crude and most of the heavy isotope methods require expensive equipment.
Until recently, an indirect procedure for assessing residual PAH activity in vivo that was used widely is the phenylalanine-load test, which is based on the determination of the rate of clearance from blood of phenylalanine after the administration to a patient of a standard dose, usually 100 mg/kg of body weight, of the amino acid. This method, for example, can readily distinguish between normal subjects and obligate heterozygotes (i.e., parents of PKU patients) presumed to have 50% or less of the normal level of enzyme (8–10).
Although the data from the phenylalanine-load test are relatively easy to come by—all that is needed is a reliable quantitative assay for phenylalanine—the usefulness of the method is limited by the lack of a suitable model of phenylalanine metabolism, one that would facilitate the interpretation of the data, and would take into account the relative contribution of hydroxylation, transamination, decarboxylation, and other pathways of phenylalanine catabolism. Without a reasonable quantitative model, impaired phenylalanine tolerance might be interpreted as a defect in either phenylalanine hydroxylation or phenylalanine transamination. Indeed, an early study of two PKU patients with impaired tolerance seemed to support the conclusion that the patients lacked phenylalanine transaminase (11). Subsequent analysis of the data provided strong arguments against this conclusion (12). Nevertheless, availability of an appropriate model of phenylalanine metabolism would extend the usefulness of phenylalanine tolerance data not only for diagnostic purposes, but also for understanding the interplay of some of the factors that affect normal phenylalanine metabolism in humans.
As a first step toward the elaboration of such a model, the reactions that determine the steady-state level of phenylalanine in plasma must be identified. The steady state is determined by those processes that lead to the net disposal of phenylalanine and those that replenish the plasma pool. After growth has stopped, the only reactions that are quantitatively important in the disposal of phenylalanine are its irreversible oxidation to tyrosine and transamination to phenylpyruvate. For growing children, a small fraction of phenylalanine is disposed of through the net increase in body protein. Even during early infancy, however, when the increment of protein per unit time is maximal (13), this route of disposal of phenylalanine is small compared with the rate of its conversion to tyrosine, especially when blood phenylalanine levels approach those seen in PKU. The evidence also indicates that reactions such as decarboxylation are too slow to affect the overall rate of disposal and can be safely ignored (12), as can urinary excretion of phenylalanine [equal to only ≈11% of the amount of total transamination products excreted (14)] and those processes that affect only the flux of phenylalanine through the various body pools of the amino acid.
As for reactions that serve to replenish the plasma pool of phenylalanine, there appear to be only two of any significance: ingestion and digestion of protein-containing foods and, under certain circumstances, breakdown of body proteins. Because the phenylalanine-loading test usually is carried out in subjects in the fasting state (15), the delivery of phenylalanine from ingested protein can be ignored.
The above considerations lead to the following relationship, where vTRANS is the rate of phenylalanine transamination (Fig. 1, reaction 2), vPAH is the rate of phenylalanine hydroxylation (Fig. 1, reaction 1), and vNPD is the rate of net protein breakdown.
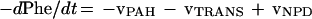 |
1 |
To transform this general equation into one that would be useful in the analysis of results of phenylalanine-loading tests, each of these velocity terms must be replaced with rate equations that describe the three metabolic processes involved. When the requisite data were unavailable, assumptions discussed below had to be made.
At the outset, it should be noted that a previous attempt to carry out such an analysis was handicapped by the lack of data on the kinetic properties of human PAH and human phenylalanine transaminase. Indeed, for the latter enzyme, even the identity of the one responsible for this activity in vivo was not known with certainty. Because the in vitro evidence indicated that phenylalanine is an excellent substrate for mitochondrial aspartate aminotransferase, it was assumed that this is the transaminase involved. Furthermore, because the properties of the human counterpart were not known, the kinetic properties of the corresponding rat enzyme were used (12). The way the problem of the human transaminase was handled in the present analysis will be discussed below.
