

Abstract
Sf9, a cell line derived from the lepidopteran insect, S. frugiperda, is widely used as a host for recombinant glycoprotein expression and purification by baculovirus vectors. Previous studies have shown that this cell line has one or more ß-N-acetylglucosaminidase activities that may be involved in the degradation and/or processing of N-glycoprotein glycans. However, these enzymes and their functions remain poorly characterized. Therefore, the goal of this study was to isolate ß-N-acetylglucosaminidase genes from Sf9 cells, over-express the gene products, and characterize their enzymatic activities. A degenerate PCR approach yielded three Sf9 cDNAs, which appeared to encode two distinct ß-N-acetylglucosaminidases, according to bioinformatic analyses. Baculovirus-mediated expression of these two cDNA products induced membrane-associated ß-N-acetylglucosaminidase activities in Sf9 cells, which cleaved terminal N-acetylglucosamine residues from the α-3 and α-6 branches of a biantennary N-glycan substrate with acidic pH optima and completely hydrolyzed chitotriose to its constituent N-acetylglucosamine monomers. GFP-tagged forms of both enzymes exhibited punctate cytoplasmic fluorescence, which did not overlap with either lysosomal or Golgi-specific dyes. Together, these results indicated that the two new Sf9 genes identified in this study encode broad-spectrum ß-N-acetylglucosaminidases that appear to have unusual intracellular distributions. Their relative lack of substrate specificity and acidic pH optima are consistent with a functional role for these enzymes in glycoprotein glycan and chitin degradation, but not with a role in N-glycoprotein glycan processing.
Introduction
Lepidopteran insect cell lines are widely used as hosts for baculovirus-mediated recombinant protein expression and purification [1–3]. One of the most widely used lines is Sf9 [3], which is a clonal derivative of IPLB-Sf21-AE, a cell line originally isolated from ovaries of the fall armyworm, Spodoptera frugiperda [4]. Sf9 cells readily support baculovirus-mediated foreign gene expression and, like other lepidopteran insect cell lines, have eukaryotic protein processing capabilities. Thus, the baculovirus-insect cell system is generally considered to be well suited for the expression and purification of recombinant glycoproteins.
ß-N-acetylglucosaminidases are enzymes that can hydrolyze the O-glycosidic bond of N-acetylglucosamine residues located in the terminal, nonreducing positions of oligosaccharides [5]. These enzymes typically have broad substrate specificities and are involved in the degradation of oligosaccharides, including glycoprotein glycans. A previous study revealed that Sf9 and other lepidopteran insect cell lines have high levels of ß-N-acetylglucosaminidase and other exoglycosidase activities, which are probably important for the normal, physiological degradation of carbohydrates in these cells and/or the insects from which they were derived [6]. However, the presence of these activities also raised the possibility that they might degrade the covalently linked oligosaccharide side-chains on recombinant glycoproteins during expression and purification of these products in the baculovirus-Sf9 cell system.
Subsequently, it was found that at least some of the ß-N-acetylglucosaminidase activity in Sf9 and other lepidopteran insect cells is membrane-bound and specific for the non-reducing terminal N-acetylglucosamine residues on the α1,3-branch of N-glycoprotein glycans [7]. This finding raised the interesting possibility that the N-glycosylation pathway in these cells might include an unusual ß-N-acetylglucosaminidase that functions in N-glycan processing. In fact, it is now widely believed that the N-glycan processing pathways of at least some insects [7], nematodes [8], and plants [9, 10] include a processing ß-N-acetylglucosaminidase and this appears to be a key distinction between the N-glycosylation pathways of lower and higher eukaryotes.
While it is clear that Sf9 cells encode and express at least one ß-N-acetylglucosaminidase that might function in glycoprotein glycan degradation, biosynthesis, or both, the nature and function of these Sf9 cell enzymes are poorly characterized. Thus, this study was designed to isolate ß-N-acetylglucosaminidase genes from Sf9 cells, express the cDNA products, and examine their functions. The results demonstrated that Sf9 cells encode and express at least two broad-spectrum ß-N-acetylglucosaminidases with acidic pH optima. Their relative lack of substrate specificity and pH optima are consistent with the idea that these enzymes function catabolically in glycoprotein and/or chitin degradation. However, they are not soluble proteins and do not appear to be lysosomal enzymes, which is an unusual set of properties for glycosylhydrolases with these degradative functions.
Materials and methods
Cells and cell culture
Sf9 cells are derived from IPLB-Sf21-AE, an established cell line originally isolated from Spodoptera frugiperda ovaries [4]. This cell line was routinely maintained as a suspension culture at densities of 0.3–3.0 X 106 cells per mL in complete TNM-FH medium, defined here as TNM-FH basal medium [3] supplemented with 10% (v/v) fetal bovine serum (HyClone Inc., Logan, UT) and 0.1% (w/v) pluronic F68 (BASF Wynandotte Corp., Parsippany, N.J.; [11]).
Isolation of SfGlcNAcase-1, a putative ß-N-acetylglucosaminidase cDNA, from Sf9 cells
Genomic DNA was isolated from Sf9 cells by a standard method [12] and used as the template for polymerase chain reactions (PCRs; [13]) with degenerate oligonucleotide primers designed against conserved regions of known hexosaminidases from F. catus (Acc. No. P49614; [14]), M. musculus (Acc. No. P20060; [15, 16] and Acc. No. NP_034551; [17, 18]), D. discoideum (Acc. No. P13723; [19]), and E. histolytica (Acc. No. P49009; [20]). The sequences of the degenerate oligonucleotide primers used for these PCRs were (5′-T/C,T,I,C,A,C/T,T,G,G,C,A,C/T,A/T/C,T,I,G,T,I,G,A-3′) and (5′-A,C/A/G,C/T,T,C,G/A,T,C,I,C,C,I,C,C,I,I,I,G/A,T,G-3′). The PCRs were performed in a total volume of 0.050 mL and contained 10 mM Tris-HCl (pH 9.0), 0.1% (v/v) Triton-X-100, 50 mM KCl, 1.5 mM MgCl2, 200 μM dNTPs, 1.25 U of Taq polymerase (Promega Corporation, Madison, WI), 40 μM of each primer, and 100 ng of Sf9 genomic DNA. The reactions were incubated for 3 min at 95°C, cycled 30 times using (i) 0.5 min at 95°C, (ii) 0.5 min at 40°C, and (iii) 1.67 min at 72°C, and finally incubated for 7 min at 72°C in a Perkin-Elmer Applied Biosystems Model 2400 GeneAmp thermal cycler (Foster City, CA). The spent reactions were harvested and analyzed on 1% Seaplaque® low melting temperature agarose gels. A specific ~750 bp amplification product detected by ethidium bromide staining was recovered from the gel, cloned into pCR®2.1-TOPO® (Invitrogen Life Technologies, Carlsbad, CA), and sequenced using universal and gene-specific primers. The resulting nucleotide sequence data were used to design exact-match primers directed against the putative Sf9 ß-N-acetylglucosaminidase gene and these primers were used, in turn, to screen an Sf9 cDNA library in λZAPII [21]; Stratagene, La Jolla, CA) by sibling selection and PCR, as described previously [22, 23]. Ultimately, a λZAPII clone was identified, plaque-purified, and the plasmid insert was excised using the ExAssist® method (Stratagene) according to the manufacturer’s protocol. Bacterial colonies containing this plasmid, which was designated pBSSf9-29.3, were identified and amplified, and then the plasmid was extracted, purified, and used to sequence both strands of the cDNA insert with universal and gene-specific primers. Bioinformatic analysis of the resulting sequence (see Results and Discussion) indicated that it was a partial ß-N-acetylglucosaminidase cDNA missing a portion of its 5′ coding sequence.
The 5′ end of this putative Sf9 ß-N-acetylglucosaminidase cDNA was isolated using a commercial rapid amplification of cDNA ends (RACE) method (SMART™ RACE; Clontech Laboratories Inc., Mountain View, CA). Briefly, total RNA was prepared from a log phase culture of uninfected Sf9 cells with Tri Reagent (Molecular Research Center, Cincinnati, OH) according to the manufacturer’s protocol. The resulting RNA was then used to prepare poly A+ RNA by oligo dT cellulose column chromatography [24] with the MessageMaker® mRNA Isolation System (Invitrogen). One microgram of the poly A+ RNA was used for first-strand cDNA synthesis, as described in the commercial SMART™ RACE protocol, to produce a 3′-anchored first strand cDNA that was used for 5′ RACE with complementary (universal primer mix A from the SMART™ RACE kit) and gene specific (5′-CTCCTGCACTTCATGGAAC-3′) primers. The 5′-RACE reaction was performed using the Advantage® 2 PCR enzyme system (Clontech), which provides high-fidelity DNA replication, and a hot-start, touchdown PCR protocol described in the commercial SMART™ RACE protocol. The spent PCR reaction was resolved on a 1% Seaplaque® low melting temperature agarose gel and then the region of the gel corresponding to the expected size range (~1.0–1.5 Kb) was sliced into five equal fractions and samples were analyzed by Southern blotting [25] with a uniformly labeled fragment of the partial cDNA clone from above (pBSSf9-29.3) as the probe. A positive fraction was identified and used for semi-nested secondary PCR with a primer complementary to the anchor sequence on the 3′ end of the first strand cDNA product (nested universal primer A from the SMART™ RACE kit) and the same gene specific primer used for the primary 5′ RACE reaction. The PCR reactions were incubated for 3 min at 95°C, cycled 30 times using (i) 0.5 min at 95°C, (ii) 0.5 min at 50°C, and (iii) 1.5 min at 72°C, and finally incubated for 7 min at 72°C, in the thermal cycler described above. The spent PCR reaction was analyzed on a 1% Seaplaque® low melting temperature agarose gel and a distinct product of ~1.0 Kb was observed, excised from the gel, recovered using the S.N.A.P.™ method (Invitrogen), and cloned into pCR®2.1-TOPO®. Three independent bacterial clones containing the recombinant plasmid were identified and amplified, and then plasmid DNA was extracted from each, purified, and used to sequence both strands of the PCR amplimer with universal and gene-specific primers. A consensus full-length coding sequence was assembled from these data plus the sequencing data from the λZAPII cDNA clone using AssemblyLIGN v. 1.0.9b (Accelrys; San Diego, CA) and designated SfGlcNAcase-1.
