

Abstract
The lack of professional afferent APCs in naive brain parenchyma contributes to the systemic immune ignorance to Ags localized exclusively within the brain. Dendritic cells (DCs) appear within the brain as a consequence of inflammation, but no molecular mechanisms accounting for this influx have been described. In this study we demonstrate that Fms-like tyrosine kinase 3 ligand (Flt3L) recruits plasmacytoid DCs (pDCs; >50-fold; p < 0.001) to the brain parenchyma. These pDCs expressed IFN-α, the hallmark cytokine produced by pDCs, indicating recruitment and activation in situ of bona fide pDCs within the brain parenchyma. Flt3L did not increase the numbers of conventional DCs, macrophages, or B, T, NK, NKT, or microglial cells within the brain. Our data demonstrate that Flt3L reconstitutes a crucial afferent component of the immune response, namely, professional APCs within the brain parenchyma, and this could counteract the intrinsic systemic immune ignorance to Ags localized exclusively within the brain.
Antigenic epitopes localized exclusively within the brain parenchyma (i.e., brain proteins, viruses, and tumors) do not prime a systemic immune response (1–3). Upon careful Ag delivery selectively to the brain parenchyma of healthy rats and mice, neither systemic activation nor tolerance against such Ags has been observed. However, systemically activated T cells display normal effector T cell function toward Ags localized in the brain, e.g., in autoimmune multiple sclerosis or in animals experimentally immunized against viral or tumor Ags localized within the brain (1, 3, 4). This implies that immune ignorance to brain Ags is due to defects in the afferent limb and not the efferent limb of the adaptive immune system.
The brain parenchyma lacks two central cellular and anatomical components necessary for priming an immune response, i.e., dendritic cells (DCs)3 and classical lymphatic outflow channels (5, 6). Therefore, the neuroanatomy of the brain immune components explains the particular characteristics of immune responses against brain Ags. DCs are unique in their ability to phagocytose foreign Ags, migrate to secondary lymphoid structures, display antigenic epitopes to naive T cells, and elicit potent T cell-dependent immune responses (7).
Conventional DCs (cDCs) and plasmacytoid DCs (pDCs) can be found in the cerebrospinal fluid (CSF), choroid plexi, and brain meninges in healthy animals and healthy humans. The noninflamed brain parenchyma, unlike the ventricles, choroid plexi, and meninges, is devoid of DCs. However, resident brain microglia, perivascular macrophages, endothelial cells, and astrocytes can all increase the expression of MHC class II (MHC II) and MHC I during brain inflammation, suggesting that they display Ags to the activated arm of the immune system through interaction with Ag-specific, primed CD4+ and CD8+ T cells, but not to naive T cells (8). None of these cells, however, has been conclusively shown to take up Ags, leave the brain parenchyma, carry the Ag to cervical lymph nodes, or present antigenic epitopes to naive T cells.
DCs are present in the brain during chronic inflammation, such as in multiple sclerosis (MS) (9, 10) and experimental autoimmune encephalitis (EAE) (11, 12), and MHC II expression on CD11c+ DCs was found to be both sufficient and necessary for complete generation of the clinical disease, underscoring the importance of CD11c+ DCs in presenting brain Ag to T cells (13). OX62+ DCs were found to infiltrate the brain parenchyma in response to an excitotoxic inflammatory lesion in the rat striatum. Depletion of bone marrow, but not perivascular- and meningeal-derived macrophages, abrogated this effect, suggesting that OX62+ DCs were derived from blood-borne immune cells (14). DCs can also infiltrate the brain parenchyma in response to the chronic presence of prions in the brain (15). However, little is currently known about the molecular mechanisms mediating DC infiltration or differentiation within the brain parenchyma during inflammation.
In this study we demonstrate that elevated expression of Fms-like tyrosine kinase 3 ligand (Flt3L) in the brain is sufficient to selectively recruit and activate pDCs, but not other immune cells, in the brain parenchyma. Flt3L is constitutively expressed by cells resident in the brain, suggesting that aberrant expression may increase the capacity of the brain to present Ag to naive, Ag-specific T cells. We have recently shown that Flt3L expression in intracranial tumors induced a robust, CD4-dependent, antitumor immune response in a rat model of brain cancer when used in combination with the conditionally cytotoxic gene thymidine kinase (16). It has been recently shown that systemic administration of Flt3L increased DCs in the brain meninges and augmented the severity of clinical symptoms in a mouse model of MS (13). Inhibition of Flt3 signaling using the small drug inhibitor of Flt3R CEP-701 significantly decreased autoreactive immune cells and improved clinical symptoms in a mouse model of established EAE (17).
In summary, our results demonstrate that Flt3L expression in the brain significantly, selectively, and specifically increases the number of pDCs in the brain parenchyma. This could overcome immune ignorance of brain Ags by promoting presentation of brain-derived Ag to T cells. The recent description of cDCs being able to migrate from the CSF to the cervical lymph nodes (18) and other supporting evidence from Carson et al. (19) and Pashenkov et al. (20) indicate that although the brain lacks classical lymphatic outflow channels, it may be able to compensate for this during disease states that cause brain inflammation.
Materials and Methods
Construction of adenoviral vectors
β-Galactosidase (β-Gal) and Flt3L were expressed in the brains of Lewis rats using first-generation, replication-defective, recombinant adenovirus type 5 (RAd5) vectors expressing transgenes under transcriptional control of the human CMV major intermediate early promoter within the E1 region. RAd0 is a first-generation adenoviral vector that does not contain any transgene and is used to control total virus load. RAd35 (an adenovirus encoding β-Gal) and RAdFlt3L (an adenovirus encoding human soluble Flt3L) have been described previously (21, 22). The methods for adenoviral generation, production, characterization, and scale up and for viral vector purification have been previously described (23, 24). The titers were 6.55 × 1011 IU/ml for RAd0, 1.64 × 1011 IU/ml for RAd35, and 6.55 × 1011 IU/ml for RAd-human serum Flt3L. All viral preparations used were free from replication-competent adenovirus and LPS contamination (24). Viruses were diluted in sterile saline for intracranial injection.
Production of anti-Flt3L Abs
We generated rabbit Abs specific for human soluble Flt3L. Briefly, New Zealand White rabbits were immunized three times (14 days apart) with purified peptide CETVFHRVSQDGLDL coupled with keyhole limpet hemocyanin (New England Peptides). This peptide sequence is present in soluble, but not bound, isoforms of human Flt3L and is not conserved with rodent Flt3L isoforms. After the third immunization, the titer of the Ab was verified by ELISA (1/64,000), and whole serum was recovered for characterization by Western blot and immunohistochemistry.
Intracranial injection of adenoviral vectors
Male Lewis rats (225–250 g in body weight) were purchased from Harlan Sprague Dawley. All sentinel rats housed in the same colony were specific pathogen free. Animals were anesthetized with ketamine and medatomidine and placed in a stereotaxic frame. The rats were then injected in the right striatum (+1 mm forward from bregma, +3 mm lateral, and −4 mm ventral from the dura) with saline or adenoviral vectors diluted in saline. A hole was drilled in the skull, and the vectors were administered in a volume of 2 μl using a 10-μl Hamilton syringe with a 26-gauge needle. A small pocket was created before the deposition of virus by holding the needle 0.2 mm below the stated ventral coordinates for 1 min before moving up to the stated coordinates and slowly injecting the virus over a period of 2 min. The needle was left in place for an additional 5 min before being carefully removed from the brain.
Flow cytometry of brain mononuclear cells
Rats were perfused with 100 ml of heparinized Tyrode’s solution 1 or 5 days after intracranial injection of virus or saline. Splenocytes were harvested, and RBC were removed using ammonium chloride and potassium solution. Brains were separated from the meninges and removed from the skull. The area around the injection site was dissected, taking care to avoid the ventricles (Fig. 1A). This tissue was then diced with a razor blade before homogenization in RPMI 1640 medium (Invitrogen Life Technologies) using a glass Tenbroeck homogenizer. Mononuclear cells were purified from brain tissue by centrifugation through a Percoll gradient; mononuclear cells migrate to the interface between 30 and 70% Percoll. Cells were counted and labeled with Abs for analysis by flow cytometry. Briefly, cells were resuspended at 5 × 105 cells/ml in 1 ml of cell surface staining buffer (0.1 M PBS without Ca2+ and Mg2+ and with 1% FBS and 0.1% sodium azide). Cells were centrifuged, and the supernatant was discarded. The cells were resuspended in 100 μl of cell surface staining buffer containing the combinations of Abs described below and incubated for 30 min at 4°C. After this incubation, the samples were washed in 1 ml of cell surface staining buffer and analyzed by flow cytometry. All Abs used were purchased from BD Pharmingen unless otherwise indicated. CD3-FITC, CD45R (B220)-PE and CD4-PE/Cy5 were used to distinguish B cells and pDCs. Samples were stained with CD11c-FITC (Serotec), CD45-PE, and CD4-PE/Cy5 to identify cDCs and resident brain microglia. CD3-FITC, CD8a-PE, and CD4-PE/Cy5 were used to label CD4+ and CD8+ T cells. CD3-FITC and CD161a-PE were used to analyze NK cells, NKT cells, and total T cells. CD11b-FITC and CD45-PE were used to stain for macrophages.