Kinetic properties of recombinant human PAH are now available (16, 17). The kinetics of PAH are somewhat complicated by the fact that phenylalanine serves not only as a substrate for the enzyme, but also as an activator (see ref. 1 and references therein). Because a previous analysis of the kinetic behavior of PAH based on a two-site model with ordered binding of phenylalanine at both a catalytic site and a regulatory site could account adequately for many peculiar aspects of the kinetic behavior of the enzyme (18), a similar two-site, ordered-binding model was used in the present analysis. The actual rate equation used (19) is shown in Eq. 2, where Km is the concentration of phenylalanine that gives half-maximum velocity and Ka is the concentration of phenylalanine that gives half-maximum activation in an experiment in which PAH was preincubated with varying concentrations of phenylalanine. For the present analysis, the following kinetic constants, determined with pure recombinant human PAH at 37°C with BH4 as the coenzyme, were used: Km for phenylalanine, 0.51 mM, and Ka for phenylalanine as activator, 0.54 mM (D. Kowlessur and S.K., unpublished data). An approximate value of Vmax for human PAH (16) (probably an underestimate) was calculated from the initial rate of decrease of serum phenylalanine levels (0.9 μmol/ml per h) in control subjects after they had received an oral load of l-phenylalanine that was sufficient to increase their serum phenylalanine levels by ≈17-fold (20).
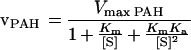 |
2 |
As indicated above, the previous problem of the identity of the enzyme in man that is responsible for phenylalanine transamination was by-passed in the present analysis. It was assumed that the major route for the net disposal of phenylalanine in classical PKU patients is via transamination. For example, as already mentioned, the urinary excretion of phenylalanine is only ≈11% of the amount that is transaminated, and, by the end of the first year of life, it can be estimated that the amount of phenylalanine disposed of via incorporation into protein is only ≈25% of that disposed of via transamination. It should be noted that with the present method for estimating the rate of phenylalanine transaminase, which is based on the rate of clearance of phenylalanine from the blood, minor reactions for disposal of phenylalanine, such as its urinary excretion and its incorporation into protein, are subsumed in the estimate of transaminase activity, resulting in a small overestimate of this activity.
To be useful in the present analysis, values for the Km and Vmax of the transaminase are needed. Attempts were made to extract a Km value for phenylalanine transamination from the results of phenylalanine-loading tests carried out on classical PKU patients (21). The approach adopted in estimating a value for Vmax for the human transaminating enzyme was to use data on the sum of all transamination-derived metabolites (i.e., phenylpyruvate, phenyllactate, and o-hydroxyphenylacetate) excreted by a group of classical PKU patients as a function of their plasma phenylalanine levels. The maximum amount excreted, expressed as mmol/mol creatinine, was 1,370, a level that appeared to plateau at plasma phenylalanine levels between 1,200 and 2,400 μmol/liter (22).
Attempts to convert this value into a rate of transamination are complicated by the wide range of ages, ≈2 years to ≈18 years, in the patient sample used in the study. For the present analysis, it was assumed that the average body weight of the patients was 50 kg and that the daily creatinine excretion was 2 g/24 h (23). A further assumption was made that the excretion of transaminase-derived metabolites occurs at a linear rate during the 24-h period and reflects the rate of formation of these metabolites. It was also assumed that these compounds equilibrate with all body fluid compartments except dense cartilage connective tissue and bone, which, together, represent 15% of the total body water (24), yielding a volume of distribution of accessible water of 500 ml/kg of body weight. On the basis of these assumptions, the maximum rate of transamination was calculated to be 0.043 μmol/ml per h.
An additional product of phenylalanine metabolism that is derived, at least in part, from phenylpyruvate that was not measured in the study of Langenbeck et al. (22) is phenylacetylglutamine (PAG). There is evidence that PAG can be formed from phenylacetate, which is derived from phenylpyruvate by oxidative decarboxylation (25). It has also been proposed that phenylacetate and, hence, PAG, can be formed from phenylalanine by a route that does not involve transamination, but instead involves decarboxylation to phenylethylamine followed by oxidation of the amine to phenylacetate (26). The finding that the amount of phenylethylamine excreted in PKU patients is small even after the oxidation of the amine was blocked by the administration of an inhibitor of amine oxidase (27), however, indicates that, as discussed previously (12), decarboxylation of phenylalanine is a quantitatively minor pathway for phenylalanine metabolism, as well as for PAG formation.
The amount of PAG excreted by normal individuals is 250–500 mg/day; PKU patients excrete twice that amount (28). For the purpose of calculating the amount of PAG formed via the transaminase pathway, the conservative assumption was made that only the “extra” amount excreted by the patients is derived from phenylpyruvate. Taking the average extra amount of PAG excreted as 350 mg/day and making the same assumptions outlined above, this excretion translates to a rate of PAG formation of 0.020 μmol/ml per h, bringing the rate of formation of all transaminated products to 0.063 μmol/ml per h.