Isolation of additional putative ß-N-acetylglucosaminidase cDNA clones from Sf9 cells
Variations of the degenerate PCR and 5′ RACE approaches described above were used to isolate two additional putative ß-N-acetylglucosaminidase cDNA clones from Sf9 cells. A new set of PCRs was performed in a gradient cycler (Eppendorf AG, Hamburg, Germany) to obtain a broad range of annealing temperatures. These replicate reactions were performed using the Advantage® 2 system and the same degenerate sense primer used to isolate SfGlcNAcase-1 plus a new degenerate antisense primer (5′-C/T,T,C,I,G/C,C/T,C,C,A,I,A,T/A/G,A/G,C,A,I,A/G,C,C/T,T,C,I,C,C,T/C/G-3′). After an initial denaturation step for 2 min at 95°C, these PCRs were cycled 20 times using (i) 0.5 min at 95°C, (ii) 0.5 min at a range of temperatures from 40–60°C, with a difference of 0.5°C between individual reactions, and (iii) 1.0 min at 72°C. This was followed by a secondary amplification with 20 cycles of (i) 0.5 min at 95°C, (ii) 0.5 min at 40°C, and (iii) 1.0 min at 72°C, and, finally, a terminal incubation step for 7 min at 72°C. The reaction products were resolved on an agarose gel, as described above, and an amplification product of about the expected size (~750 bp) was observed, recovered, purified by the S.N.A.P. method, and cloned into pCR®2.1-TOPO®. Sequence analysis of several clones revealed that the amplimer was composed of a mixture of two nearly identical sequences and BLAST [26] analysis revealed that each was most closely related to known hexosaminidases . These sequences were designated SfGlcNAcase-2 and SfGlcNAcase-3 and specific antisense primers were used in combination with universal primer mix A for 5′ RACE reactions with the SMART RACE kit, as described above. The reaction products were resolved on agarose gels, as described above, and specific amplification products of about the expected size (~1360 bp) were observed, recovered, purified, and cloned into pCR®2.1-TOPO®, as described above. The sequences of the amplimers in four independent subclones were determined in each case to obtain initial consensus sequences of the 5′ two-thirds of the SfGlcNAcase-2 and -3 genes. Finally, the 5′ end of each consensus sequence was used to design specific sense primers, which were used in combination with universal primer mix A for 3′RACE reactions with the SMART RACE kit, as described above. The reaction products were resolved on agarose gels and amplification products of about the expected size (~1750 bp) were recovered, purified, and cloned into pCR®2.1-TOPO®, as described above. The sequences of the amplimers in three independent clones of SfGlcNAcase-2 and -3 were determined and all the sequence data obtained with the 3′ and 5′ RACE products, plus the sequence data from three clones obtained in independent 3′ RACE reactions similar to the one described above, were used to generate the final consensus sequences of SfGlcNAcase-2 and -3.
Construction of baculovirus transfer plasmids
A pCR®2.1-TOPO® clone encoding the full-length, untagged SfGlcNAcase-1 consensus sequence was constructed by recombining the StuI fragments from a 5′ RACE clone and the partial cDNA clone (pBSSf9-29.3) described above. This TOPO clone was used to construct immediate early baculovirus transfer plasmids [27], which were used to perform preliminary gene expression and enzyme activity assay experiments as part of this study (data not shown). Subsequently, a 2 kb BamHI-NotI fragment that included the full-length SfGlcNAcase-1 open reading frame was subcloned from one of these plasmids, pAcP(−)IEGlcNAcase-1, into the corresponding sites of pVL1393 [28]. This yielded a conventional baculovirus transfer plasmid, pAcGlcNAcase-1, which was used to produce a recombinant baculovirus, as described below.
An immediate early transfer plasmid encoding the full-length, untagged, consensus SfGlcNAcase-3 sequence was constructed by subcloning a ~1.67 kb BamHI-NotI fragment from a pCR®2.1-TOPO®-3′ RACE clone into the BglII and NotI sites of pAcP(+)IE1TV3 [27]. After being used for preliminary gene expression and enzyme activity assays (data not shown), this plasmid was used as the template for a PCR reaction with sense (5′-CACCATGTTACGGCACGTAATATTGTTATTCGCTGTTGTTG-3′) and antisense (5′-GGCCCTGGATTTTGCGTAACAGGGAATTACTTTTAG-3′) primers to amplify the full-length, untagged SfGlcNAcase-3 coding sequence. The resulting amplification product was gel-purified, cloned into pENTR™/D-TOPO®, and an error-free clone was identified, designated pENTR-SfGlcNAcase-3, and used to produce a recombinant baculovirus by the BaculoDirect™ method (Invitrogen), as described below.
A PCR-based approach was used to construct an immediate early baculovirus transfer plasmid encoding the full-length, consensus SfGlcNAcase-1 amino acid sequence fused in-frame to a red-shifted variant of the Aequoria victoria green fluorescent protein (GFP; [29]). Sense (5′-GCTCAGCCACCCAATGGGCCAGGATTCTGTGTTGGAGCAATGGTGAGCAAGGGCGAGGAGCTG-3′) and antisense (5′-GCGGCCGCTTTACTTGTAC-3′) primers were used with pEGFP-N1 (Clontech) as the template to produce a DNA fragment that encoded a short portion of the 3′ end of the SfGlcNAcase-1 sequence, without the termination codon, fused in-frame to the full-length GFP coding sequence. This fragment was gel-purified, cloned into pCR®2.1-TOPO®, and an error-free clone was identified by sequencing. Subsequently, the coding sequence was excised using a natural BlpI site in the SfGlcNAcase-1 portion and a NotI site that had been introduced downstream of the GFP portion of the amplimer with the antisense primer. The BlpI-NotI fragment was then gel-purified and subcloned into the corresponding sites pAcP(−)IEGlcNAcase-1. This yielded a daughter immediate early baculovirus transfer plasmid designated pAcP(−)IEGlcNAcase-1-GFP, which was used to produce an immediate early recombinant baculovirus, as described below.
A PCR-based approach also was used to construct an immediate early baculovirus transfer plasmid encoding the full-length, consensus SfGlcNAcase-3 sequence fused in-frame to GFP. The SfGlcNAcase-3 open reading frame was amplified using a pCR®2.1-TOPO®-3′ RACE clone as the template, together with sense (5′-GGATCCGCGGCCGCACCATGTTACGGCACGTAATATTG-3′) and antisense (5′-GGATCCCCAAAGTAATTCCCTGTTACGC-3′) primers that introduced BamHI and NotI sites on the 5′end and a BamHI site on the 3′ end of the amplimer. This product was gel-purified, cloned into pCR®2.1-TOPO®, and an error-free clone was identified by sequencing. Subsequently, the amplimer was excised with BamHI, gel-purified, and subcloned into the unique BamHI site of pEGFP-N1. Finally, the resulting chimeric gene encoding the SfGlcNAcase-3-GFP fusion protein was excised with NotI, gel-purified, and subcloned into the unique NotI site of pAcP(+)IE1TV3 [27]. This yielded the immediate early baculovirus transfer plasmid designated pAcP(+)IEGlcNAcase-3-GFP, which was used to produce an immediate early recombinant baculovirus, as described below.
Isolation of baculovirus expression vectors
Each of the baculovirus transfer plasmids described in the preceding section was extracted from large-scale E. coli cultures and purified by isopycnic ultracentrifugation on ethidium bromide-cesium chloride gradients, as described previously [12]. Each plasmid except pENTR-SfGlcNAcase-3 was then separately mixed with Bsu36I-digested BakPak6 baculoviral DNA [30] and the mixtures were co-transfected onto Sf9 cells using a modified calcium phosphate method [3]. Recombinant baculovirus expression vectors, designated AcGlcNAcase-1, AcP(+)IEGlcNAcase-1-GFP, and AcP(+)IEGlcNAcase-3-GFP were then isolated by at least two rounds of plaque-purification, amplified in Sf9 cells, and titered by plaque assay on Sf9 cells, as described previously [3]. pENTR-SfGlcNAcase-3 was used to produce a recombinant baculovirus by the BaculoDirect™ method (Invitrogen), according to the manufacturer’s protocol. The recombinant baculovirus vector produced by this protocol, designated AcSfGlcNAcase-3, was isolated by one round of plaque-purification, amplified, and titered by plaque assay on Sf9 cells, as described above.
Expression of SfGlcNAcase cDNAs in insect cells
Sf9 cells were seeded into 50 mL of complete TNM-FH in 100 mL DeLong flasks (Corning Glass Works, Corning, NY) at a density of about 0.5 X 106 cells/mL and incubated overnight at 28°C and 125 rpm in a Forma Model 4580 rotary platform shaker-incubator (Forma Scientific, Inc., Marietta, OH). The cells were then pelleted by low speed centrifugation and mock-infected or infected at a multiplicity of 5 plaque-forming units per cell with a wild type baculovirus, AcGlcNAcase-1, or AcSfGlcNAcase-3 in a small volume of complete TNM-FH. After a 1 h adsorption period at 28°C on a rocking platform, the cells were pelleted by low speed centrifugation, re-suspended in fresh complete TNM-FH, returned to the DeLong flasks, and incubated for another 48 h at 28°C and 125 rpm in the shaker-incubator.
Isolation of purified microsomal fractions
A combination of previously described methods was used to isolate microsomal fractions from the baculovirus-infected Sf9 cells [7, 31, 32]. Briefly, the cells were harvested by low speed centrifugation, washed once with 5 mM imidazole buffer (pH 7.0), and re-suspended in a homogenization buffer (5 mM imidazole, 0.25 M sucrose, 0.5 mM β-mercaptoethanol, 1 mM EDTA, 1mM MgCl2; pH 7.0) supplemented with Complete™ protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). The cells were then transferred to a Dounce homogenizer, placed on ice, and homogenized with pestle A until ~90% of the cells were broken, as monitored by phase-contrast microscopy. The homogenate was centrifuged at 2000 g for 10 min at 4°C and then the supernatant was collected and loaded onto a 1.3 M sucrose cushion prepared in 5 mM imidazole and 0.1 mM EDTA (pH 7.0). The sample was then overlaid with 0.125 M sucrose in the same buffer and centrifuged in a Beckman SW28 rotor at 100,000 g for 1 h at 4°C. The microsomal band on top of the cushion was harvested, diluted with 0.125 M sucrose buffer, pelleted by ultracentrifugation at 110,000 g for 1 h at 4°C, and re-suspended in storage buffer (100 mM citrate-phosphate buffer, 0.1 mM EDTA, 125 mM sucrose; pH 6.0). A sample of the microsomal fraction was used to measure the total protein concentration with a commercial bicinchoninic acid assay kit (Pierce Biotechnology Inc., Rockford, IL). The remainder was aliquoted and stored at −85°C until needed for enzyme activity assays. For some experiments, samples of the microsomes were either held or sonicated on ice with ten pulses from a Branson Model 450 Sonifier (Danbury, CT) adjusted to 50% output. The microsomes were pelleted by centrifugation for 10 min at top speed in a microcentrifuge (Hermle Model Z180M) and then re-suspended in ß-N-acetylglucosaminidase assay buffer (100 mM citrate-phosphate buffer, 0.1 mM EDTA, 125 mM sucrose; pH 6.0). The sonication and centrifugation steps were repeated, the final pellets were re-suspended in assay buffer containing 0.5% (v/v) Triton-X-100 (Sigma Chemical Company, St. Louis, MO), and then the solubilized microsomes were assayed for ß-N-acetylglucosaminidase activity, as described below.
ß-N-acetylglucosaminidase activity assays
Frozen microsomal fractions were quick-thawed, clarified for 15 min at top speed in a microcentrifuge at 4°C, and the pellets were re-suspended in ß-N-acetylglucosaminidase assay buffer containing 0.5% (v/v) Triton-X-100. For experiments designed to examine the pH optima of the SfGlcNAcases, the pH of the assay buffer was varied from 4–8, as detailed in the legend for Figure 10. Samples containing equal amounts of total protein were then used for assays with 25 pmol of various pyridylamine (PA)-tagged oligosaccharides in a total volume of 0.050 mL. The synthetic oligosaccharide substrates used in this study included GlcNAcß2Manα6(GlcNAcß2Manα3)Manß4GlcNAcß4GlcNAc-PA (GnGn; CalBiochem, La Jolla, CA), GlcNAcß2Manα6(Manα3)Manß4GlcNAcß4GlcNAc-PA (GnM, kindly provided by Dr. Friedrich Altmann, University of Vienna), and Manα6(GlcNAcß2Manα3)Manß4GlcNAcß4GlcNAc-PA (MGn; kindly provided by Dr. Altmann). After being incubated for various times at 37°C, these reactions were diluted to 0.150 mL with ß-N-acetylglucosaminidase reaction buffer and analyzed by reverse phase high performance liquid chromatography, as described previously [33]. GnGn, GnM, MGn, and Manα6(Manα3)Manß4GlcNAcß4GlcNAc-PA (MM; CalBiochem), were used as standards for the chromatographic analyses.