FIGURE 1.
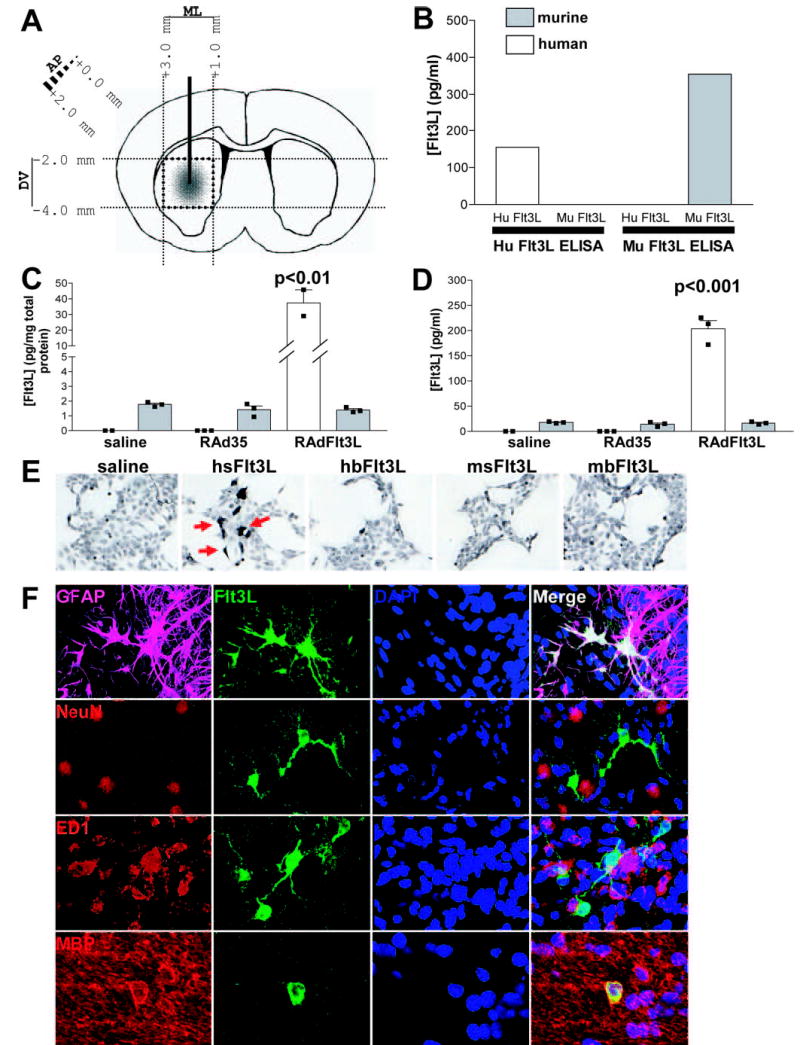
Expression of Flt3L in rat brain using RAdFlt3L. A, Schematic diagram outlining the injection of RAdFlt3L into rat brain striatum and the subsequent dissection of brain tissue around the injection site while carefully avoiding ventricles and meninges. B, Human Flt3L ELISA is specific for recombinant human Flt3L and does not cross-react with recombinant mouse Flt3L. Rodent Flt3L ELISA does not cross-react with recombinant human Flt3L. C, Human Flt3L was elevated 20-fold compared with endogenous rat Flt3L in the brain when animals were injected with RadFlt3L. However, endogenous Flt3L did not change in response to viral injection into the brain. D, Injection of RAdFlt3L in the brain elevated serum concentrations of Flt3L 10-fold over those of endogenous rat serum Flt3L, the concentration of which was not found to change in response to viral injection. E, HEK 293 cells were plated on coverslips in 6-well plates at a density of 1.5 × 105 cells/well. The following day, medium was changed, and cells were transfected with 1 μg of plasmid DNA (pAL119-hsFlt3L, pAL119-hbFlt3L, pAL119-msFlt3L, or pAL119-mbFlt3L) using GeneJuice (Novatech) according to the manufacturer’s instructions. After 48 h, cells were fixed in 4% paraformaldehyde for 5 min and stained for human soluble Flt3L using rabbit anti-Flt3L Ab. Goat anti-rabbit-HRP secondary Ab and 3,3′-diaminobenzidine were used to visualize Flt3L-expressing cells. F, Confocal image showing the expression of human Flt3L (green) and GFAP (magenta), NeuN (red), ED1 (red), or MBP (red), as indicated, within the brain striatum. Double labeling of Flt3L with GFAP- (white) and MBP (yellow)-expressing cells indicates that the majority of RAdFlt3L infected astrocytes and oligodendrocytes. Flt3L was not found to colocalize with NeuN (yellow), suggesting that RAdFlt3L was not expressed in rat neurons. We also observed double labeling of ED1 and RAdFlt3L by confocal microscopy (yellow). However, transverse slices revealed that Flt3L does not colocalize with ED1 and was, instead, indicative of contacts or close association between activated microglia and RAdFlt3L-expressing cells, rather than actual expression of Flt3L by ED1-positive microglia. Results are representative of five animals analyzed in each group.
Immunohistochemistry
Animals were killed by perfusion/fixation with heparinized Tyrode’s solution (132 mM NaCl, 1.8 mM CaCl2, 0.32 mM NaH2PO4, 5.56 mM glucose, 11.6 mM NaHCO3, and 2.68 mM KCl), followed by 4% paraformaldehyde in PBS at the desired time points. Brains were removed for immunohistochemical analysis. Fifty-micrometer-thick coronal sections of fixed brains were cut using a Vibratome (Leica VT10005), and free-floating immunohistochemistry was performed as described elsewhere (22, 25). Briefly, rabbit anti-Flt3L was used at a concentration of 1/1000 for immunohistochemistry and immunofluorescence. An Axioplan II microscope and imaging software (Zeiss) were used to obtain images of 3,3′-diaminobenzidine-stained cells. For immunofluorescence labeling, rabbit anti-Flt3L was detected using anti-rabbit Alexa 488 (1/1000; Molecular Probes). Guinea pig anti-glial fibrillary acidic protein (anti-GFAP; 1/1000, Advanced Immunochemical) was detected using anti-guinea pig Alexa 647 (1/800; Molecular Probes). Mouse anti-ED1 (1/1000; Serotec), mouse antineuronal-specific nuclear protein (1/1000; Chemicon International), and mouse anti-myelin basic protein (anti-MBP; 1/1000; Chemicon International) were detected with anti-mouse Alexa 594 (1/800; Molecular Probes). Confocal images were taken using a Leica DMIRE2 microscope with a ×63 oil objective and Leica confocal software. Confocal images of each section were taken at a 0.5-μm-thick resolution and were overlaid to produce the final images.
RT-PCR from rat brain tissue homogenate
RNA was extracted from rat brain tissue, isolated from striatum tissue dissected as indicated in Fig. 1A and injected 1 or 5 days previously with saline, RAd35, or RAdFlt3L using RNABee reagent (Tel-Test). An RT reaction was set up using 0.5 μg of RNA in each sample and oligo(dT) primer with Moloney murine leukemia virus reverse transcriptase. Primers specific for GAPDH (forward, 5′-ACC ACA GTC CAT GCC ATC AC-3′; reverse, 5′-TCC ACC ACC CTG TTG CTG TA-3′), IFN-α (forward, 5′-CAG CAG ATC CAG AAG DCT CAA RC-3′; reverse, 5′-CAG GCA CAG GGR CTG TGT TT-3′; D = A, G, or T; R = A or G), IFN-α (forward, 5′-TGC TGT GCT TCT CCA CCA CT-3′; reverse, 5′-TAG TCT CAT TCC ACC CAG TGC T-3′), TLR7 (forward, 5′-TTT CCC AGA GCA TAC AGC TCA G-3′; reverse 5′-CAC TCA AGG ACA GAA CTG CTG C-3′), TLR9 (forward, 5′-CCT GGC ACA CAA TGA CAT TCA-3′; reverse, 5′-TAA AGG TCC TCC TCG TCC CA-3′), MHC II (forward, 5′-CCC CAG ATG CCA ATG TGA-3′; reverse, 5′-TCA GAT GCT GTC ACA GCA-3′), and CD80 (forward, 5′-CTG CTG GTT GGT CTA TTC CA-3′; reverse, 5′-TCA GAT TCA GGA TCC TGG GAA-3′) were subsequently used to amplify target sequences from the cDNA using PCR. An annealing temperature of 55°C was used, and the number of cycles is indicated in the figure legends. DNA bands were separated using agarose gel electrophoresis and were visualized using a gel documentation system (Alpha Innotech).