With the use of this value for Vmax, results of the phenylalanine-loading test carried out on classical PKU patients (21) were used to calculate a value of 1.37 ± 0.14 mM (mean ± SD, n = 3) for the Km of phenylalanine transaminase.
Because in the present analysis, PAH and transaminase activities are calculated as a function of blood phenylalanine levels, it is important that these levels reflect tissue levels of the amino acid. Relevant to this point, phenylalanine levels in liver tissue from a PKU patient (29), as well as in liver and kidney tissue from hyperphenylalanemic rats (30), have been reported to be comparable to the corresponding levels in blood.
The third term in Eq. 2, the rate of net protein degradation, was estimated from the data of Waterlow and Jackson (31), showing that in the fasting state, the state under which the phenylalanine-loading test is carried out, net protein breakdown (i.e., the amount of protein broken down minus the amount synthesized) equals 0.30 g/kg of body weight per 12 h. Because skeletal muscle constitutes ≈40% of body mass (24) and protein catabolism in this tissue plays a major role in the delivery of amino acids to the periphery, protein degradation in skeletal muscle was taken as the predominant event in the degradation of protein that occurs during fasting.
Human skeletal muscle contains ≈46 μmol phenylalanine/g tissue (32). On the basis of that value and the finding that adult human muscle contains 19.8% protein (33), it can be estimated that muscle contains 232 μmol phenylalanine/g of muscle protein. If this value is taken as representative of the body protein stores, it would indicate that ≈70 μmol phenylalanine/kg body weight per 12 h would be liberated during the fasting period. On the basis of the same assumptions as those made above in estimating the rate of phenylalanine transamination, the last value would translate into an hourly rate of net protein degradation (and of release of phenylalanine from this process) of 0.012 μmol/ml per h. Because the substrate for this reaction, namely, the body stores of protein, would probably remain relatively constant during a short period of fasting, protein degradation was assumed to follow zero-order kinetics.
Substituting the values estimated for the kinetic constants for the three reactions shown in Eq. 1 yields Eq. 3:
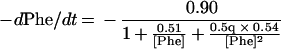 |
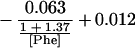 |
3 |
RESULTS AND DISCUSSION
The general validity of Eq. 3 can be assessed in several ways. First, with the use of the expression for the velocity of the PAH-catalyzed reaction, including the kinetic constants shown in the equation, the basal rate of the hydroxylation reaction was calculated to be 0.010 μmol/ml per h. This value agrees well with the following reported values for normal subjects on the basis of experiments in which subjects were infused with l-[ring-2H5]phenylalanine: 0.013 μmol/ml per h; 0.008 μmol/ml per h (34); 0.012 μmol/ml per h (5); 0.010 μmol/ml per h (6). A value of 0.020 μmol/ml per h was found in the last study when subjects were infused with l-[1-13C]phenylalanine (6). The cited in vivo rates for the conversion of phenylalanine to tyrosine all were reported as μmol/h per kg. They were converted to μmol/ml per h on the basis of the same assumptions used previously, i.e., that the volume distribution of metabolites such as phenylalanine is 500 ml/kg of body weight. These results show that the calculated rate of phenylalanine hydroxylation agrees well with the experimentally determined rates.
Another test of the validity of the model is to calculate the steady-state blood phenylalanine level for both control subjects and for PKU heterozygotes who are presumed to have 50% of normal PAH activity, as well as the t1/2 for the clearance of a load of phenylalanine (i.e., the time required for the initial concentration of phenylalanine to decrease to one-half its original value) from the blood for these two groups. The steady-state phenylalanine level for controls, calculated from Eq. 3 (by setting the term “−dPhe/dt” equal to zero and calculating the concentration of phenylalanine), is 0.059 mM and that for subjects with 50% residual PAH activity is 0.079 mM, 1.34-fold higher than the control level. Although the value of 0.059 mM for normal subjects agrees well with the accepted value of 0.058 ± 0.015 mM (mean and SD) (35), the value of 0.079 mM for heterozygotes who might be expected to have 50% of the normal PAH level, appears to be too low. The ratio of blood phenylalanine levels for controls and for obligate PKU heterozygotes has been reported to be in the range of 1.57–1.61 (36–38) rather than the ratio of 1.34 that was predicted by the model.