Fig. 10.
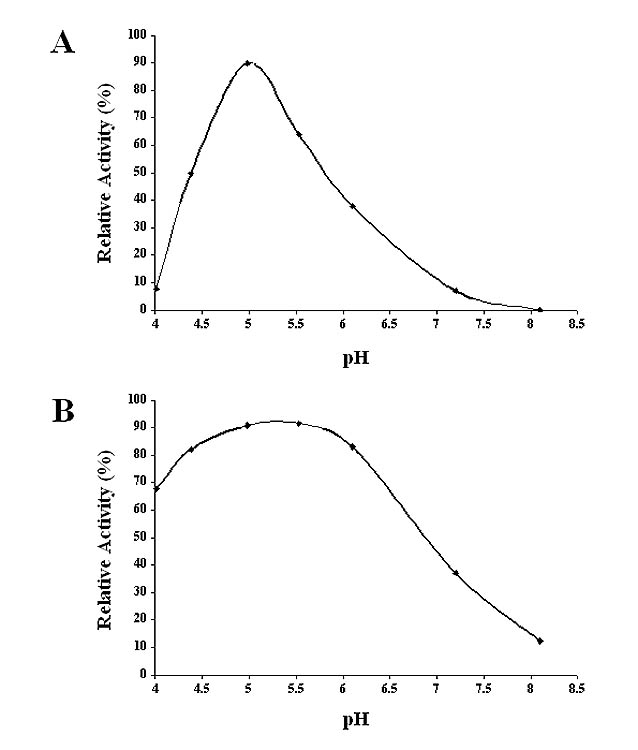
pH optima for SfGlcNAcase activities towards GnGn. Microsomal fractions containing (A) 10 ug of total protein from AcGlcNAcase-1 or (B) 1.5 ug of total protein from AcGlcNAcase-3-infected Sf9 cells were incubated for about 16 h or 4 h, respectively, with GnGn at pH values ranging from 4.0–8.0, and then the reaction products were recovered and analyzed by reverse-phase HPLC, as described in Materials and Methods. The plots show the relative amounts of products observed at each pH, as a percentage of the sum of the areas under all product peaks divided by the sum of the areas under all observed substrate and product peaks.
Living cell fluorescence assays
Sf9 cells were seeded into 35 mm uncoated glass bottom culture plates (MatTek Corp., Ashland, MA) at a density of about 0.5 X 106 cells/plate and allowed to attach for at least 1 h at 28°C. The cells were then infected with either AcP(+)IEGlcNAcase-1-GFP or AcP(+)IEGlcNAcase-3-GFP and incubated at 28°C for 18 h. The growth media were removed and then the infected cells were washed with fresh Grace’s medium [34] containing 0.5% fetal bovine serum and stained with the same medium containing 250 nM BODIPY®TR ceramide (Molecular Probes Inc., Eugene, OR) or 25 mM LysoTracker™ Red DND-99 (Molecular Probes) for 0.5 h or 5 min, respectively. After staining, the cells were rinsed with fresh Grace’s medium containing 0.5% fetal bovine serum and examined under a Leica TCS SP2 confocal laser-scanning microscope (Leica Microsystems, Heidelberg, Germany) equipped with an imaging system.
Results and Discussion
Isolation of putative ß-N-acetylglucosaminidase cDNAs from Sf9 cells
Our attempts to isolate an insect ß-N-acetylglucosaminidase cDNA began with a previously described degenerate PCR approach [35]. Three conserved amino acid sequences were identified in a multiple sequence alignment of known hexosaminidases from several different species, as described in Materials and Methods. These were used to design degenerate oligonucleotides, which were used, in turn, as primers for PCRs with genomic DNA from Sf9 cells as the template. One primer pair yielded a 750 bp amplification product, which appeared to be specific because it was not amplified in the absence of Sf9 DNA, with any single degenerate oligonucleotide, or with any other combination of the degenerate oligonucleotide primers (data not shown). This amplification product was cloned, sequenced, and the computer-derived translation product was found to be similar to known hexosaminidases, with amino acid identities ranging from about 30% to 50%, relative to E. histolytica hexosaminidase (Acc. No. P49009; [20]) and M. musculus hexosaminidase A (Acc. No. NP_034551; [17, 18]), respectively (data not shown). These results suggested that the amplified DNA fragment might be derived from an Sf9 ß-N-acetylglucosaminidase gene, which prompted us to determine its full-length sequence.
The sequence of the Sf9 amplification product was used to design exact-match primers, which were used, in turn, to identify a positive clone in an Sf9 cell cDNA library by sibling selection and PCR, as described previously [22, 23]. The cDNA insert was excised from this clone, sequenced, and the resulting 1761 bp sequence was used to derive an amino acid sequence. A multiple sequence alignment revealed that the theoretical translation product was similar to known hexosaminidases, but truncated at its N-terminus (data not shown). Thus, 5′-RACE was used to isolate the 5′ end of the putative Sf9 ß-N-acetylglucosaminidase coding sequence, as described in Materials and Methods. The sequence of the 5′-RACE product overlapped with the original cDNA sequence, extended it by 170 bp in the 5′ direction, and encoded an additional 35 amino acids and a potential translational initiation site (data not shown). These data were combined with the partial cDNA sequence data to obtain a full-length open reading frame of 1647 bp and derive the entire amino acid sequence of a putative ß-N-acetylglucosaminidase, which was designated SfGlcNAcase-1 (Fig. 1).
Fig. 1.

Nucleotide and amino acid sequences of SfGlcNAcase-1. The nucleotide and computer-derived amino acid sequences of SfGlcNAcase-1 are shown together with the transmembrane domain (underlined) predicted by TopPred2 [36] and consensus N-glycosylation sites (double-underlined). The GenBank Accession Number for SfGlcNAcase-1 is DQ249307.
The degenerate PCR experiments described above were repeated with some modifications to isolate two additional putative ß-N-acetylglucosaminidase cDNA clones from Sf9 cells, as described in Materials and Methods. The key procedural modification involved the use of a different primer pair for the degenerate PCR reactions. The sense primer was identical to the one used to isolate the SfGlcNAcase-1 amplimer, but the antisense primer was designed to exclude the SfGlcNAcase-1 sequence and more closely accommodate the conserved amino acid sequences in three putative D. melanogaster hexosaminidases identified in GenBank (Acc. Nos. NM079200, NM165909, and AAM11018). This new primer pair yielded another ~750 bp amplimer, which was gel-purified, cloned, and sequenced. The results revealed two nearly identical nucleotide sequences, which encoded theoretical translation products that were highly similar to known hexosaminidases (data not shown). These DNA sequences were used to isolate multiple 5′- and 3′-RACE products, which were cloned and sequenced. Ultimately, this effort yielded two new full-length, consensus nucleotide sequences, which were designated SfGlcNAcase-2 and SfGlcNAcase-3.
The open reading frames of SfGlcNAcase-2 and -3 consisted of 1665 and 1668 bp, respectively, and were 98% identical to each other, suggesting that these sequences represented very closely related or identical genes. This conclusion was supported by a comparison of the computer-derived translation products of these two genes, which revealed only two differences. The differences were that SfGlcNAcase-3 has an additional valine at position seven and it also has a glutamate residue at position 325, in place of the aspartate residue at position 324 of SfGlcNAcase-2. Based on these comparisons and preliminary enzyme activity assay results (data not shown), SfGlcNAcase-2 and -3 appeared to be functionally identical and, therefore, SfGlcNAcase-3 (Fig. 2) was used as the representative of both of these genes and gene products for the remainder of this study. In contrast, we could not assume that SfGlcNAcase-1 and -3 were functionally identical, as the open reading frames were only about 67% and the theoretical polypeptides were only about 63% identical. Therefore, the purpose of the remainder of this study was to examine the potential roles of both SfGlcNAcase-1 and SfGlcNAcase-3 as degradative and/or processing ß-N-acetylglucosaminidases in Sf9 cells.
Fig. 2.

Nucleotide and amino acid sequences of SfGlcNAcase-3. The nucleotide and computer-derived amino acid sequences of SfGlcNAcase-3 are shown together with the transmembrane domain (underlined) predicted by TopPred2 [36] and consensus N-glycosylation sites (double-underlined). The GenBank Accession Number for SfGlcNAcase-3 is DQ249309.
In silico analyses of SfGlcNAcase-1 and-3
The computer-derived SfGlcNAcase-1 (Fig. 1) and SfGlcNAcase-3 (Fig. 2) primary translation products consist of 548 and 555 amino acids and have calculated molecular masses of 62,915 and 63,475 daltons, respectively. SfGlcNAcase-1 has seven and SfGlcNAcase-3 has four consensus N-glycosylation sites. An analysis of the theoretical proteins using several different transmembrane domain prediction algorithms yielded mixed results. TopPred2 [36] predicted that both proteins have N-terminal transmembrane domains extending from amino acids 4 to 24 with in/out topology. TMAP [37] predicted that SfGlcNAcase-1 and -3 have N-terminal transmembrane domains extending from amino acids 3 to 31 and 3–22, respectively. Finally, TMHMM [38] predicted that SfGlcNAcase-1 has an N-terminal transmembrane domain extending from amino acids 5 to 22 with in/out topology, but that SfGlcNAcase-3 has none. Signal-P, an algorithm designed to detect potential signal peptides, predicted that both proteins have cleavable signal peptides from amino acids 1–18. Thus, the majority of the computer predictions suggested that both SfGlcNAcase-1 and SfGlcNAcase-3 have signals that can mediate their entry into the secretory pathway and, therefore, that they are at least transiently associated with membranes as either soluble or transmembrane proteins. This was consistent with the anticipated functions of these gene products as either lysosomal enzymes involved in glycoprotein glycan degradation or Golgi enzymes involved in glycoprotein glycan processing.
A search of the Pfam database [39] revealed that SfGlcNAcase-1 and -3 are most closely related to members of glycosylhydrolase Family 20, which includes the ß-N-acetylglucosaminidases [40]. This result was confirmed and extended by a BLAST-P [26] analysis of the non-redundant database. The two most significant hits were predicted honeybee (A. mellifera) and mosquito (A. gambiae) hexosaminidases (Table 1). Both gene products were also highly similar to known human alpha [41] and beta [42] hexosaminidases, which function in oligosaccharide degradation. In addition, the Sf9 gene products had lower, but significant levels of similarity to predicted or known hexosaminidases from a wide range of lower eukaryotes, including C. elegans, A. thaliana, several different lepidopteran insect species, and the dipteran insect, D. melanogaster. Interestingly, some of the lepidopteran insect enzymes, including those from C. fumiferana (Y. Zheng, P.J. Krell, and Q. Feng, unpublished), B. mori [43], and M. sexta [44], have been functionally implicated in molting, which involves chitin degradation by both chitinases and ß-N-acetylglucosaminidases. A Clustal W lineup of the new Sf9 gene products and the BLAST-P hits with e values of at least 1e-100 showed that these proteins share some highly conserved peptides (Fig. 3), as expected from the original lineups used to design the degenerate oligonucleotides. Together, the results of these in silico analyses supported the idea that SfGlcNAcase-1 and -3 are ß-N-acetylglucosaminidases that might be involved in glycoprotein degradation, glycoprotein biosynthesis, and/or chitin degradation in Sf9 cells and/or in the fall armyworm, S. frugiperda.