Purification of pDCs from brain and spleen
Brain tissue from 30 rat brain hemispheres injected 5 days previously with RAdFlt3L was processed as described above to isolate mononuclear cells. These cells were pooled and stained with CD3-FITC, CD45R-PE, and CD4-PE/Cy5. Spleen cells from a single rat injected with RAdFlt3L 5 days previously were also purified and treated with ammonium chloride and potassium solution to remove erythrocytes before staining with CD3-FITC, CD45R-PE, or CD4-PE/Cy5. The pDCs were sorted using an Ario cell sorter (DakoCytomation), gating live leukocytes for CD3−, CD45+, and CD4+. We isolated 10,000 pDCs from the pooled brain mononuclear cells. We also isolated 10,000 pDCs from the spleen as a positive control sample. Five thousand spleen pDCs and 5000 brain pDCs were stimulated with 20 μg/ml CpG for 3 h in X-Vivo10 medium (Cambrex). The remaining 5000 pDCs from spleen and brain were incubated for 3 h in X-Vivo10 medium without CpG. Total RNA was isolated from samples, as outlined above, using RNAbee reagent, and 50 μg of tRNA was used as the carrier nucleic acid during isopropanol and ethanol precipitation steps. RT-PCR was performed as outlined previously.
Flt3L ELISA
Blood was collected and allowed to coagulate before centrifugation to isolate serum. Animals were killed by terminal perfusion with heparinized Tyrode’s solution. Brains were separated from the meninges and removed from the skull. Blocks of brain tissue around the injection site were homogenized into a single-cell suspension. Multiple freeze-thaw cycles were used to extract cellular protein, which was separated from insoluble tissue by centrifugation and stored on ice until use. Quantikine ELISA kits for rat Flt3L and human Flt3L were purchased from R&D Systems and were used as recommended in the manufacturer’s instructions.
Statistical analyses
Statistical analysis was performed using one-way nonparametric ANOVA to compare all samples. Grubb’s test for outliers was used to remove extraneous data points, and Dunn’s multiple comparison was used to compare all samples against saline-injected animals. A difference of p < 0.05 was considered significant.
Results
Overexpression of Flt3L within brain: levels and cell type-specific expression
It was important to assess the levels as well as the cell types within the brain that expressed human Flt3L after injection of RAdFlt3L and compare them with the levels of endogenous rodent Flt3L. We used purified recombinant human and mouse Flt3L to confirm that human and rodent Flt3L ELISA kits (R&D Systems) were species specific (Fig. 1B). We assessed by ELISA the concentrations of transgene and endogenous Flt3L expressed in both brain and serum of rats 5 days after intracranial injection of adenoviral vectors. As expected, a high concentration of human Flt3L (>30 pg/mg total brain protein) was observed in the brains of rats injected with RAdFlt3L, whereas no expression was detected in saline- and control virus-injected samples. The concentration of endogenous rat Flt3L expressed in the brain of saline-injected animals (<2 pg/mg total brain protein) was calculated to be >15 times lower than that of human Flt3L transgene in the brains of rats infected with RAdFlt3L, and endogenous levels of Flt3L expression did not change in response to the injection of either RAd35 or RAdFlt3L (Fig. 1C). We also detected >200 pg/ml human Flt3L transgene in rat sera 5 days after infection with RAdFlt3L, indicating that significant quantities of soluble human Flt3L expressed within the brain diffused across the blood-brain barrier and entered the systemic circulation. Endogenous Flt3L expression was not found to change in response to either RAdFlt3L or RAd35, and serum concentrations of human Flt3L in RAdFlt3L-infected animals were >10-fold higher than those of endogenous rat Flt3L (<20 pg/ml; Fig. 1D). These results suggest that increasing the serum concentration of Flt3L by 10-fold (200 pg/ml) is insufficient to significantly expand populations of either pDCs or cDCs in the rat spleen.
Next, we determined which cell types within the brain expressed human Flt3L after intracranial injection of RAdFlt3L. We raised an Ab against human soluble (hs) Flt3L using an epitope not present on any rodent or bound human isoform of Flt3L. We tested the specificity of the antiserum for human soluble Flt3L in HEK 293 cells transfected with the human and mouse isoforms of Flt3L (Fig. 1E). Preincubation with a blocking peptide completely blocked immunoreactivity, confirming that the antiserum was specifically binding the Ag against which it was raised (data not shown). We used confocal microscopy to demonstrate that human Flt3L expression in the brain was only evident within the striatum and was predominantly associated with GFAP, a marker of astrocytes (~90%), and MBP, a protein selectively expressed by oligodendrocytes in the brain (~10%; Fig. 1F).
Flt3L increases a putative pDC population (CD3−CD45R+CD4+), but not cDCs, in brain parenchyma
The brain parenchyma is usually devoid of mature DCs except during brain inflammation, e.g., injection of LPS (26), intracerebral viral infections, or autoimmune diseases (27). It is believed that the absence of a local lymphatic system and resident DCs capable of phagocytosing local Ags and migrating to lymphatic structures, where they can present antigenic epitopes to naive T cells, is a major factor that prevents the induction of Ag-specific immune responses after careful injection of infectious particulate Ag into brain parenchyma (2, 4). We hypothesized that the expression of Flt3L in the striatum would induce the migration and differentiation of DC populations within the brain, because Flt3L is sufficient to promote all stages of DC differentiation from precursors. Infiltrating mononuclear cells in the brain were quantified in a 2-mm3 cube of brain tissue surrounding the injection site, carefully dissected to avoid including the ventricular system that constitutively contains DCs (Fig. 1A).
We found that the average number of pDCs (CD3−CD4+CD45R+) in brain parenchyma increased >50-fold after Flt3L was expressed in the brain for 5 days compared with that after saline only ( p < 0.001; Fig. 2, A and C). The number of pDCs 5 days after the expression of β-Gal was modestly elevated compared with that after saline; this difference was not statistically significantly ( p > 0.05). The number of pDC in Flt3L-injected animals was significantly higher than that in β-Gal-injected animals ( p < 0.001). This indicates that Flt3L, and not viral particles or other virally expressed proteins, mediated the increase in pDCs in the brain.
FIGURE 2.