This calculated value raises the possibility that PKU heterozygotes may have less than 50% of control PAH activity. Substituting a value of 40% of control PAH activity for heterozygotes into Eq. 3 yields a steady-state phenylalanine concentration of 0.093 mM; with the use of this value and the value of 0.058 mM for controls, a ratio of 1.60 is obtained, which is close to the range reported for heterozygotes and controls (see above). In this regard, it should be noted that the residual PAH activity in liver biopsy samples found for six HPA obligate heterozygotes ranged between 5.8 and 31% of control values (39). These results provided the first indication that HPA heterozygotes have significantly less than 50% of control activity. Two subsequent larger studies of parents of patients with PKU were in accord with these earlier results: one study reported a mean value of 29.3% of controls (n = 9) (40) and another reported a mean value of 28.1% (n = 8) (41).
The model also predicts t1/2 values for clearance of phenylalanine from blood for both normals and heterozygotes that are in accord with actual clinical results. For normals, a value of 65 min is obtained, which is lower than the reported mean value of 89 min but well within the range of 60–120 min (10). For heterozygotes with 50 and 40% residual PAH activity, the t1/2 values calculated from Eq. 3 are 144 and 180 min, respectively, compared with a reported mean value of 159 min [range, 112–245 min (10)].
Reference has been made earlier to a report of two HPA patients whose inability to metabolize phenylalanine appeared to be a result of a deficiency of transaminase (11) and the evidence against this conclusion (12). The present model provides an additional reason for viewing this claim with skepticism. Fig. 2 shows the time course of disappearance of 1 mM phenylalanine from plasma of a control subject (curve A) as well as from one lacking the transaminase but with normal levels of PAH (curve B). As can be seen, the two rates are almost the same, making it extremely unlikely that pronounced HPA can be caused by a lack of transaminase. The reason for the near identity of the two rates is that the rate of disappearance of phenylalanine in the total absence of PAH (curve D) is very small, the initial rate being only 2.6% of that of a control with normal PAH levels. Fig. 2 (curve C) also displays the rate of disposal of phenylalanine in an individual with 40% of the normal level of PAH, a deficit of PAH activity that, as discussed above, may represent the average for PKU heterozygotes.
Figure 2.

Calculated rates of clearance of a load of phenylalanine for controls and for individuals with different genotypes. A, controls; B, subject with zero transaminase activity; C, subject with 40% of control PAH activity; D, subject with 0% of control PAH activity.
Recently, PKU patients were classified by assigning them to phenotype categories on the basis of their dietary phenylalanine tolerance. Patients with classical PKU tolerate less than 20 mg/kg of phenylalanine per day to maintain their blood phenylalanine levels at the accepted level of 0.3 mM, those with “moderate PKU” tolerate 20–25 mg/kg per day, and those with “mild PKU” tolerate 25–50 mg/kg per day (42).
To see whether these dietary phenylalanine tolerance values are coherent with the predictions made by Eq. 3, it was assumed that the intake of the allowed amount of phenylalanine was divided equally into three “meals.” For classical PKU patients with an intake of phenylalanine of 15 mg/kg per day, each meal would contain 5 mg/kg per day and would add 0.06 μmol/ml to the baseline value of 0.30 μmol/ml for a total plasma phenylalanine level of 0.30 + 0.06 = 0.36 μmol/ml. Substituting this value into Eq. 3 (assuming Vmax for a classical PKU patient is equal to zero), −dPhe/dt is equal to 0.001 μmol/ml per h, i.e., at this level of phenylalanine, the velocity of the disappearance of phenylalanine via the transamination reaction just barely exceeds the velocity of entry of phenylalanine into the plasma pool via net protein degradation. Therefore, Eq. 3 predicts that these PKU patients could tolerate a phenylalanine intake of 15 mg/kg per day.
Calculated in the same way, “moderate PKU” patients with a dietary phenylalanine tolerance of 25 mg/kg per day would require a residual PAH activity equal to 15% of that of wild type to metabolize it in 3.5 h. Similarly, “mild PKU” patients with a dietary phenylalanine tolerance of 50 mg/kg per day would require a residual PAH level of 25% of the wild-type level to metabolize the added phenylalanine in about 3.5 h. These results indicate that Eq. 3 can account for the tolerance of dietary phenylalanine seen in these different patient groups.