Table I.
Selected results of BLAST-Pa analyses for SfGlcNAcase-1 and -3
| SfGlcNAcase-1
|
SfGlcNAcase-3
|
|||||
|---|---|---|---|---|---|---|
| BLAST-P Hit | Bit Score | E-value | Bit Score | E-value | Genbank Acc. No. | References |
|
Apis mellifera
Predicted glycosylhydrolase 20 family member |
501 | 1e-140 | 496 | 9e-139 | XM_392543 | [53] |
|
Anopheles gambiae
Predicted glycosylhydrolase 20 family member |
498 | 1e-139 | 492 | 8e-138 | XM_319210 | [53] |
|
Homo sapiens
ß-hexosaminidase A |
410 | 4e-113 | 426 | 9e-118 | NM_000520 | [41, 54] |
|
Homo sapiens
ß-hexosaminidase B |
409 | 1e-112 | 426 | 1e-117 | NM_000521 | [42] |
|
Caenorhabditis elegans
Predicted ß-N-acetylhexosaminidase |
315 | 2e-84 | 309 | 1e-82 | NM_076008 | [53] |
|
Arabidopsis thaliana
Predicted glycosylhydrolase family 20 member |
281 | 3e-74 | 286 | 9e-76 | BT000831 | [53] |
|
Choristoneura fumiferana
Molt-related ß-N-acetylglucosaminidase Trichoplusia ni |
241 | 5e-62 | 221 | 5e-56 | DQ005717 | b |
| ß-N-acetylglucosaminidase | 229 | 2e-58 | 209 | 1e-52 | AY078172 | c |
|
Drosophila melanogaster
CG1318 (Hexo1) Predicted ß-hexosaminidase |
227 | 5e-58 | 221 | 4e-56 | NM_079200 | [55] |
|
Bombyx mori
Chitooligosaccharidolytic ß-N-acetylglucosaminidase |
226 | 1e-57 | 226 | 8e-58 | S77548 | [43] |
|
Manduca sexta
ß-N-acetylglucosaminidase |
222 | 2e-56 | 208 | 3e-52 | AY368703 | [44] |
|
Drosophila melanogaster
CG8824 (Fdl) Predicted ß-N-acetylhexosaminidase |
207 | 4e-52 | 207 | 7e-52 | NM_165909 | [55] |
|
Drosophila melanogaster
CG1787 (Hexo2) Predicted ß-hexosaminidase |
204 | 4e-51 | 203 | 9e-51 | AAM11018 | [55] |
[56].
Y. Zheng, P.J. Krell, and Q. Feng. 2005. Expression, localization and RH5992-regulated expression of molt-related beta-N-acetylglucosaminidase in the spruce budworm, Choristoneura fumiferana. Unpublished.
S. Watanabe, T. Kohuko, T. Kubota, and S. Inumaru. 2002. Direct NCBI submission.
Fig. 3.

Clustal-W comparison of the SfGlcNAcase-1 and -3 proteins with known or predicted hexosaminidases. This Figure shows a Clustal-W multiple sequence alignment between the two putative ß-N-acetylglucosaminidase proteins and selected known or predicted hexosaminidases from other species, including Apis mellifera, Anopheles gambiae, and Homo sapiens.
SfGlcNAcase expression and enzyme activity
Preliminary gene expression and enzyme activity assays were performed using p-nitrophenyl N-acetyl-ß-D-glucosaminide (pNP-GlcNAc) as an artificial substrate. The results revealed that intracellular and extracellular fractions isolated from AcSfGlcNAcase-1- or AcSfGlcNAcase-3-infected Sf9 cells both had significantly higher levels of ß-N-acetylglucosaminidase activity than wild type baculovirus-infected controls (data not shown). These results indicated that both of the newly isolated Sf9 genes encode functional ß-N-acetylglucosaminidases, as predicted. Hence, we designed and constructed a new set of baculovirus expression vectors encoding full-length SfGlcNAcase-1 or its computer-derived soluble domain with generic purification tags to facilitate expression and purification for detailed biochemical studies. Unfortunately, the resulting fusion proteins, which had either 6X-HIS or protein C epitope (EDQVDPRLIDGK) tags on either their amino or carboxy-terminal ends, in various combinations, either could not be effectively purified or had very low or no detectable hydrolytic activities (data not shown). These results suggested that at least these initial conventional molecular engineering strategies were not useful for the expression and purification of these enzymes. Additional preliminary work showed that the intracellular ß-N-acetylglucosaminidase activities induced by SfGlcNAcase-1 and -3 were membrane associated, as predicted (data not shown). Therefore, we resorted to the use of microsomal membrane preparations from AcSfGlcNAcase-1 or -3-infected Sf9 cells as a crude, but enriched source of the over-expressed, native gene products for enzyme activity assays. Recognizing the limitations of this approach, we focused these analyses on the hydrolytic specificities of the crude enzyme preparations towards synthetic oligosaccharide substrates.
Microsomal membranes were isolated from wild type baculovirus-, AcSfGlcNAcase-1-, or AcGlcNAcase-3-infected Sf9 cells, incubated with GnGn, and the reaction products were analyzed by HPLC, as described in Materials and Methods. The results showed that the microsomes from wild type baculovirus-infected Sf9 cells had relatively low levels of endogenous ß-N-acetylglucosaminidase activity, hydrolyzing about half of the GnGn substrate to GnM (Fig. 4B). Microsomes from uninfected Sf9 cells also converted GnGn to GnM, but no further, and had about 30% higher activity than an equivalent amount of microsomes from the wild type virus-infected infected cells (data not shown). In contrast, the microsomes from AcSfGlcNAcase-1-infected cells hydrolyzed nearly all of the GnGn and produced small amounts of GnM and MGn plus large amounts of MM (Fig. 4C). Microsomes from AcSfGlcNAcase-3-infected cells completely hydrolyzed GnGn to MM (Fig. 4D). These results confirmed that both of the newly isolated Sf9 genes encode functional ß-N-acetylglucosaminidases, as expression of these genes by recombinant baculoviruses induced these activities above endogenous levels. The presence of GnM and MGn peaks in the digestion profile obtained with SfGlcNAcase-1 revealed that this enzyme hydrolyzes GnGn to MM through GnM and MGn intermediates. Although SfGlcNAcase-3 completely hydrolyzed GnGn to MM in the experiment shown in Fig. 4D, a subsequent time-course experiment showed that it also hydrolyzes GnGn to MM through these same intermediates (Fig. 5). A microsome preparation from SfGlcNAcase-3-infected Sf9 cells hydrolyzed small amounts of GnGn to GnM and MGn within a 1 h incubation period (Fig. 5A–B). The initial appearance of larger amounts of GnM suggested that AcSfGlcNAcase-3 has a slight preference for the non-reducing terminal N-acetylglucosamine residue on the α3-branch of GnGn. The GnM and MGn peaks increased as the GnGn peak decreased with increasing incubation times (Fig. 5C–D). MM was produced after a 6 h incubation period (Fig. 5C) and the amounts of MM increased from 6–24 h at the expense of GnGn, GnM, and MGn (Fig. 5C–D). Together, all of the enzyme activity results obtained using GnGn suggested that SfGlcNAcase-1 and SfGlcNAcase-3 hydrolyze the non-reducing ß-N-acetylglucosamine residues from both branches of this synthetic oligosaccharide without any pronounced branch preferences. Accordingly, neither of these enzymes can account for the processing ß-N-acetylglucosaminidase activity originally detected in Sf9 cells by Altmann and coworkers (1995), as that activity had a clear preference for the non-reducing terminal N-acetylglucosamine residue on the α3-branch of GnGn [7].
Fig. 4.
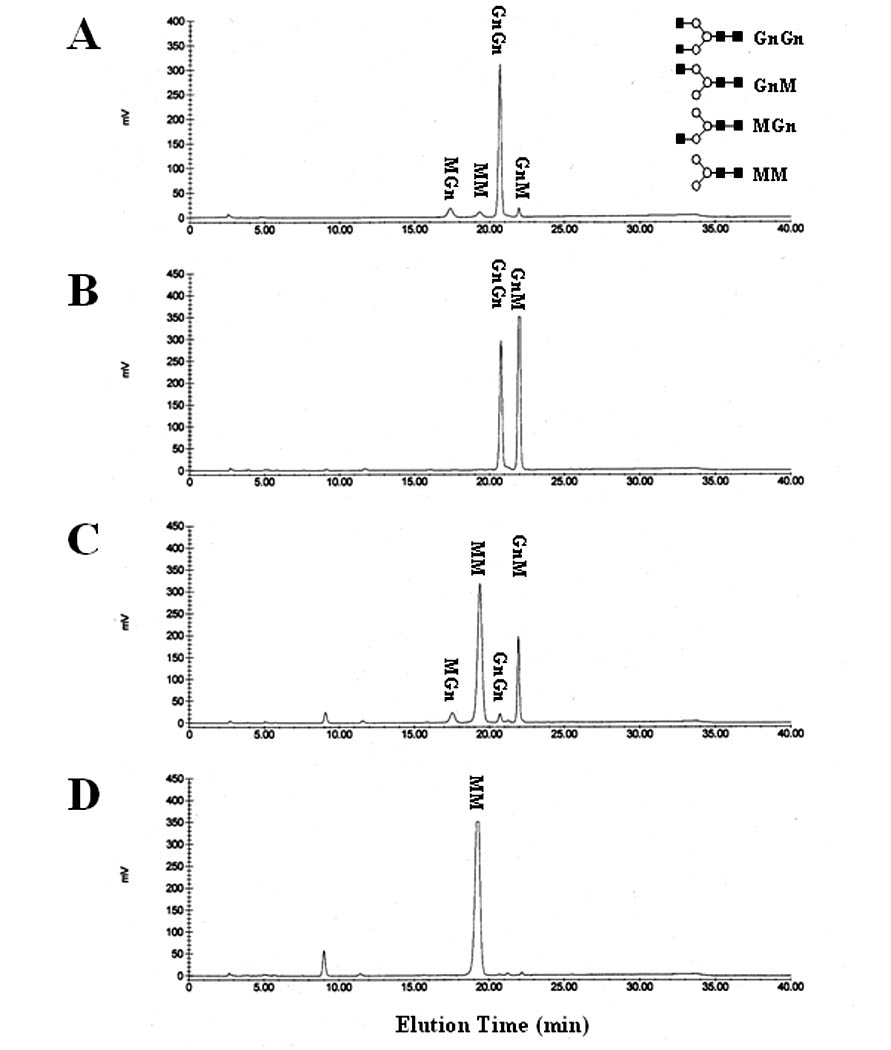
GnGn hydrolysis by SfGlcNAcase-1 and -3. The synthetic oligosaccharide substrate GnGn was incubated for about 16 h with microsomal fractions containing 40 ug of total protein from Sf9 cells infected with (B) wild type baculovirus, (C) AcGlcNAcase-1, or (D) AcGlcNAcase-3. The reaction products were then recovered and analyzed by reverse-phase HPLC, as described in Materials and Methods. (A) Elution of untreated synthetic oligosaccharides MGn, MM, GnGn and GnM, from left to right. The structures are shown in standard oligosaccharide drawing format [5] on the right.