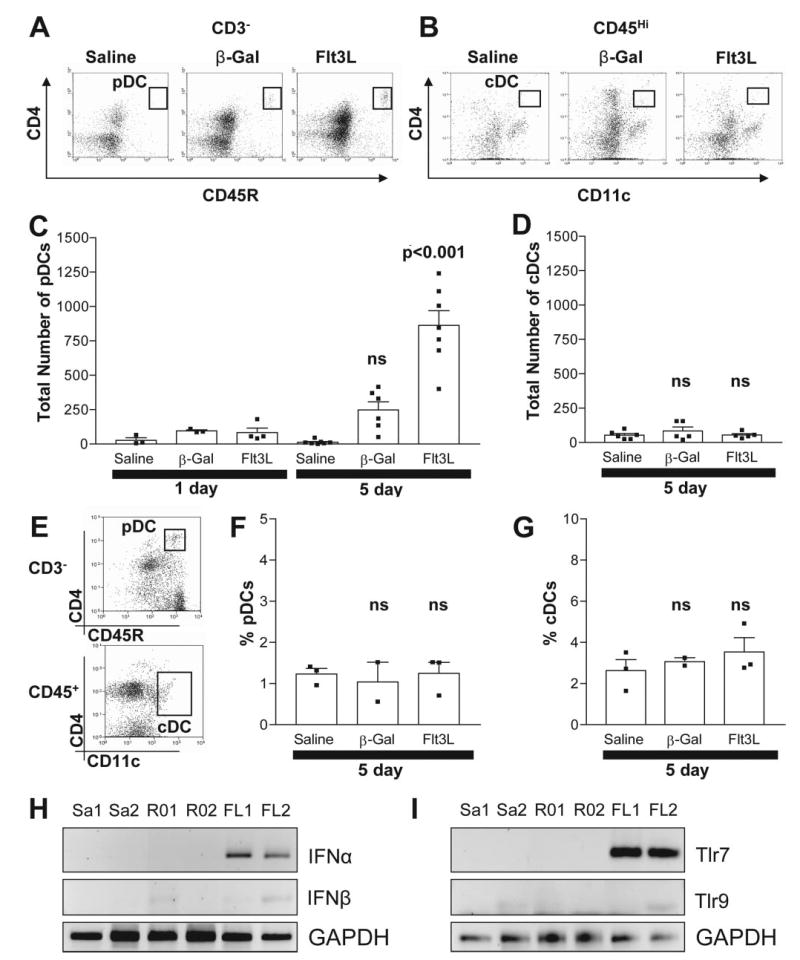
Intracranial injection of RAdFlt3L increased infiltrating pDC (CD3−CD45R+CD4+), IFN-α and TLR7 in brain parenchyma. A, Detection of pDC (CD3−CD4+CD45R+) in brain parenchyma using flow cytometry. B, Detection of CD4+ cDC (CD4+CD11c+CD45high) in brain parenchyma using flow cytometry. C, Absolute numbers of pDC in brain parenchyma are elevated 5 days after the injection of RAdhsFlt3L compared with those in control animals (p < 0.001). D, CD4+ cDCs are not elevated in brain parenchyma of animals 5 days after intracranial delivery of RAdFlt3L compared with controls (NS, p > 0.05). E, Detection of pDCs (CD3−CD4+CD45R+; upper panel) and cDCs (CD4+CD11c+CD45high; lower panel) in spleen using flow cytometry. The percentage of pDCs (F) or cDCs (G) in the spleen did not change in response to either Flt3L expression or adenoviral delivery into the brain. Rats were injected in the striatum with saline (Sa), RAd0 (R0), or RAdFlt3L (FL). RNA was extracted from brain tissue surrounding the injection site after either 1 or 5 days, and RT-PCR was used to determine type I IFN (H) and TLR (I) expression. We found that IFN-α (H) and TLR7 (I) were transiently increased in response to Flt3L expression in the brain compared with saline and RAd0 controls after 1 day. IFN-β (H) and TLR9 (I) were not altered. Twenty-five cycles of PCR were used to detect GAPDH, and 30 cycles of PCR were used to detect IFN-α, IFN-β, TLR7, and TLR9 expression. Flow cytometry dot plots (A, B, and E) indicate the cell populations quantified and are representative of staining observed in 5-day samples. Error bars in C, D, F, and G depict the SEM.
No change in the total number of pDCs was observed after the expression of either Flt3L or β-Gal for 1 day compared with saline (p > 0.05). Low levels of CD4+ cDC (CD4+CD11c+CD45+) were found in the brains of rats injected in the striatum 5 days previously with saline, and this was not increased in rats expressing either β-Gal or hsFlt3L (Fig. 2, B and D), indicating that elevated expression of Flt3L in the brain specifically recruited pDCs, and not cDCs. No CD4− cDCs were detected in any brain samples (Fig. 2B and data not shown). A large population of CD11c+ CD4low immune cells was detected in the brains of animals treated with saline, and this corresponds to a subpopulation of macrophages, but treatment with Flt3L did not elevate these immune cells (Fig. 2B and data not shown).
To determine whether leakage of Flt3L from the brain into the general circulation could be implicated in the increase in pDCs in the brain, we analyzed DC subpopulations in the spleen after intracranial expression of Flt3L. Although we did detect a significant increase in the serum levels of Flt3L, we did not detect changes in total pDC numbers (Fig. 2, E and F) or cDC numbers (Fig. 2, E and G) in the spleen compared with control animals. This suggests that a high level of intraparenchymal Flt3L, rather than an increase in serum Flt3L, is necessary to increase pDCs in the brain. Thus, our data indicate that pDCs infiltrating from the CSF or brain meninges or differentiation of precursor cells within the brain parenchyma are more likely sources of pDCs observed within the brain parenchyma in response to Flt3L.
Global expression of IFN-α and TLR7 transiently increases in brain parenchyma in response to RAdFlt3L
Plasmacytoid pDCs, unlike cDCs, secrete very high concentrations of type I IFN in response to viral infections and TLR stimulation, 100- and 1000-fold greater than other cells, including immune cells infected with virus (28). We determined whether the injection of adenoviral vectors alone into the brain was sufficient to induce the expression of IFN-α in the brain. We isolated total RNA from the brain striatum 1 and 5 days after the injection of saline, RAd0, or RAdFlt3L and analyzed the expression of IFN-α. We found that Flt3L expression for 1 day, but not the control vectors, increased the expression of IFN-α (Fig. 2H) and TLR7 (Fig. 2I). IFN-β and TLR9 did not increase (Fig. 2, H and I). This global increase in brain expression of IFN-α and TLR7 in response to Flt3L was transient in nature and returned to basal levels after 5 days (data not shown). However, pDCs were absent from the brain 1 day after the expression of Flt3L. Thus, the global increase in IFN-α and TLR7 expression 1 day after the expression of Flt3L originated from a different cell type, e.g., brain perivascular macrophages or microglia, rather than pDCs.
Putative pDCs (CD3−CD45R+CD4+) isolated from brain constitutively express IFN-α
We verified whether pDCs identified by immunocytochemistry and isolated from the brain after treatment with Flt3L were capable of producing IFN-α. IFN-α expression in response to viral or TLR stimulation (100–1,000 times more than normal cells) is a hallmark of immature pDCs. We used cell sorting to purify CD3−CD45R+CD4+ cells from brain homogenates of rats 5 days after intracranial Flt3L delivery (Fig. 3A). A total of 10,000 pDCs was isolated from 30 brain hemispheres carefully dissected to avoid including any ventricular tissue. We also purified 10,000 pDCs from the spleen of a rat expressing Flt3L in the brain for 5 days as a positive control for pDC function. The pDCs isolated from brain and spleen expressed similar levels of MHC II mRNA, but did not express the costimulatory molecule CD80, indicating that pDCs in the brain and those isolated from the spleen were immature pDCs (Fig. 3B). MHC II-expressing cells in the brain were detected in response to Flt3L expression using confocal microscopy, but CD45R cells did not express detectable levels of MHC II by immunocytochemistry, suggesting that brain pDCs express MHC II mRNA, but do not express enough protein at the membrane to be detected by immunocytochemistry. Most likely this reflects the immature nature of brain pDCs detected (Fig. 3, C and D). Using immunofluorescent labeling of CD45R to identify the anatomical location of pDCs within the brain, we found that pDCs were present throughout the striatum of animals treated with Flt3L. Furthermore, we did not observe any CD45R immunoreactivity in intact meninges from animals 5 days after expression of either β-Gal (Fig. 3E) or Flt3L (Fig. 3F), indicating that the expression of Flt3L in brain parenchyma does not induce pDC infiltration or expansion in brain meninges.
FIGURE 3.