It would be useful to try to correlate these estimates of the residual PAH activity for the “mild PKU” and “moderate PKU” patients with the residual hydroxylase activity measured in vitro for the mutant PAH species harbored by the patients. At the present time, however, such an attempt is hampered because there is too much scatter in the in vitro data. Thus, several patients classified as having “moderate PKU” (42) have been shown to harbor the following three mutant forms of PAH (with their in vitro residual PAH activities expressed as a percentage of wild-type activities, shown in parenthesis): L348V (25%), R261Q (30%, 47%), and R158Q (10%) (43). It can be seen that these values vary by almost 5-fold. As discussed previously (2, 43), in general, in vitro estimates of residual hydoxylase activity of PAH mutants tend to be higher than those observed in liver biopsies. At least one reason for this tendency is that in vitro PAH activities are customarily measured by using saturating concentrations of phenylalanine and BH4, as was done for mutant R261Q (44). Given this situation, it is possible that residual PAH activities estimated with the use of Eq. 3 may prove to be a better reflection in in vivo activities than those measured in vitro.
The present model of phenylalanine metabolism is relevant to the conclusion reached by Thompson and his colleagues (45, 46), on the basis of results obtained by infusion of subjects with deuterium-labeled phenylalanine and tyrosine, that classical PKU patients have “substantial” PAH activity that is equal to about 76% that of control subjects. This startlingly high-phenylalanine-hydroxylating activity was attributed to tyrosine hydroxylase (45). As discussed already, the results summarized in Fig. 2 show that in the absence of PAH, a dose of phenylalanine is cleared from the blood at less than 3% of the rate seen in controls. There is no indication from the present analysis that any alternate pathway exists in humans that can dispose of large amounts of phenylalanine. Recently, van Spronsen et al. (34) have pointed out a potential methodological problem with the method used by Thompson and coworkers.
In summary, the quantitative results obtained with the model for PAH metabolism are coherent with data that indirectly reflect the in vivo activity of PAH, such as steady-state blood phenylalanine levels, rates of clearance (conventionally expressed as t1/2 values) of phenylalanine from the blood after a load of phenylalanine, and dietary tolerance for phenylalanine. The model has the potential for quantitatively estimating residual PAH activity from any of these values, particularly from the measured rates of clearances of a load of phenylalanine. The predicted residual PAH levels or the values derived from it may be helpful in making decisions about how stringent the dietary restriction of phenylalanine must be to achieve the desired blood phenylalanine level. Table 1 summarizes the t1/2 values and steady-state blood phenylalanine levels calculated from Eq. 3 (assuming no intake of phenylalanine during the test period) for different levels of residual PAH activity, as well as comparable values from relevant clinical data.
Table 1.
Steady-state phenylalanine blood levels and t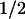 values for clearance of phenylalnine calculated from Eq. 3 for various levels of PAH
values for clearance of phenylalnine calculated from Eq. 3 for various levels of PAH
| Residual PAH activity, % | Steady-state level of Phe, mM |
t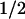 of clearance of a load of Phe, min of clearance of a load of Phe, min |
|---|---|---|
| 100 | 0.057 (0.059) | 65 (89, 60–120) |
| 50 | 0.079 (0.092)* | 144 (159, 112–245)* |
| 40 | 0.093 | 180 |
| 20 | 0.129 | 360 |
| 10 | 0.154 | 705 |
| 3 | 0.226 | 1,410 |
| 0 | 0.314† | 3,240 |
Values in parentheses for steady-state levels of phenylalanine are published mean values. For the t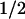 of clearance, values in parentheses are mean values followed by the range of values reported for normals and obligate heterozygotes. See text for references. Phe, phenylalanine.
of clearance, values in parentheses are mean values followed by the range of values reported for normals and obligate heterozygotes. See text for references. Phe, phenylalanine.
For obligate heterozygotes.
This steady-state concentration of Phe was reached in 295 h, during which time it was assumed that no food (i.e., no Phe) was consumed. Clearly, it is not a value that would be reached in any clinical study.