Fig. 5.
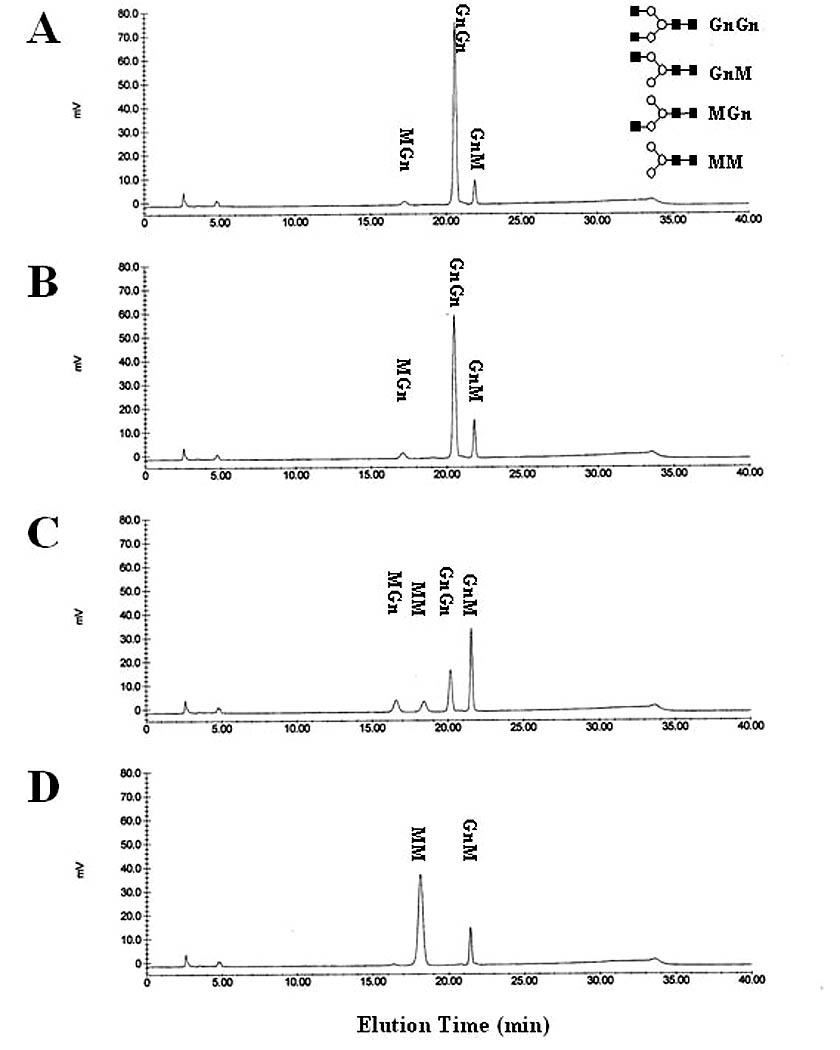
Time course of GnGn hydrolysis by SfGlcNAcase-3. GnGn was incubated for (A) 0.5 h, (B) 1 h, (C) 4 h, or (D) 24 h with a microsomal fraction containing 1 ug of total protein from Sf9 cells infected with AcGlcNAcase-3. The reaction products were then recovered and analyzed by reverse-phase HPLC, as described in Materials and Methods. Oligosaccharide structures are shown in standard oligosaccharide drawing format [5] on the right-hand side of panel (A).
To extend this substrate specificity analysis, enzyme assays were performed using synthetic oligosaccharide substrates with a single non-reducing terminal N-acetylglucosamine residue on either their α3- (MGn) or α6-branch (GnM). Microsomes from AcSfGlcNAcase-1- (Fig. 6C) or AcSfGlcNAcase-3- (Fig. 6D) infected Sf9 cells completely hydrolyzed the terminal N-acetylglucosamine residue from the α3 branch of MGn to produce MM. Both activities were induced relative to the wild-type baculovirus-infected cell control, as expected (Fig. 6B). Similarly, microsomes from Sf9 cells infected with either recombinant virus nearly completely or completely hydrolyzed the terminal N-acetylglucosamine residue from the α6 branch of GnM to produce MM (Fig. 7C–D) and both activities were clearly induced relative to the wild-type control (Fig. 7B). These results confirmed and extended our evidence that the two new Sf9 cell cDNAs described in this study encode functional ß-N-acetylglucosaminidases that can hydrolyze the non-reducing terminal N-acetylglucosamine residues from both branches of a synthetic, biantennary oligosaccharide and that neither one has a clear branch preference. Thus, these results supported the conclusion that neither SfGlcNAcase-1 nor SfGlcNAcase-3 accounts for the processing ß-N-acetylglucosaminidase activity originally detected in Sf9 cells by Altmann and coworkers (1995). The broad activities of these enzymes are more consistent with the conclusion that they are involved in glycoprotein glycan degradation.
Fig. 6.
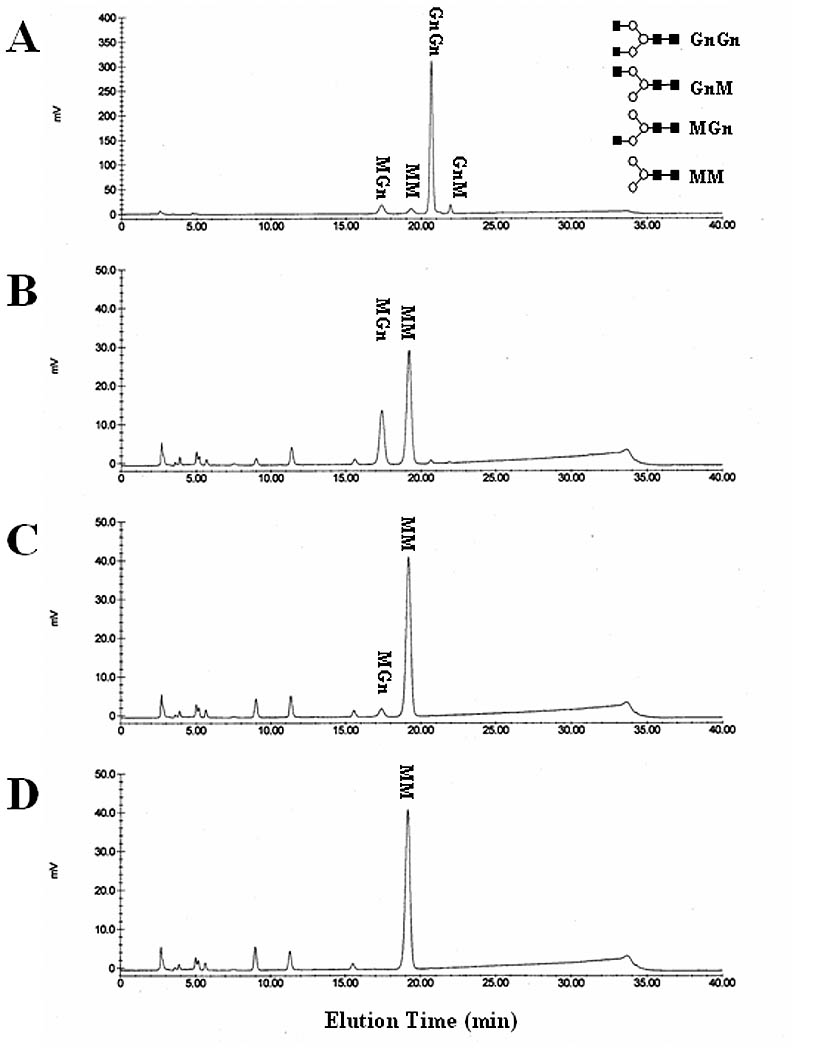
Hydrolysis of MGn by SfGlcNAcase-1 and -3. MGn was incubated for about 16 h with microsomal fractions containing 40 ug of total protein from Sf9 cells infected with (B) wild type baculovirus, (C) AcGlcNAcase-1, or (D) AcGlcNAcase-3. The reaction products were then recovered and analyzed by reverse-phase HPLC, as described in Materials and Methods. (A) Elution of untreated synthetic oligosaccharides and their structures, as in Figure 4.
Fig. 7.
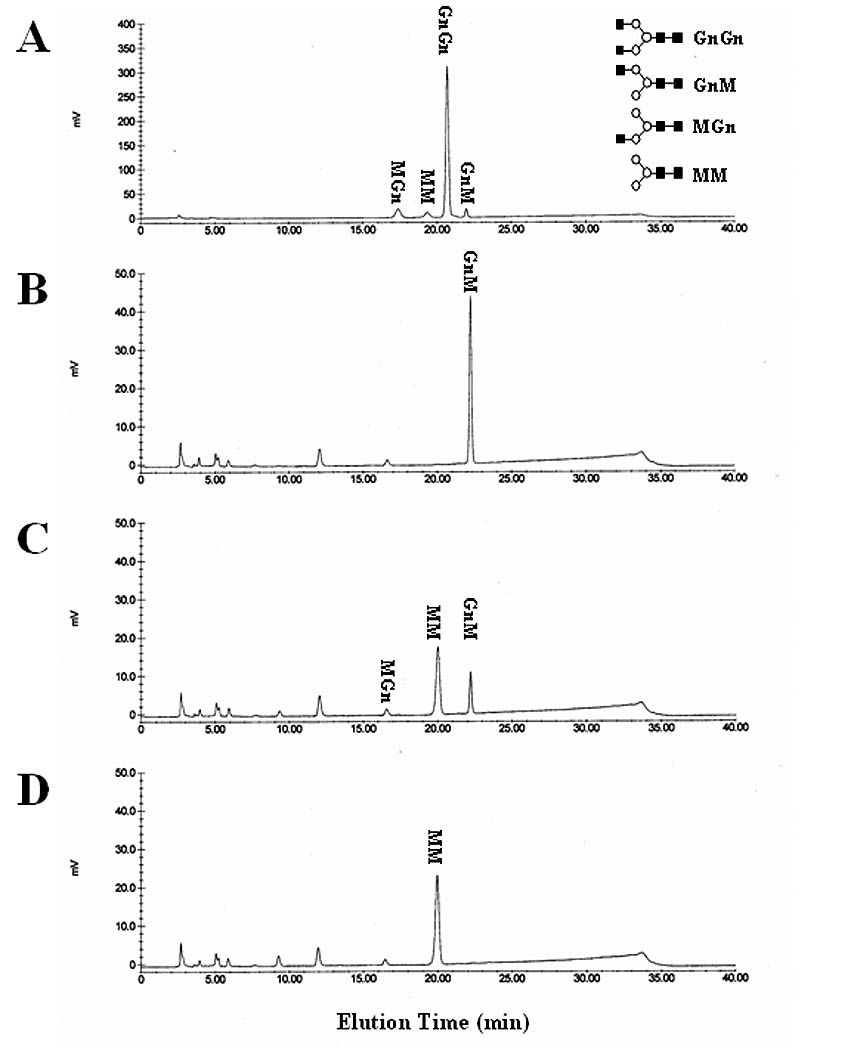
Hydrolysis of GnM by SfGlcNAcase-1 and -3. GnM was incubated for about 16 h with microsomal fractions containing 40 ug of total protein from Sf9 cells infected with (B) wild type baculovirus, (C) AcGlcNAcase-1, or (D) AcGlcNAcase-3. The reaction products were then recovered and analyzed by reverse-phase HPLC, as described in Materials and Methods. (A) Elution of untreated synthetic oligosaccharides and their structures, as in Figure 4.