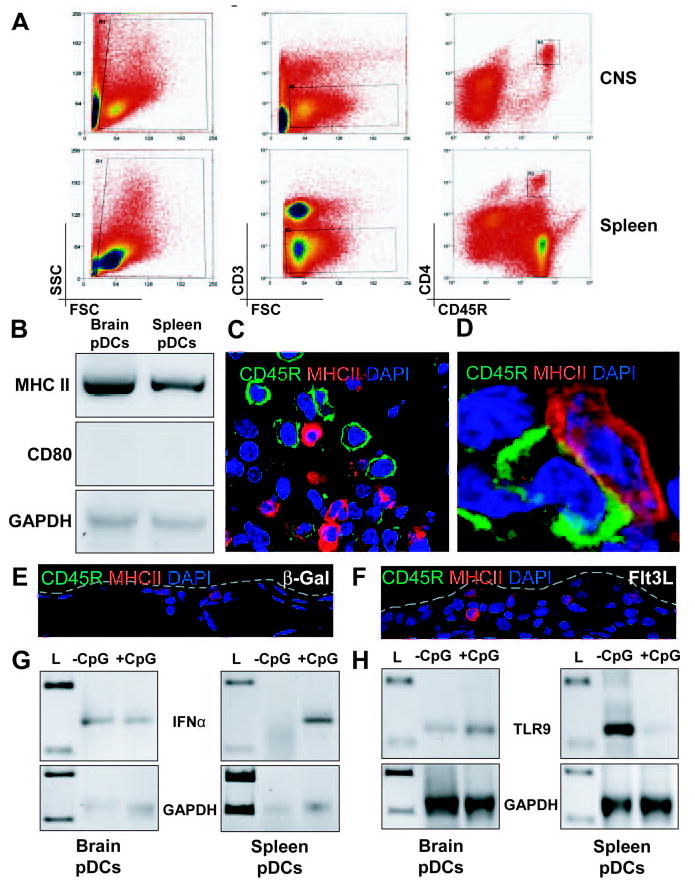
Brain-infiltrating pDCs are IFN-producing cells and constitutively express high levels of IFN-α. A, Brain mononuclear cells from 30 brain hemispheres treated with RAdFlt3L were pooled and stained, and 10,000 CD3−CD45R+CD4+ cells were collected. Ten thousand CD3−CD45R+CD4+ cells were also collected from the spleen. B, We used 40 cycles of PCR to detect the expression of MHC II and CD80 in unstimulated pDCs isolated from brain or spleen. Both brain and spleen pDCs expressed MHC II (top panel), but we did not detect any expression of CD80 (middle panel). GAPDH was also analyzed to demonstrate equal loading of samples (bottom panel). C, Cells expressing CD45R and MHC II were present in the brains of rats 5 days after the injection of RAdFlt3L. However, CD45R cells were not found to express MHC II. D, Some overlap between CD45R and MHC II was found at cell junctions, indicating close association of the cell membranes. CD45R+/CD4+ cells infiltrated the brain. E and F, Cells expressing MHC II (red), but not CD45R (green), were observed in intact meninges of animals 5 days after the expression of either RAd35 (E) or RAdFlt3L (F). The borders of the meninges are outlined in gray. G, Thirty-five cycles of PCR were used to detect IFN-α and GAPDH expression in cDNA copies of total RNA isolated from 10,000 purified brain or splenic pDCs incubated with 20 μg/ml CpG for 3 h or left untreated. IFN-α was detected in spleen pDCs only after treatment with CpG (right panel), but was constitutively expressed in pDCs purified from brain mononuclear cells (left panel). H, Forty cycles of PCR were used to detect TLR9 and GAPDH expression in purified pDC samples isolated from brain and spleen. pDCs were incubated with or without 20 μg/ml CpG for 3 h before total RNA was isolated for RT-PCR analysis. TLR9 was expressed at lower levels in brain pDCs (left panel) than in unstimulated spleen-derived pDCs, but was reduced in spleen pDCs after incubation with CpG (right panel). L, lanes containing DNA ladder.
Brain-derived pDCs constitutively expressed IFN-α mRNA (Fig. 3G, left), and incubation with the TLR9 agonist CpG did not further increase the level of IFN-α expression (Fig. 3G, left). As a control, pDCs isolated from the spleen did not express detectable levels of IFN-α, and stimulation with CpG increased the expression of IFN-α mRNA (Fig. 3G, right) as previously reported in spleen-derived pDCs from humans, mice, and rats (29–31). Our data suggest that brain-derived pDCs isolated from Flt3L-treated rats had already been activated in vivo, and TLR9 activation by CpG was unable to further increase the expression of IFN-α.
We used RT-PCR to confirm that both brain- and spleen-derived pDCs expressed TLR9 and confirmed that TLR9 was expressed in the absence of CpG stimulation (Fig. 3H). Surprisingly, we found that incubation of spleen-derived pDCs with CpG reduced the expression of TLR9 mRNA (Fig. 3H, right), indicating a possible mechanism by which pDCs can become refractory to additional stimulation with CpG by down-modulating the expression of components of the TLR9 signal transduction pathway, including TLR9. In addition, the expression of TLR9 in unstimulated, brain-derived pDCs (Fig. 3H, left) was much lower than that of spleen-derived pDCs (Fig. 3H, right), and this may reflect a previous encounter with TLR agonist in the brain that induced IFN-α expression and a subsequent refractory phenotype to additional stimulation with the TLR9 agonist CpG.
pDCs (CD3−CD45R+CD4+) recruited to the brain by Flt3L do not express B cell markers
CD45R/B220 is expressed by pDCs, but not cDCs, and can be used to separate these two subclasses of DC. CD45R is also expressed on many B cells throughout their development from the pro-B cell stage onward. Although most B cells do not express CD4, we tried to establish whether the CD45R+ CD4+ population infiltrating the brain in response to RAdFlt3L was pDC or a sub-population of B cells. We counterstained CD45R+ cells purified from Flt3L-expressing brains with Abs that bind to CD4 and either Igκ or Igλ. Igκ and Igλ are L chain components of the BCR, and cell surface expression of either Igκ or Igλ is first detected on immature B cells and is thereafter present throughout their subsequent development. We did not detect any significant population of CD4+CD45R+ cells (Fig. 4A) that also expresses either Igκ (Fig. 4B) or Igλ (Fig. 4C). This confirms that the infiltrating population of CD3−CD45R+CD4+ cells was not B cells. Plasma cells are terminally differentiated B cells that have low surface expression of BCR and stain negatively for Igκ and Igλ by flow cytometry. However, it is extremely unlikely that we were detecting plasma cells in the brain in response to Flt3L, because plasma cells are usually sequestered in T cell zones of secondary lymphoid organs, do not express CD4, require a longer incubation time to develop (i.e., Ag-specific T cells must first be activated before an Ab-dependent immune response can occur), and do not express CD45R.
FIGURE 4.
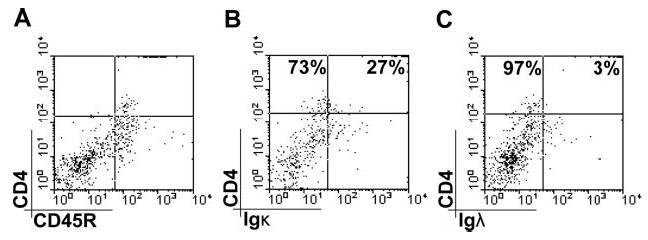
Infiltrating CD4+/CD45R+ cells do not express other BCR markers. A, Cells were analyzed for cell surface Igκ (B) and Igλ (C) expression using flow cytometry. CD45R+CD4+ cells did not express either Igκ or Igλ. However, some CD4−CD45R+ cells (B cells) were identified that expressed Igκ(B) or Igλ(C). Data shown are representative of four separate animals.
Expression of Flt3L in brain specifically does not increase the numbers of other immune cells in brain parenchyma
To determine whether increased expression of Flt3L could also promote the expansion of other immune cell populations within the brain, we analyzed the numbers of B cells, T cells, NK cells, NKT cells, macrophages, and microglia within the CNS before and after the expression of Flt3L. No significant increase in other immune cells was observed in response to Flt3L after expression in the brain for either 1 or 5 days (Fig. 5). Acute inflammation due to injection trauma or viral vectors was evident 1 day after injection; however, this increase was transient in nature, and all populations of immune cells in the brain, except NK cells (Fig. 5, C and J; p < 0.01), had returned to background levels 5 days after injection into the brain. Furthermore, Flt3L did not significantly alter any immune cell population in the spleen (Fig. 6), indicating that increased expression of Flt3L in the brain acts exclusively on local populations of pDCs.
FIGURE 5.