ABBREVIATIONS
- PAH
phenylalanine hydroxylase
- PKU
phenylketonuria
- HPA
hyperphenylalaninemia
- PAG
phenylacetylglutamine
References
- 1.Kaufman S. Adv Enzymol Relat Areas Mol Biol. 1993;67:77–264. doi: 10.1002/9780470123133.ch2. [DOI] [PubMed] [Google Scholar]
- 2.Kaufman S. Tetrahydrobiopterin. Basic Biochemistry and Role in Human Disease. Baltimore: Johns Hopkins Univ. Press; 1997. pp. 1–342. [Google Scholar]
- 3.Scriver C R, Kaufman S, Woo S L C. In: The Metabolic Basis of Inherited Disease. 7th Ed. Scriver C R, Beaudet A L, Sly W S, Valles D, editors. New York: McGraw-Hill; 1989. pp. 495–546. [Google Scholar]
- 4.Trefz F K, Burgard P, König T, Goebel-Schreiner B, Lichter-Konecki U, Konecki D, Schmidt E, Schmidt H, Bickel H. Clin Chim Acta. 1993;217:15–21. doi: 10.1016/0009-8981(93)90233-t. [DOI] [PubMed] [Google Scholar]
- 5.Clarke J T R, Bier D M. Metabolism. 1982;31:999–1005. doi: 10.1016/0026-0495(82)90142-1. [DOI] [PubMed] [Google Scholar]
- 6.Marchini J S, Castillo L, Chapman T E, Vogt J A, Ajami A, Young V R. Metabolism. 1993;42:1316–1322. doi: 10.1016/0026-0495(93)90131-7. [DOI] [PubMed] [Google Scholar]
- 7.Milstien S, Kaufman S. J Biol Chem. 1975;250:4782–4785. [PubMed] [Google Scholar]
- 8.Hsia D Y Y, Driscoll K, Troll W, Knox W E. Nature (London) 1956;178:1239–1240. doi: 10.1038/1781239a0. [DOI] [PubMed] [Google Scholar]
- 9.Knox W E, Messenger E. Amer J Hum Genet. 1958;10:53–60. [PMC free article] [PubMed] [Google Scholar]
- 10.Bremer H J, Neumann W. Klin Wochenschr. 1966;44:1076–1081. [Google Scholar]
- 11.Auerbach V H, DiGeorge A M, Carpenter G G. In: Amino Acid Metabolism and Genetic Variation. Nyhan W L, editor. New York: McGraw-Hill; 1967. pp. 11–68. [Google Scholar]
- 12.Kaufman S. In: Advances in Neurochemistry. Agranoff B W, Aprison M H, editors. New York: Plenum; 1977. pp. 1–132. [Google Scholar]
- 13.Diem K, editor. Documenta Geigy Scientific Tables. 6th Ed. Ardsley, NY: Geigy Pharmaceuticals; 1962. p. 608. [Google Scholar]
- 14.Jervis G A. Proc Soc Exp Biol Med. 1950;75:83–86. doi: 10.3181/00379727-75-18108. [DOI] [PubMed] [Google Scholar]
- 15.Güttler F. Acta Paediatr Scand. 1980;280:7–80. [PubMed] [Google Scholar]
- 16.Kowlessur D, Citron B A, Kaufman S. Arch Biochem Biophys. 1996;333:85–95. doi: 10.1006/abbi.1996.0367. [DOI] [PubMed] [Google Scholar]
- 17.Martinez A, Knappskog P M, Olafsdottir S, Døskeland A P, Eiken H G, Svebak R M, Bozzini M, Apold J, Flatmark T. Biochem J. 1995;306:589–597. doi: 10.1042/bj3060589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips R S, Parniak M A, Kaufman S. J Biol Chem. 1984;259:271–277. [PubMed] [Google Scholar]
- 19.Segel I H. Enzyme Kinetics. New York: Wiley; 1975. [Google Scholar]
- 20.Güttler F, Hansen G. Scand J Clin Lab Invest. 1977;37:717–722. doi: 10.3109/00365517709101855. [DOI] [PubMed] [Google Scholar]
- 21.Guldberg P, Mikkelsen I, Henriksen K F, Lou H C, Güttler F. Eur J Pediatr. 1995;154:551–556. doi: 10.1007/BF02074833. [DOI] [PubMed] [Google Scholar]
- 22.Langenbeck U, Behbehani A, Mench-Hoinowski A. J Inherit Metab Dis. 1992;15:136–144. doi: 10.1007/BF01800355. [DOI] [PubMed] [Google Scholar]
- 23.Kuhl W J, Jr, Beck E M, Gershberg H, Street E, Ralli E P. Metabolism. 1952;4:143–152. [PubMed] [Google Scholar]
- 24.Mountcastle V B. Med Phys. 1968;1:288. [Google Scholar]