Finally, the substrate specificity of these enzymes was examined using chitotriose, which is a linear oligosaccharide chain of three ß1,2-linked N-acetylglucosamine residues (Fig. 8). The endogenous activity in microsomes from wild-type baculovirus-infected cells removed a single N-acetylglucosamine residue from a fraction of the chitotriose substrate. However, microsomes from AcSfGlcNAcase-1- or -3-infected cells nearly completely or completely hydrolyzed chitotriose to its constituent monomers (Fig. 8C–D). These results underscored the relatively broad substrate specificities of SfGlcNAcase-1 and -3, supported the idea that these enzymes might degrade glycoprotein glycans and other carbohydrates in Sf9 cells, and indicated that they also might play a role in chitin degradation.
Fig. 8.
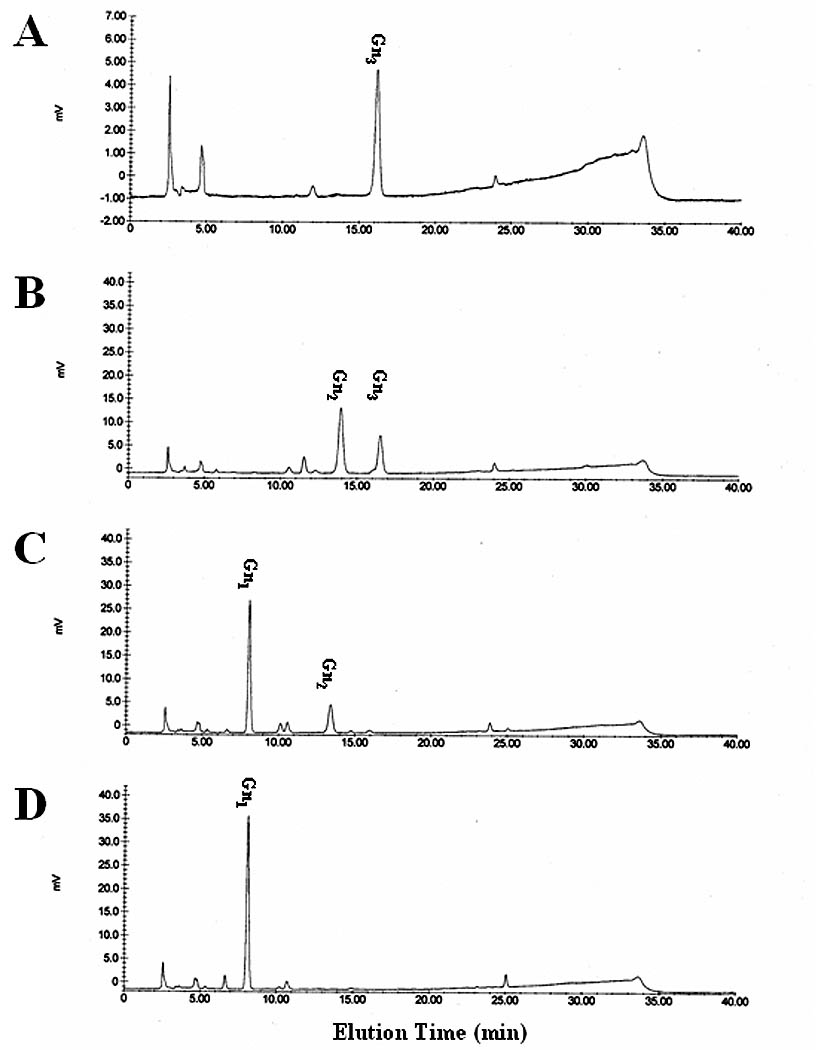
Hydrolysis of chitotriose by SfGlcNAcase-1 and -3. Chitotriose was incubated for about 16 h with microsomal fractions containing 40 ug of total protein from Sf9 cells infected with (B) wild type baculovirus, (C) AcGlcNAcase-1, or (D) AcGlcNAcase-3. The reaction products were then recovered and analyzed by reverse-phase HPLC, as described in Materials and Methods. (A) Elution of chitotriose. Elution position of monomeric N-acetylglucosamine was determined in a parallel HPLC analysis.
SfGlcNAcase membrane association and pH optima
In addition to their substrate specificities, ß-N-acetylglucosaminidases alternatively involved in either glycoprotein glycan degradation or processing generally might be expected to have two other distinguishing features [5]. A degradative enzyme should be a soluble, lysosomal enzyme with an acidic pH optimum of about 5.0 [45], while a processing enzyme should be a transmembrane, Golgi protein with a more neutral pH optimum of about 6.5–7.0 [46]. With these considerations in mind, we more closely examined the nature of the association between SfGlcNAcase-1, SfGlcNAcase-3, and membranes and we also determined their pH optima.
The microsome preparations used for our enzyme activity assays included a mixture of lysosome- and Golgi-derived vesicles. Therefore, “membrane associated” proteins in these preparations included both soluble and transmembrane proteins. To separate these two different groups of membrane associated proteins, we subjected microsomes from AcSfGlcNAcase-1- or -3-infected cells to two rounds of vigorous sonication, dilution, and centrifugation before assaying their enzyme activity, as described in Materials and Methods. Sonication was used to transiently break and reform the microsomal vesicles in order to release their soluble contents. Centrifugation was used to recover the empty microsomes, together with their transmembrane proteins, for the enzyme assays. Thus, ß-N-acetylglucosaminidase activity would be lost only if the SfGlcNAcases were soluble proteins, not if they were transmembrane proteins. The results of these experiments showed that this treatment did not remove the induced ß-N-acetylglucosaminidase activities in microsomal membranes from the recombinant baculovirus-infected Sf9 cells (Fig. 9). The same microsomal membrane preparations were used to obtain the data shown in Figs. 4 and 9 and a quantitative analysis of these data showed that about 73%, 88% and 86% of the activity in wild type virus-, AcSfGlcNAcase-1- and AcSfGlcNAcase-3-infected cell microsome preparations, respectively, were recovered after two rounds of sonication and centrifugation. These results indicated that SfGlcNAcase-1 and -3 are transmembrane, not soluble proteins in the microsome preparations, which is inconsistent with the idea that these enzymes are lysosomal hydrolases.
Fig. 9.
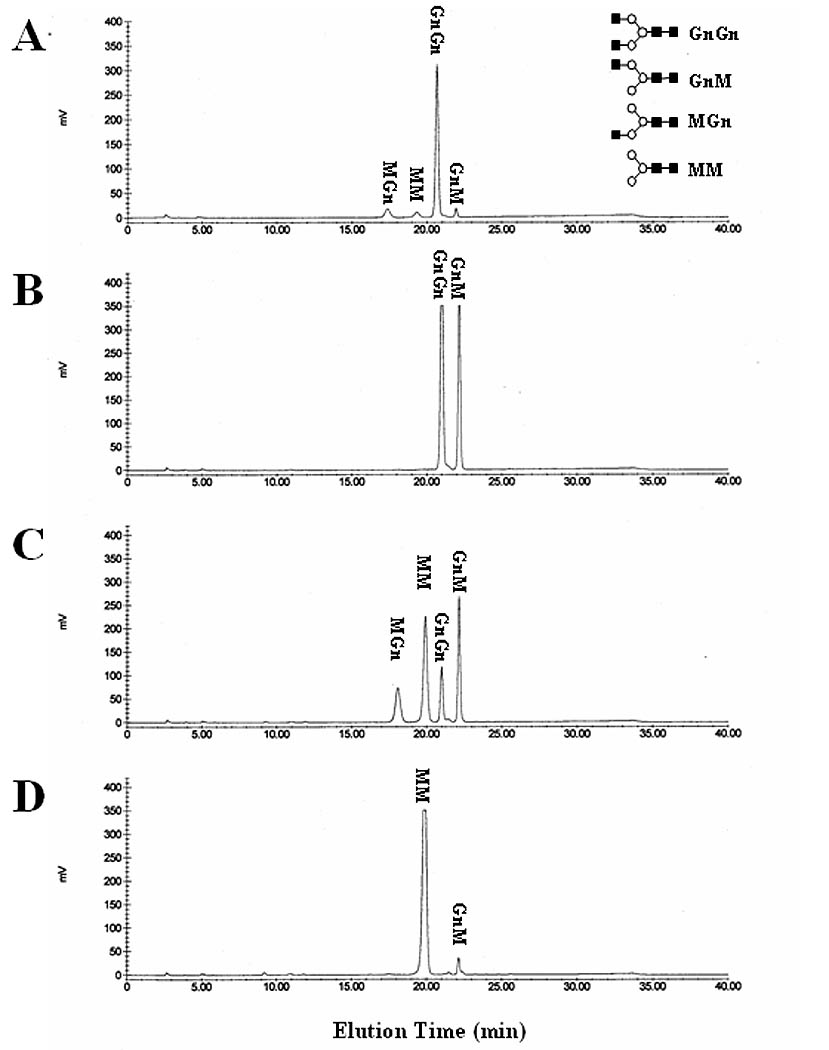
Nature of SfGlcNAcase-1 and -3 membrane association. Microsomal fractions containing 40 ug of total protein from Sf9 cells infected with (B) wild type baculovirus, (C) AcGlcNAcase-1, or (D) AcGlcNAcase-3 were subjected to two rounds of dilution, sonication, and centrifugation, as described in Materials and Methods. The microsomal pellets were then solubilized, incubated for about 16 h with GnGn, and the reaction products were recovered and analyzed by reverse-phase HPLC, as described in Materials and Methods. (A) Elution of untreated synthetic oligosaccharides and their structures, as in Figure 4.
The pH optima for GnGn hydrolysis by SfGlcNAcase-1 and -3 were examined by measuring the relative amounts of MM produced in enzyme activity assays performed at pH values ranging from 4.0 to 8.0. The results showed that the optimal pH for both of these enzymes was 5.0–5.5 (Fig. 10), which was consistent with the conclusion that these enzymes are lysosomal hydrolases.
Considered together, these results indicating that SfGlcNAcase-1 and -3 are transmembrane proteins with acidic pH optima provided a rather mixed and obviously indirect hint regarding their intracellular distribution and function. Thus, additional experiments were undertaken to address this issue more directly.