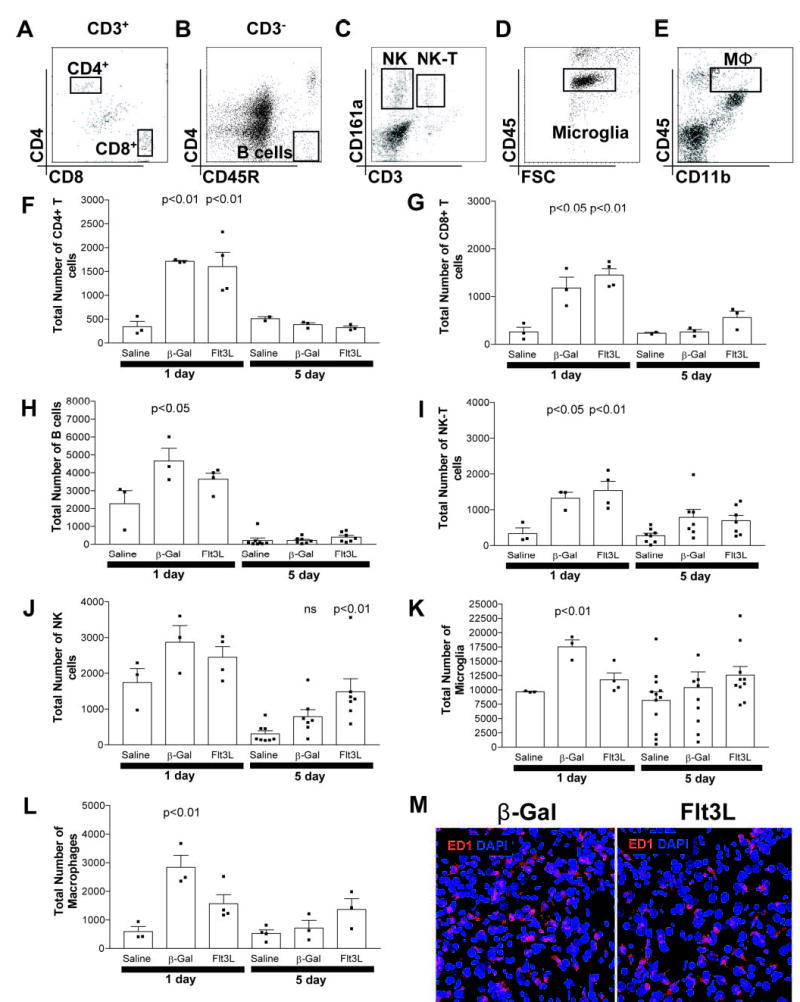
Flt3L does not induce infiltration of other immune cells into brain parenchyma. Detection of CD4+ T cells (CD3+CD4+CD8a−) and CD8+T cells (CD3+CD4−CD8a+; A), B cells (CD3−CD4−CD45R+; B), NK cells (CD3−CD161a+), and NK-T cells (CD3+CD161a+; C); microglia (CD45dim; D); and macrophages (CD11b+CD45high; E) in brain parenchyma was performed using flow cytometry. Acute inflammation 1 day after the injection of either virus resulted in a significant increase in CD4+ T cells (p < 0.01; F), CD8+ T cells (p < 0.05; G), B cells (p < 0.05; H), and NK-T cells (p < 0.05; I), suggesting that virus delivery alone transiently increases immune cells in the brain. NK cells were elevated in all samples 1 day after the injection of virus or saline (J), suggesting that infiltration occurred in response to tissue damage associated with injection trauma. The numbers of microglia (K; p < 0.01) and macrophages (L; p < 0.01) were elevated in RAd35-injected animals 1 day after injection compared with saline-injected animals. RAdFlt3L also induced an increase in total macrophages, although this was not significant (p > 0.05). This acute inflammatory response was transient, and absolute numbers of CD4+ T cells (F), CD8+ T cells (G), B cells (H), NK-T cells (I), and microglia (J) had all returned to normal levels after 5 days. NK cells remained slightly elevated in both RAd35- and RAdFlt3L-injected animals (J), and the number of macrophages was modestly elevated (NS, p > 0.05) in Flt3L-injected groups after 5 days (L). M, ED1 expression in the brain (red) was visualized using confocal microscopy. We saw parenchymal macrophages or activated microglia staining in the brains of rats 5 days after injection of either RAdFlt3L or RAd35, but not in saline-injected animals (not shown). However, no significant difference was observed between RAdFlt3L or RAd35, indicating that Flt3L does not increase the total number of ED1-immunoreactive cells in the brain. The flow cytometry graphs shown in A--E indicate the cell populations quantified and are representative of staining observed in 5-day samples injected with RAdFlt3L. Three animals per group were used for all 1 day points and 5 day points in (F, G, and L). At least seven animals per group were used for all other 5 day points (H–K). Error bars in F–L depict the SEM. The confocal images shown in M are representative of five animals analyzed per group.
FIGURE 6.
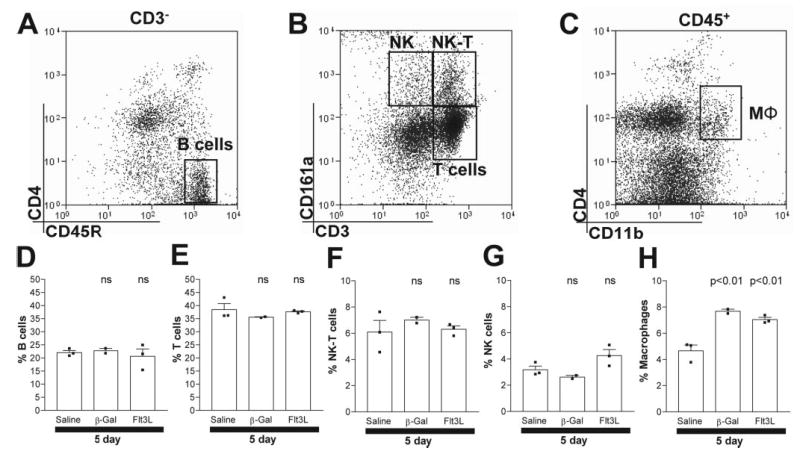
Flt3L expression in the brain does not alter populations of immune cells in the spleen. Animals were injected with saline, RAd35, or RAdFlt3L. After 5 days, the percentages of B cells (CD3−CD45R+CD4−; A and D), T cells (CD3+CD161a−; B and E), NKT cells (CD3+CD161a+; B and F), NK cells (CD3−CD161a+; B and G), and macrophages (C and H; CD4+CD11b+CD45+) within the spleen were assessed. We found that only macrophages significantly increased in response to RAdFlt3L compared with saline-injected animals. However, these cells were also elevated in response to RAd35 injected into the brain, indicating that this was a response to viral delivery and not Flt3L-induced proliferation of macrophages.
Discussion
Ags localized exclusively within the brain parenchyma, including tumor Ags, fail to prime a systemic immune response (1–3). This failure of infectious, particulate, or tumor Ags to prime the immune system is due in part to the absence of afferent professional APCs, such as DCs, within the naive, noninflamed brain parenchyma. However, DCs have been found in the brains of MS patients, and this was associated with elevated CCR7 production by myeloid cells (10). Elevated DC levels in the brain promote the clinical severity of EAE (13), implicating DCs in the maintenance of chronic immune responses in the brain. Additionally, MHC II expression exclusively on CD11c+ DCs is permissive for complete EAE induction, indicating a crucial role for CD11c+ DCs in initiating immune responses against brain Ags. Prolonged exposure of the immune system to myelin Ag was required for the development of EAE (8), suggesting that the immune system encounters brain Ag in the absence of chronic inflammation. This, in turn, implies that DCs or other APCs can encounter brain Ag before chronic inflammation. A cell type located within the naive brain, able to pick up Ag and transport it to the cervical lymph nodes, has not yet been indisputably described.
Flt3L is a cytokine related to c-Kit and stem cell factor that binds to the Flt3R expressed on immature hemopoietic cells, especially DC precursors (32, 33). We have recently shown that the expression of Flt3L in an intracranial tumor dramatically improved rat survival in an immune-mediated manner (22). Addition of the cytotoxic gene thymidine kinase was able to improve long-term survival still further, with 70% of animals surviving for up to 1 year, when all other viruses tested alone or in combination failed. Depletion of CD4+ T cells and macrophages completely abrogated these therapeutic effects, highlighting the fundamental role of Flt3L expression in the brain in stimulating innate and adaptive immune responses against brain tumor Ag (16). The normal function of Flt3L in the brain is to promote neuronal survival in combination with nerve growth factor (34) and is expressed by microglia in vitro, although whether other cells also contribute to the level of endogenous brain Flt3L is not known (35). We hypothesized that the expression of Flt3L within the brain stimulates the migration of DCs to brain parenchyma.
Previous work revealed that DCs in the brain are located in the CSF, meninges, and subarachnoid space (36). We found that the expression of Flt3L in the striatum induced the infiltration of pDCs into brain parenchyma. We did not observe any evidence of cDCs in brain parenchyma in response to Flt3L using markers for cDCs by either flow cytometry or confocal microscopy. Furthermore, other immune cells, including T cells, B cells, NK cells, NKT cells, and macrophages, were not significantly elevated, suggesting that Flt3L expression within the brain is acting specifically on the pDC subpopulation. The population of brain-infiltrating pDCs after the expression of Flt3L appeared to be an immature, lineage-committed pDC. In support of this, MHC II expression was low and could not be detected in CD45R+ cells infiltrating the brain when analyzed by confocal microscopy. This is consistent with previously published descriptions of pDCs, where it has been shown that pDCs isolated from the bone marrow lack expression of MHC II protein (37). We did, however, detect MHC II mRNA extracted from purified, brain-derived pDCs, and the levels of expression closely correlated with those of spleen pDCs. In addition, these cells were not found to express the costimulatory molecule CD80, indicating that pDCs in the brain had not fully matured into APCs. Immature, IFN-α-producing pDCs do not express the co-stimulatory molecules CD80 and CD86 and have been shown to be incapable of driving Ag-specific T cell proliferation (28).