- 25.Meister A. Biochemistry of the Amino Acids. New York: Academic; 1965. p. 907. [Google Scholar]
- 26.Seakins J W T. Clin Chim Acta. 1971;35:121–131. doi: 10.1016/0009-8981(71)90302-0. [DOI] [PubMed] [Google Scholar]
- 27.Oates J A, Nirenberg P Z, Jepson J B, Sjoerdsma A, Udenfriend S. Proc Soc Exp Biol Med. 1963;112:1078–1081. doi: 10.3181/00379727-112-28256. [DOI] [PubMed] [Google Scholar]
- 28.Knox W E. In: The Metabolic Basis of Inherited Disease. Stanbury J B, Wyngaarden J B, Fredricksons D S, editors. New York: McGraw-Hill; 1972. pp. 266–295. [Google Scholar]
- 29.Friedman P A, Fisher D B, Kang E S, Kaufman S. Proc Natl Acad Sci USA. 1973;70:552–556. doi: 10.1073/pnas.70.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstein F B. J Biol Chem. 1961;236:2656–2661. [PubMed] [Google Scholar]
- 31.Waterlow J C, Jackson A A. Brit Med Bulletin. 1981;37:5–10. doi: 10.1093/oxfordjournals.bmb.a071676. [DOI] [PubMed] [Google Scholar]
- 32.Clowes G H A, Jr, Randall H T, Cha C-J. J Parenter Enteral Nutr. 1980;4:195–203. doi: 10.1177/014860718000400225. [DOI] [PubMed] [Google Scholar]
- 33.White D R. In: Encyclopedia of Human Biology. Dulbecco R, editor. Vol. 6. San Diego: Academic; 1991. p. 437. [Google Scholar]
- 34.van Spronsen F J, Reijngoud D-J, Smit G P A, Nagel G T, Stellaard F, Berger R, Heymans H S A. J Clin Invest. 1998;101:2875–2880. doi: 10.1172/JCI737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scriver C R, Gregory D M, Sovetts D, Tissenbaum G. Metabolism. 1985;34:868–873. doi: 10.1016/0026-0495(85)90112-x. [DOI] [PubMed] [Google Scholar]
- 36.Stegink L D, Filer L J, Jr, Brummel M C, Baker G L, Krause W L, Bell E F, Ziegler E E. Am J Clin Nutr. 1991;53:670–675. doi: 10.1093/ajcn/53.3.670. [DOI] [PubMed] [Google Scholar]
- 37.Hilton M A, Sharpe J N, Hicks L G, Andrews B F. J Pediatr. 1986;109:601–604. doi: 10.1016/s0022-3476(86)80220-7. [DOI] [PubMed] [Google Scholar]
- 38.Freehauf C L, Lezotte D, Goodman S I, McCabe E R B. Am J Hum Genet. 1984;36:1180–1189. [PMC free article] [PubMed] [Google Scholar]
- 39.Kaufman S, Max E E, Kang E S. Pediatr Res. 1975;9:632–634. doi: 10.1203/00006450-197508000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Bartholomé K. Hum Genet. 1979;5:1241–1245. [Google Scholar]
- 41.Grimm U, Knapp A, Schlenzka K, Hesse R. Acta Biol Med Germ. 1977;36:1179–1182. [PubMed] [Google Scholar]
- 42.Guldberg P, Rey F, Zschocke J, Romano V, Francois B, Michiels L, Ullrich K, Hoffmann G F, Burgard P, Schmidt H, et al. Am J Hum Genet. 1998;63:71–79. doi: 10.1086/301920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waters P J, Parniak M A, Nowacki P, Scriver C R. Hum Mutat. 1998;11:4–17. doi: 10.1002/(SICI)1098-1004(1998)11:1<4::AID-HUMU2>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 44.Knappskog P M, Eiken H G, Martinez A, Olafsdottir S, Haavik J, Flatmark T, Apold J. Adv Exp Med Biol. 1993;338:59–62. doi: 10.1007/978-1-4615-2960-6_11. [DOI] [PubMed] [Google Scholar]
- 45.Thompson G N, Halliday D. J Clin Invest. 1990;86:317–322. doi: 10.1172/JCI114701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Treacy E, Pitt J J, Seller K, Thompson G N, Ramos S, Cotton RGH. J Inher Metab Dis. 1996;19:595–602. doi: 10.1007/BF01799832. [DOI] [PubMed] [Google Scholar]