Intracellular distribution of GFP-tagged SfGlcNAcase fusion proteins
The intracellular distribution of SfGlcNAcase-1 and -3 was more directly examined by expressing GFP-tagged versions of the full-length gene products in Sf9 cells with immediate early baculovirus expression vectors [27]. We routinely use this approach for protein localization studies because the immediate early promoter in these vectors provides relatively lower expression levels during the early phase of infection, which circumvents potential mislocalization problems associated with higher levels of expression and/or the onset of baculoviral cytotoxicity [27, 47]. Sf9 cells were infected with AcP(−)IESfGlcNAcase-1-GFP or AcP(+)IESfGlcNAcase-3-GFP, stained with Golgi- or lysosome-specific dyes, and examined with a confocal fluorescence microscope. The SfGlcNAcase-3-GFP-infected cells exhibited punctate green fluorescence throughout the cytoplasm (Fig. 11A, C). Surprisingly, however, there was no significant overlap between this pattern and the red, cytoplasmic fluorescence emanating from dyes specific for the Golgi apparatus (Fig. 11B) or lysosomes (Fig. 11D). This same general result was obtained with SfGlcNAcase-1-GFP-infected cells (data not shown). These results suggested that SfGlcNAcase-1 and -3 reside in the cytoplasm, but not in the Golgi apparatus or lysosomes of Sf9 cells. Considering the rather surprising nature of this result, we must emphasize that the GFP tag could have interfered with proper localization of the SfGlcNAcase proteins. However, our previous co-localization experiments have revealed substantial overlap between GFP-tagged ß4-galactosyltransferase family members, which are known Golgi enzymes, and the Golgi dye used in this study in baculovirus-infected Sf9 cells [48–50]. Similarly, we have previously observed substantial overlap between a GFP-tagged, lepidopteran insect lysosomal α-mannosidase and the lysosomal dye used in this study using this same experimental design (J. Loveridge and D.L. Jarvis, unpublished). Interestingly, the optimal pH of the insect lysosomal α-mannosidase was about 4.2 (L. Paquin and D.L. Jarvis, unpublished), which is significantly lower than the optimal pH of 5.0 for SfGlcNAcase-1 and -3. Thus, it is possible that the ß-N-acetylglucosminidases described in this study reside in a pre-lysosomal compartment with a higher pH than the lysosomes in Sf9 cells [45]. But, if this compartment has an environmental pH reflecting the optimal pH for these enzymes, it, too, should have been stained with LysoTracker Red, which is an acidotrophic probe that can be used to stain any acidic organelle in living cells.
Fig. 11.
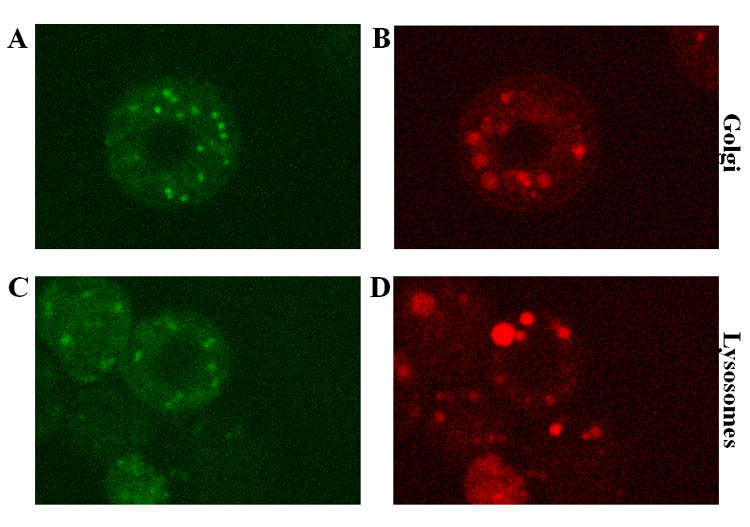
Intracellular distribution of GFP-tagged SfGlcNAcase-3. Sf9 cells were infected with AcP(+)IESfGlcNAcase-3-GFP and stained with BODIPY®TR ceramide (A–B) or LysoTracker™ Red DND-99 (C–D), as described in Materials and Methods. Panels A and C show the green fluorescence emitted by the SfGlcNAcase-1-GFP fusion protein. Panels B and D show the red fluorescence emitted by BODIPY®TR ceramide and LysoTracker™ Red DND-99, respectively.
Summary
Various ß-N-acetylglucosaminidases have been implicated in at least four fundamental physiologic processes in insects, including the interaction between sperm and egg [51, 52], degradation of chitin during molting (Zheng, P.J. Krell, and Q. Feng, unpublished, [43, 44]), glycoprotein glycan processing [7], and glycoprotein glycan degradation [6]. The existence of one or more ß-N-acetylglucosaminidases in Sf9 cells and their functional roles in glycoprotein glycan processing and/or degradation have thus far been garnered from enzyme activity assays alone. Prior to this study, no ß-N-acetylglucosaminidase gene(s) had been isolated from Sf9 cells, despite the prevalent use of this cell line for recombinant glycoprotein expression and purification. For the past decade, our group has consistently argued that molecular genetics is the best way to examine recombinant protein glycosylation issues in the baculovirus-insect cell system. Thus, the present study was undertaken to isolate ß-N-acetylglucosaminidase cDNAs from Sf9 cells, which enabled expression of the gene products and functional characterization of the overexpressed enzyme activities.
This gene hunt was successful, as enzyme activity assays clearly showed that we had isolated cDNAs encoding broad-spectrum ß-N-acetylglucosaminidases that can hydrolyze non-reducing terminal N-acetylglucosamine residues from either branch of synthetic, biantennary N-glycans with no strong branch preference. This relative lack of substrate specificity was underscored by the ability of these enzymes to hydrolyze chitotriose to its constituent monosaccharides. Together, these results clearly indicated that neither SfGlcNAcase-1 nor SfGlcNAcase-3 could account for the Sf9 processing ß-N-acetylglucosaminidase activity originally described by Altmann and coworkers (1995) because that activity had a clear branch preference. In fact, our enzyme activity assay results were more consistent with the idea that SfGlcNAcase-1 and -3 have a functional role in glycoprotein glycan, oligosaccharide, and chitin degradation in Sf9 cells and S. frugiperda.
This conclusion was, in turn, consistent with computer predictions indicating that SfGlcNAcase-1 and SfGlcNAcase-3 are equipped to enter the secretory pathway and with experimental data demonstrating that both are associated with an intracellular membrane fraction and are released into the extracellular fraction. These findings, together with the very high level of similarity between these insect gene products and the human hexosaminidases, led us to believe that SfGlcNAcase-1 and SfGlcNAcase-3 might be insect lysosomal hexosaminidases. The pH optima of SfGlcNAcase-1 and -3 were consistent with this conclusion, but our evidence that both enzymes are transmembrane proteins was inconsistent with this conclusion. As an aside, the apparent transmembrane nature of these enzymes suggests that their ultimate appearance in the extracellular fraction is either an artifact of over-expression or the result of proteolytic processing. Direct examination of the intracellular distribution of GFP-tagged forms of these enzymes revealed that they reside in discrete sites throughout the cytoplasm, but these sites do not correspond to lysosomes or the Golgi apparatus. An important caveat of the last result was that the GFP tag could have interfered with the normal localization of SfGlcNAcase-1 and -3, but our previous experience tagging lysosomal and Golgi proteins in this system argued against this interpretation. Future studies should be directed towards the production of specific antibodies that can be used to localize the untagged forms of these proteins and clarify the nature of the compartment in which they reside.
In the final analysis, this study demonstrated that Sf9 cells encode and express at least two ß-N-acetylglucosaminidases with a mixed set of properties, including both properties of lysosomal enzymes involved in oligosaccharide degradation and properties of Golgi enzymes involved in glycoprotein biosynthesis. The relative lack of substrate specificity is the most definitive functional property of these gene products and, as such, this property strongly suggests that neither of these enzymes is involved in glycoprotein glycan processing. Similarly, the high degree of sequence similarity between the SfGlcNAcases and the human hexosaminidases is striking and further suggests that the insect enzymes are involved in glycan degradation. Based on available data, one might speculate that Sf9 cells have two functionally redundant degradative GlcNAcases. Thus, it is interesting to note that humans have two lysosomal hexosaminidases, which function as homo- and heterodimers and, in the future, it will be of interest to determine if the SfGlcNAcases form homo- and/or heterodimers, as well, and to determine if the functions of these proteins are influenced by their oligomeric state(s).
In context of the previous results of Altmann and coworkers (1995), the broad substrate specificities of SfGlcNAcase-1 and -3 and their acidic pH optima indicate that Sf9 cells encode yet another ß-N-acetylglucosaminidase that functions biosynthetically in N-glycoprotein glycan processing. The most confusing, but interesting property was the unusual intracellular distributions of the GFP tagged versions of SfGlcNAcase-1 and -3. In the future, it will be of great interest to more clearly define the intracellular compartment occupied by these enzymes and to try to relate their presence in this compartment to their apparent functions.
Acknowledgments
We thank Ziad Kawar for performing the original multiple sequence alignments, designing the degenerate oligonucleotide primers, and performing some of the early PCR reactions for this project. We thank Angela Harte, Jonathan Hartley, Jeremiah Hensley, Daniel Hill, and Jonathan Olsen for general technical assistance during the course of this project. Finally, we thank Dr. Friedrich Altmann and other colleagues at the University of Vienna for providing some of the synthetic N-glycans used in this study and for communicating their results prior to publication. This work was supported by NSF Grant BES-9814157 and NIH Grant #GM49734.