We found that brain-derived pDCs purified from brain tissue homogenate 5 days after the injection of RAdFlt3L constitutively expressed high levels of IFN-α, a hallmark of activated, immature pDCs. These cells were refractory to subsequent stimulation with CpG, whereas IFN-α expression in naive spleen pDCs from the same animals required stimulation with CpG. It has recently been reported that TLR9 is required for IFN-α expression in pDCs, but after 24 h, IFN-α synthesis becomes refractory to additional stimulation by the same or different viruses (H. Kanzler, W. Cao, and Y.-J. Liu, unpublished observations; reported in Ref. 28). This was consistent with our observations and suggests that pDCs infiltrating brain parenchyma in response to Flt3L are activated to constitutively produce IFN-α (Fig. 7). However, future studies are required to confirm whether TLR9 stimulation in brain pDCs can create a refractory phenotype to further TLR9 stimulation.
FIGURE 7.
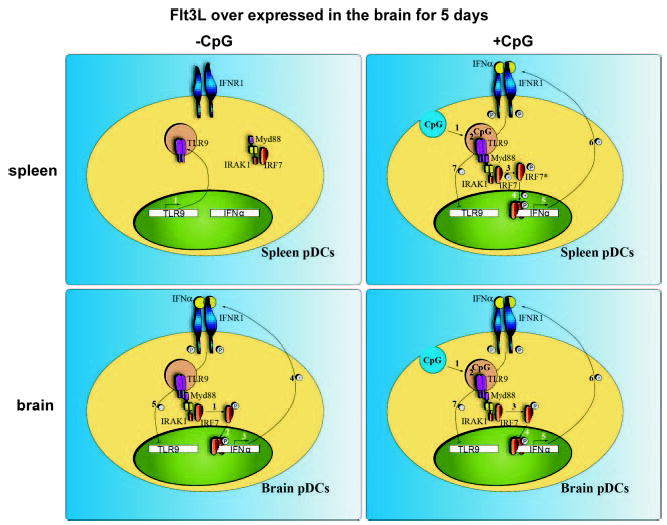
TLR9 signaling in brain- and spleen-derived pDCs. In the absence of CpG, unstimulated pDCs isolated from the spleen (upper left) expressed TLR9 (1). However, IFN-α was not expressed in these cells (A, left). After incubation with CpG (upper right), CpG was endocytosed by spleen pDCs (1) and bound with TLR9 in the endosomal compartment (2). This recruited the TLR9 signaling complex, comprised of MyD88, IRAK1, and IFN-regulatory factor 7 (IRF7), to TLR9. Activation of IRAK1 resulted in IRF7 phosphorylation (3) and translocation to the nucleus (4). IRF7 activates transcription of IFN-α (5), which, in turn, is secreted from the cell and binds with IFNR1 (6). IFNR1 signaling appears to down-regulate TLR9 expression in spleen cells shortly after stimulation with CpG (7). The pDCs isolated from the brain after overexpression of Flt3L (lower left) had lower expression of TLR9 than the naive spleen counterparts. IFN-α was constitutively expressed (3), presumably through MyD88 and IRF7 (1, 2) and was secreted by these cells (4). After incubation with CpG (lower right) (1, 2), IFN-α was not further increased (3), indicating that TLR9 signaling was refractory to superstimulation using CpG.
The presence of immature pDCs has been associated with promoting tolerance to brain Ags. Freshly isolated human pDCs induce CD4+ T cell anergy (38), and it has been reported that immature pDCs activated by CpG can differentiate naive T cells into regulatory T cells (39). Immature pDCs are present in early stages of breast cancer and have been associated with a marked reduction in survival. These cells are nonactivated pDCs and appear to promote immune tolerance against tumor Ag (40). However, activated pDCs possess the capacity to enhance cDC-mediated T cell activation (28) and can mature into APCs by increasing the expression of CD40 and CD86 costimulatory molecules and MHC II in response to TLR7 or TLR9 stimulation (28, 41). Mature pDCs with increased MHC II and costimulatory molecule expression (42) can induce the expansion of CD8+ T cells and Th1-polarized CD4+ T cells against endogenous Ag (43). Our results indicate that Flt3L alone recruits IFN-α-producing, immature pDCs into the brain.
Infiltrating pDCs within the brain parenchyma in response to Flt3L may come from a number of potential sources, including peripheral sites, such as the spleen, or local sites, including the CSF and meninges, or from differentiation of precursor cells within brain parenchyma. We did not observe any expansion of peripheral populations of pDCs, suggesting that infiltration of blood-derived pDCs across the blood-brain barrier was not responsible for the elevated number of brain DCs in response to Flt3L expression (14). Therefore, local sources of DCs, such as the meninges and choroid plexus, are more likely sources of pDCs infiltrating the brain (44). Another potential local source of pDC precursor cells is resident brain microglia. Brain microglia are derived from precursor monocytes that migrate into the brain during fetal development (45) and have a very low turnover in adults. Microglia express markers common to macrophages and DCs and treatment of enriched microglial cultures from healthy adult mouse brains with GM-CSF and CD40L induced the expansion of DCs, suggesting that cells that can serve as DC precursors were present in the enriched microglial culture (12) unless the culture contained the choroids plexus and ventricles, which it most certainly did. Mouse neonatal microglial cells were demonstrated to express c-Kit, but not Flt3R (46). In addition, Flt3L neither induced the proliferation of neonatal microglial cells (46) nor stimulated the differentiation of DCs from adult microglial cells in vitro (12). Consistent with these data, we did not find any evidence for increased proliferation of microglia within the brain in response to Flt3L, and the evidence from the literature suggests that microglia are not capable of differentiating into pDCs in response to Flt3L. However, this does not rule out the presence of progenitor immune cells within the brain as the source of pDCs.
In conclusion, we have demonstrated that elevated expression of Flt3L induces an increase in the number of IFN-α-producing, immature pDCs within brain parenchyma. This finding has important implications in the field of neuroimmunology. By increasing the level of Flt3L within the brain, we can increase the number of pDCs and stimulate immune responses against foreign Ags located within the brain, such as tumor Ag (16, 22). Our data suggest that it is possible to recruit pDCs into brain parenchyma by manipulating the brain microenvironment. This opens up the possibility of manipulating the priming of adaptive immune responses against foreign Ags from the brain in situ and could, in turn, give rise to novel therapeutic approaches to treat brain infections or tumors.
Acknowledgments
We are grateful for the graphical and editorial contributions of Ivan DiStefano, and we also thank the academic and administrative leadership of Dr. Shlomo Melmed, Richard Katzman, and David Meyer.
Footnotes
Gene therapy projects for neurological diseases are funded by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant 1R01NS44556.01; National Institute of Diabetes and Digestive and Kidney Diseases Grant 1RO3TW006273-01 (to M.G.C.); National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant 1RO1NS42893.01, U54NS045309-01, and 1R21NS047298-01 and Bram and Elaine Goldsmith Chair in Gene Therapeutics (to P.R.L.); and The Linda Tallen and David Paul Kane Annual Fellowship (to M.G.C. and P.R.L.). This work was also supported by the Board of Governors, Cedars Sinai Medical Center.
Abbreviations used in this paper: DC, dendritic cell; cDC, conventional DC; CFS, cerebrospinal fluid; EAE, experimental autoimmune encephalitis; Flt3L, Fms-like tyrosine kinase 3 ligand; hs, human soluble; β-Gal, β-galactosidase; GFAP, glial fibrillary acidic protein; MBP, myelin basic protein; MHC I, MHC class I; MS, multiple sclerosis; pDC, plasmacytoid DC; RAd5, recombinant adenovirus type 5.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Thomas CE, Schiedner G, Kochanek S, Castro MG, Lowenstein PR. Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with high-capacity, adenovirus vectors: toward realistic long-term neurological gene therapy for chronic diseases. Proc Natl Acad Sci USA. 2000;97:7482–7487. doi: 10.1073/pnas.120474397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowenstein PR. Immunology of viral-vector-mediated gene transfer into the brain: an evolutionary and developmental perspective. Trends Immunol. 2002;23:23–30. doi: 10.1016/s1471-4906(01)02063-4. [DOI] [PubMed] [Google Scholar]
- 3.Thomas CE, Schiedner G, Kochanek S, Castro MG, Lowenstein PR. Preexisting antiadenoviral immunity is not a barrier to efficient and stable transduction of the brain, mediated by novel high-capacity adenovirus vectors. Hum Gene Ther. 2001;12:839–846. doi: 10.1089/104303401750148829. [DOI] [PubMed] [Google Scholar]
- 4.Perry VH. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J Neuroimmunol. 1998;90:113–121. doi: 10.1016/s0165-5728(98)00145-3. [DOI] [PubMed] [Google Scholar]
- 5.Fabry Z, Raine CS, Hart MN. Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol Today. 1994;15:218–224. doi: 10.1016/0167-5699(94)90247-X. [DOI] [PubMed] [Google Scholar]
- 6.Owens T, Babcock A. Immune response induction in the central nervous system. Front Biosci. 2002;7:d427–d438. doi: 10.2741/A786. [DOI] [PubMed] [Google Scholar]
- 7.McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. 2005;79:17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 9.Plumb J, Armstrong MA, Duddy M, Mirakhur M, McQuaid S. CD83-positive dendritic cells are present in occasional perivascular cuffs in multiple sclerosis lesions. Mult Scler. 2003;9:142–147. doi: 10.1191/1352458503ms890oa. [DOI] [PubMed] [Google Scholar]
- 10.Kivisakk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B, Wujek J, Ravid R, Staugaitis SM, Lassmann H, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- 11.Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157:1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol. 2001;166:2717–2726. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- 13.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 14.Newman TA, Galea I, van Rooijen N, Perry VH. Blood-derived dendritic cells in an acute brain injury. J Neuroimmunol. 2005;166:167–172. doi: 10.1016/j.jneuroim.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 15.Rosicarelli B, Serafini B, Sbriccoli M, Lu M, Cardone F, Pocchiari M, Aloisi F. Migration of dendritic cells into the brain in a mouse model of prion disease. J Neuroimmunol. 2005;165:114–120. doi: 10.1016/j.jneuroim.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 16.Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, Puntel M, Cheng Q, Prieto J, Ribas A, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005;65:7194–7204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whartenby KA, Calabresi PA, McCadden E, Nguyen B, Kardian D, Wang T, Mosse C, Pardoll DM, Small D. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc Natl Acad Sci USA. 2005;102:16741–16746. doi: 10.1073/pnas.0506088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatterer E, Davoust N, Didier-Bazes M, Vuaillat C, Malcus C, Belin MF, Nataf S. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood. 2006;107:806–812. doi: 10.1182/blood-2005-01-0154. [DOI] [PubMed] [Google Scholar]
- 19.Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Disproportionate recruitment of CD8+ T cells into the central nervous system by professional antigen-presenting cells. Am J Pathol. 1999;154:481–494. doi: 10.1016/S0002-9440(10)65294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pashenkov M, Huang YM, Kostulas V, Haglund M, Soderstrom M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001;124:480–492. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- 21.Wilkinson GW, Akrigg A. Constitutive and enhanced expression from the CMV major IE promoter in a defective adenovirus vector. Nucleic Acids Res. 1992;20:2233–2239. doi: 10.1093/nar/20.9.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali S, Curtin JF, Zirger JM, Xiong W, King GD, Barcia C, Liu C, Puntel M, Goverdhana S, Lowenstein PR, et al. Inflammatory and anti-glioma effects of an adenovirus expressing human soluble Fms-like tyrosine kinase 3 ligand (hsFlt3L): treatment with hsFlt3L inhibits intracranial glioma progression. Mol Ther. 2004;10:1071–1084. doi: 10.1016/j.ymthe.2004.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shering AF, Bain D, Stewart K, Epstein AL, Castro MG, Wilkinson GW, Lowenstein PR. Cell type-specific expression in brain cell cultures from a short human cytomegalovirus major immediate early promoter depends on whether it is inserted into herpesvirus or adenovirus vectors. J Gen Virol. 1997;78:445–459. doi: 10.1099/0022-1317-78-2-445. [DOI] [PubMed] [Google Scholar]
- 24.Southgate T, Kingston P, Castro MG. Gene transfer into neural cells in vivo using adenoviral vectors. In: Gerfen CR, McKay R, Rogawski MA, Sibley DR, Skolnick P, editors. Current Protocols in Neuroscience. 4.23.1–4.23.40. Wiley & Sons; New York: 2000. pp. 4.23.21–24.23.40. [Google Scholar]
- 25.Dewey RA, Morrissey G, Cowsill CM, Stone D, Bolognani F, Dodd NJ, Southgate TD, Klatzmann D, Lassmann H, Castro MG, et al. Chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus-mediated gene therapy: implications for clinical trials. Nat Med. 1999;5:1256–1263. doi: 10.1038/15207. [DOI] [PubMed] [Google Scholar]
- 26.Perry VH, Andersson PB. The inflammatory response in the CNS. Neuropathol Appl Neurobiol. 1992;18:454–459. doi: 10.1111/j.1365-2990.1992.tb00811.x. [DOI] [PubMed] [Google Scholar]
- 27.Pashenkov M, Teleshova N, Link H. Inflammation in the central nervous system: the role for dendritic cells. Brain Pathol. 2003;13:23–33. doi: 10.1111/j.1750-3639.2003.tb00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 29.Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O’Garra A, Biron C, Briere F, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 30.Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 31.Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 32.Lyman SD, James L, Vanden Bos T, de Vries P, Brasel K, Gliniak B, Hollingsworth LT, Picha KS, McKenna HJ, Splett RR, et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell. 1993;75:1157–1167. doi: 10.1016/0092-8674(93)90325-k. [DOI] [PubMed] [Google Scholar]
- 33.Lyman SD, Jacobsen SE. c-Kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood. 1998;91:1101–1134. [PubMed] [Google Scholar]
- 34.Brazel CY, Ducceschi MH, Pytowski B, Levison SW. The FLT3 tyrosine kinase receptor inhibits neural stem/progenitor cell proliferation and collaborates with NGF to promote neuronal survival. Mol Cell Neurosci. 2001;18:381–393. doi: 10.1006/mcne.2001.1033. [DOI] [PubMed] [Google Scholar]
- 35.Meeuwsen S, Bsibsi M, Persoon-Deen C, Ravid R, van Noort JM. Cultured human adult microglia from different donors display stable cytokine, chemokine and growth factor gene profiles but respond differently to a pro-inflammatory stimulus. Neuroimmunomodulation. 2005;12:235–245. doi: 10.1159/000085655. [DOI] [PubMed] [Google Scholar]
- 36.Matyszak MK, V, Perry H. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74:599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- 37.Diao J, Winter E, Chen W, Cantin C, Cattral MS. Characterization of distinct conventional and plasmacytoid dendritic cell-committed precursors in murine bone marrow. J Immunol. 2004;173:1826–1833. doi: 10.4049/jimmunol.173.3.1826. [DOI] [PubMed] [Google Scholar]
- 38.Kuwana M. Induction of anergic and regulatory T cells by plasmacytoid dendritic cells and other dendritic cell subsets. Hum Immunol. 2002;63:1156–1163. doi: 10.1016/s0198-8859(02)00754-1. [DOI] [PubMed] [Google Scholar]
- 39.Moseman EA, Liang X, Dawson AJ, Panoskaltsis-Mortari A, Krieg AM, Liu YJ, Blazar BR, Chen W. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J Immunol. 2004;173:4433–4442. doi: 10.4049/jimmunol.173.7.4433. [DOI] [PubMed] [Google Scholar]
- 40.Treilleux I, Blay JY, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla JP, Bremond A, Goddard S, Pin JJ, Barthelemy-Dubois C, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res. 2004;10:7466–7474. doi: 10.1158/1078-0432.CCR-04-0684. [DOI] [PubMed] [Google Scholar]
- 41.Asselin-Paturel C, Trinchieri G. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005;202:461–465. doi: 10.1084/jem.20051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. 2000;1:305–310. doi: 10.1038/79747. [DOI] [PubMed] [Google Scholar]
- 43.Krug A, Veeraswamy R, Pekosz A, Kanagawa O, Unanue ER, Colonna M, Cella M. Interferon-producing cells fail to induce proliferation of naive T cells but can promote expansion and T helper 1 differentiation of antigen-experienced unpolarized T cells. J Exp Med. 2003;197:899–906. doi: 10.1084/jem.20021091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol. 1999;405:553–562. [PubMed] [Google Scholar]
- 45.Davis EJ, Foster TD, Thomas WE. Cellular forms and functions of brain microglia. Brain Res Bull. 1994;34:73–78. doi: 10.1016/0361-9230(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 46.Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R. Developmental plasticity of CNS microglia. Proc Natl Acad Sci USA. 2001;98:6295–6300. doi: 10.1073/pnas.111152498. [DOI] [PMC free article] [PubMed] [Google Scholar]