References
- 1.Jarvis DL. Baculovirus expression vectors. In: Miller LK, editor. The Baculoviruses. Plenum Press; New York: 1997. pp. 389–431. [Google Scholar]
- 2.O’Reilly DR, Miller LK, Luckow VA. W.H. Freeman and Company; New York: 1992. Baculovirus expression vectors. [Google Scholar]
- 3.Summers MD, Smith GE. A manual of methods for baculovirus vectors and insect cell culture procedures. Tx Ag Expt Stn Bull No 1555. 1987 [Google Scholar]
- 4.Vaughn JL, Goodwin RH, Thompkins GJ, McCawley P. The establishment of two insect cell lines from the insect Spodoptera frugiperda (Lepidoptera:Noctuidae) In Vitro. 1977;13:213–217. doi: 10.1007/BF02615077. [DOI] [PubMed] [Google Scholar]
- 5.Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J. Cold Spring Harbor; New York: 1999. Essentials of Glycobiology, Cold Spring Harbor Press. [PubMed] [Google Scholar]
- 6.Licari PJ, Jarvis DL, Bailey JE. Insect cell hosts for baculovirus expression vectors contain endogenous exoglycosidase activity. Biotechnol Progr. 1993;9:146–152. doi: 10.1021/bp00020a005. [DOI] [PubMed] [Google Scholar]
- 7.Altmann F, Schwihla H, Staudacher E, Glossl J, Marz L. Insect cells contain an unusual, membrane-bound ß-N-acetylglucosaminidase probably involved in the processing of protein N-glycans. J Biol Chem. 1995;270:17344–17349. doi: 10.1074/jbc.270.29.17344. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W, Cao P, Chen S, Spence AM, Zhu S, Staudacher E, Schachter H. Synthesis of paucimannose N-glycans by Caenorhabditis elegans requires prior actions of UDP-N-acetyl-D-glucosamine:alpha-3-D-mannoside beta1,2-N-acetylglucosaminyltransferase I, alpha3,6-mannosidase II and a specific membrane-bound beta-N-acetylglucosaminidase. Biochem J. 2003;372:53–64. doi: 10.1042/BJ20021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vitale A, Chrispeels MJ. Transient N-acetylglucosamine in the biosynthesis of phytohemagglutinin: attachment in the Golgi apparatus and removal in protein bodies. J Cell Biol. 1984;99:133–140. doi: 10.1083/jcb.99.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sturm A. N-glycosylation of proteins in plants. In: Montreuil J, Vliegenthart JFG, Schachter H, editors. Glycoproteins. Elsevier; Amsterdam: 1995. pp. 521–541. [Google Scholar]
- 11.Murhammer DW, Goochee CF. Scaleup of insect cell cultures: protective effects of pluronic F-68. Bio/Technology. 1988;6:1411–1418. [Google Scholar]
- 12.Sambrook J, Fritsch EF, Maniatis T. Cold Spring Harbor Press; Cold Spring Harbor, New York: 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 13.Innis MA, Gelfand DH. Optimization of PCRs. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR Protocols: A Guide to Methods and Applications. Academic Press; San Diego: 1990. pp. 3–12. [Google Scholar]
- 14.Muldoon LL, Neuwelt EA, Pagel MA, Weiss DL. Characterization of the molecular defect in a feline model for type II GM2-gangliosidosis (Sandhoff disease) Am J Pathol. 1994;144:1109–1118. [PMC free article] [PubMed] [Google Scholar]
- 15.Bapat B, Ethier M, Neote K, Mahuran D, Gravel RA. Cloning and sequence analysis of a cDNA encoding the beta-subunit of mouse beta-hexosaminidase. FEBS Lett. 1988;237:191–195. doi: 10.1016/0014-5793(88)80199-6. [DOI] [PubMed] [Google Scholar]
- 16.Yamanaka S, Johnson ON, Norflus F, Boles DJ, Proia RL. Structure and expression of the mouse beta-hexosaminidase genes, Hexa and Hexb. Genomics. 1994;21:588–596. doi: 10.1006/geno.1994.1318. [DOI] [PubMed] [Google Scholar]
- 17.Wakamatsu N, Benoit G, Lamhonwah AM, Zhang ZX, Trasler JM, Triggs-Raine BL, Gravel RA. Structural organization, sequence, and expression of the mouse HEXA gene encoding the alpha subunit of hexosaminidase A. Genomics. 1994;24:110–119. doi: 10.1006/geno.1994.1587. [DOI] [PubMed] [Google Scholar]
- 18.Beccari T, Hoade J, Orlacchio A, Stirling JL. Cloning and sequence analysis of a cDNA encoding the alpha-subunit of mouse beta-N-acetylhexosaminidase and comparison with the human enzyme. Biochem J. 1992;285:593–596. doi: 10.1042/bj2850593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham TR, Zassenhaus HP, Kaplan A. Molecular cloning of the cDNA which encodes beta-N-acetylhexosaminidase A from Dictyostelium discoideum. Complete amino acid sequence and homology with the human enzyme. J Biol Chem. 1988;263:16823–16829. [PubMed] [Google Scholar]
- 20.Beanan MJ, Bailey GB. The primary structure of an Entamoeba histolytica beta-hexosaminidase A subunit. J Eukaryot Microbiol. 1995;42:632–636. doi: 10.1111/j.1550-7408.1995.tb05919.x. [DOI] [PubMed] [Google Scholar]
- 21.Short JM, Fernandez JM, Sorge JA, Huse WM. Lambda ZAP: a bacteriophage lambda expression vector with in vivo excision properties. Nucl Acids Res. 1988;16:7583–7600. doi: 10.1093/nar/16.15.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarvis DL, Bohlmeyer DA, Liao YF, Lomax KK, Merkle RK, Weinkauf C, Moremen KW. Isolation and characterization of a class II alpha-mannosidase cDNA from lepidopteran insect cells. Glycobiology. 1997;7:113–127. doi: 10.1093/glycob/7.1.113. [DOI] [PubMed] [Google Scholar]
- 23.Kawar Z, Herscovics A, Jarvis DL. Isolation and characterization of an alpha 1,2-mannosidase cDNA from the lepidopteran insect cell line Sf9. Glycobiology. 1997;7:433–443. doi: 10.1093/glycob/7.3.433. [DOI] [PubMed] [Google Scholar]
- 24.Aviv H, Leder P. Purification of biologically active globin messenger RNA by chromatography on oligothymidylic acid-cellulose. Proc Natl Acad Sci USA. 1972;69:1408–1412. doi: 10.1073/pnas.69.6.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 26.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 27.Jarvis DL, Weinkauf C, Guarino LA. Immediate early baculovirus vectors for foreign gene expression in transformed or infected insect cells. Prot Expr Purif. 1996;8:191–203. doi: 10.1006/prep.1996.0092. [DOI] [PubMed] [Google Scholar]
- 28.Webb NR, Summers MD. Expression of proteins using recombinant baculoviruses. Technique. 1990;2:173–188. [Google Scholar]
- 29.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 30.Kitts PA, Possee RD. A method for producing recombinant baculovirus expression vectors at high frequency. Biotechniques. 1993;14:810–817. [PubMed] [Google Scholar]
- 31.Velardo MA, Bretthauer RK, Boutaud A, Reinhold B, Reinhold VN, Castellino FJ. The presence of UDP-N-acetylglucosamine:alpha-3-D-mannoside beta 1,2-N-acetylglucosaminyltransferase I activity in Spodoptera frugiperda cells (IPLB-SF-21AE) and its enhancement as a result of baculovirus infection. J Biol Chem. 1993;268:17902–17907. [PubMed] [Google Scholar]
- 32.Chatterton JE, Hirsch D, Schwartz JJ, Bickel PE, Rosenberg RD, Lodish HF, Krieger M. Expression cloning of LDLB, a gene essential for normal Golgi function and assembly of the ldlCp complex. Proc Natl Acad Sci USA. 1999;96:915–920. doi: 10.1073/pnas.96.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altmann F, Kornfeld G, Dalik T, Staudacher E, Glossl J. Processing of asparagine-linked oligosaccharides in insect cells. N-acetylglucosaminyltransferase I and II activities in cultured lepidopteran cells. Glycobiology. 1993;3:619–625. doi: 10.1093/glycob/3.6.619. [DOI] [PubMed] [Google Scholar]
- 34.Grace TDC. Establishment of four strains of cells from insect tissues grown in vitro. Nature. 1962;195:788–789. doi: 10.1038/195788a0. [DOI] [PubMed] [Google Scholar]
- 35.Moremen KW. Isolation of a rat liver Golgi mannosidase II clone by mixed oligonucleotide-primed amplification of cDNA. Proc Natl Acad Sci USA. 1989;86:5276–5280. doi: 10.1073/pnas.86.14.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 37.Milpetz F, Argos P, Persson B. TMAP: a new email and WWW service for membrane-protein structural predictions. Trends Biochem Sci. 1995;20:204–205. doi: 10.1016/s0968-0004(00)89009-x. [DOI] [PubMed] [Google Scholar]
- 38.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 39.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucl Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falquet L, Pagni M, Bucher P, Hulo N, Sigrist CJ, Hofmann K, Bairoch A. The PROSITE database, its status in 2002. Nucl Acids Res. 2002;30:235–238. doi: 10.1093/nar/30.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Myerowitz R, Piekarz R, Neufeld EF, Shows TB, Suzuki K. Human beta-hexosaminidase alpha chain: coding sequence and homology with the beta chain. Proc Natl Acad Sci USA. 1985;82:7830–7834. doi: 10.1073/pnas.82.23.7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Dowd BF, Quan F, Willard HF, Lamhonwah AM, Korneluk RG, Lowden JA, Gravel RA, Mahuran DJ. Isolation of cDNA clones coding for the beta subunit of human beta-hexosaminidase. Proc Natl Acad Sci USA. 1985;82:1184–1188. doi: 10.1073/pnas.82.4.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagamatsu Y, Yanagisawa I, Kimoto M, Okamoto E, Koga D. Purification of a chitooligosaccharidolytic beta-N-acetylglucosaminidase from Bombyx mori larvae during metamorphosis and the nucleotide sequence of its cDNA. Biosci Biotechnol Biochem. 1995;59:219–225. doi: 10.1271/bbb.59.219. [DOI] [PubMed] [Google Scholar]
- 44.Zen KC, Choi HK, Krishnamachary N, Muthukrishnan S, Kramer KJ. Cloning, expression, and hormonal regulation of an insect beta-N-acetylglucosaminidase gene. Insect Biochem Mol Biol. 1996;26:435–444. doi: 10.1016/0965-1748(95)00111-5. [DOI] [PubMed] [Google Scholar]
- 45.Pillay CS, Elliott E, Dennison C. Endolysosomal proteolysis and its regulation. Biochem J. 2002;363:417–429. doi: 10.1042/0264-6021:3630417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci USA. 1998;95:6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jarvis DL, Summers MD. Glycosylation and secretion of human tissue plasminogen activator in recombinant baculovirus-infected insect cells. Mol Cell Biol. 1989;9:214–223. doi: 10.1128/mcb.9.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vadaie N, Hulinsky R, Jarvis DL. Expression and functional characterization of Drosophila ß4-galactosyltransferase VII. Glycobiology. 2002;12:589–597. doi: 10.1093/glycob/cwf074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vadaie N, Jarvis DL. Molecular cloning of a lepidopteran insect ß1,4N-acetylgalactosaminyltransferase with an unusually broad substrate specificity, a functional role in N-glycoprotein biosynthesis, and a potential functional role in glycolipid biosynthesis. J Biol Chem. 2004;279:33501–33518. doi: 10.1074/jbc.M404925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawar ZS, Jarvis DL. Biosynthesis and intracellular localization of a lepidopteran insect alpha 1,2-mannosidase. Insect Biochem Mol Biol. 2001;31:289–297. doi: 10.1016/s0965-1748(00)00121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perotti ME, Cattaneo F, Pasini ME, Verni F, Hackstein JH. Male sterile mutant casanova gives clues to mechanisms of sperm-egg interactions in Drosophila melanogaster. Mol Repr Dev. 2001;60:248–259. doi: 10.1002/mrd.1085. [DOI] [PubMed] [Google Scholar]
- 52.Cattaneo F, Ogiso M, Hoshi M, Perotti ME, Pasini ME. Purification and characterization of the plasma membrane glycosidases of Drosophila melanogaster spermatozoa. Insect Biochem Mol Biol. 2002;32:929–941. doi: 10.1016/s0965-1748(02)00031-0. [DOI] [PubMed] [Google Scholar]
- 53.Tatusova TA, Karsch-Mizrachi I, Ostell JA. Complete genomes in WWW Entrez: data representation and analysis. Bioinformatics. 1999;15:536–543. doi: 10.1093/bioinformatics/15.7.536. [DOI] [PubMed] [Google Scholar]
- 54.Korneluk RG, Mahuran DJ, Neote K, Klavins MH, O’Dowd BF, Tropak M, Willard HF, Anderson MJ, Lowden JA, Gravel RA. Isolation of cDNA clones coding for the alpha-subunit of human beta-hexosaminidase. Extensive homology between the alpha- and beta-subunits and studies on Tay-Sachs disease. J Biol Chem. 1986;261:8407–8413. [PubMed] [Google Scholar]
- 55.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, de Pablos B, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RD, Scheeler F, Shen H, Shue BC, Siden-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang ZY, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, Woodage T, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh RF, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 56.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]