

Abstract
The development of highly efficient analytical methods capable of probing biological systems at system level is an important task that is required in order to meet the requirements of the emerging field of systems biology. Optical molecular imaging (OMI) is a very powerful tool for studying the temporal and spatial dynamics of specific biomolecules and their interactions in real time in vivo. In this article, recent advances in OMI are reviewed extensively, such as the development of molecular probes that make imaging brighter, more stable and more informative (e.g., FPs and semiconductor nanocrystals, also referred to as quantum dots), the development of imaging approaches that provide higher resolution and greater tissue penetration, and applications for measuring biological events from molecule to organism level, including gene expression, protein and subcellular compartment localization, protein activation and interaction, and low-mass molecule dynamics. These advances are of great significance in the field of biological science and could also be applied to disease diagnosis and pharmaceutical screening. Further developments in OMI for systems biology are also proposed.
Keywords: Optical molecular imaging, Fluorescent protein, Quantum dots, Systems biology, Review
Introduction
Due to the rapid progress in the field of molecular biology that has occurred over the last century, we have come to see that life involves thousands of genes, proteins, metabolites, etc. However, life is not invoked by simply collecting these together; an integrated system formed from those components is required. It is vital to understand biological systems such as molecular networks, cells, tissues and even the organism itself at the system level. This shift in biological paradigm from reductionism to integration has made the field of systems biology [1–5] a new challenge that is currently a hot topic. Although attempts to systematically understand biological systems can be dated back to Wiener’s time [6], research into systems biology represent the first attempts to achieve this based on detailed knowledge of molecules. It promises to reveal the relationships among elements of systems that may include just a few proteins that together perform a defined task or more complex molecular machines, cells and groups of cells, with the goal of understanding their emergent properties [3].
It is widely agreed that systems biology is a large scientific field that relies greatly on collaborations between disciplines such as the life sciences, information science, system engineering and analytical technology. One of the keys to successful research in this field is considered to be work being done in analytical chemistry [7, 8] to develop innovative analytical methods that meet the needs of systems biology research. For example, capillary electrophoresis has been recognized as being the “gold standard” in genomics for DNA sequencing. Similar “gold standard” methods are available in transcriptomics (DNA microarray), proteomics (multidimensional separation coupling with mass spectrometry) and metabolomics (microseparation coupled with mass spectrometry and nuclear magnetic resonance). However, more of these methods are needed. As well as revealing genes, proteins and metabolites from those omic investigations, which relate to the basic structures of systems, it is important to have methods capable of monitoring their localizations, connections, and in particular their dynamics over time under various physiological or pathological conditions.
Optical molecular imaging (OMI) is a versatile technique that can be used to investigate the dynamics of biological events in molecules, cells, tissues and organisms in real time and in vivo (Fig. 1) [9–11]. Compared with other imaging approaches like magnetic resonance imaging and positron emission tomography etc., OMI exhibits the great advantages of high temporal (picosecond) and spatial (submicron) resolutions, high sensitivity (single-molecule level) and minimal invasion, and shows high potential for systems biology. This review highlights recent advances in OMI, focusing especially on the development of fluorescent probes such as fluorescent proteins and semiconductor nanocrystals (also referred to as quantum dots, QDs), OMI instrumentation and approaches (techniques). The application of OMI to studies of localization, conformations and interactions of biomolecules in vivo are also highlighted. In particular, this review describes the application of OMI for tracing the dynamics of single molecules and whole-body living organisms, which strongly suggest that OMI is an informative systems biology method that may be used to uncover biological events at different system levels. The future development of OMI is also predicted.
Fig. 1.
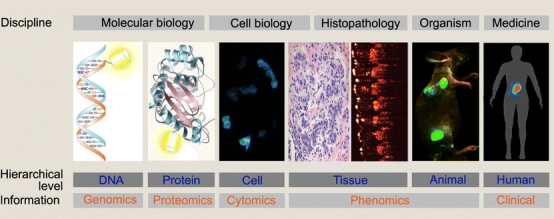
Utilization of OMI in life science research. OMI can investigate the dynamics of biological events in real time from molecules (left), cells, tissues and organisms (right) digitally and quantitatively. It can handle a wide range of intensities (about 12 orders of magnitude), times (femtoseconds to years) and spatial dimensions (nanometers to centimeters), and it gives high spatial and temporal resolution of the targeted cellular structures—better than any other method
Advances in fluorescent probes
Aequorea fluorescent protein (AFP)-derived mutants
Labeling of biomolecules with fluorescent probes or other dyes has facilitated in vitro or in vivo studies of biomolecular structures and dynamics, as well as their interactions, which is critical if we are to understand the biomolecular mechanisms of cellular function. However, traditional methods of chemical labeling based on fluorescent dyes are often inadequate for biomolecular labeling, repurification and reintroduction into cells by invasive methods like microinjection. These limitations have spawned efforts to noninvasively and site-specifically label protein in living cells or tissues by using green fluorescent protein (GFP) and its variants. The fluorescent protein presents a relatively small size and a compact, single-domain structure, which allow it to fuse to other targeted proteins with little or no interference in native protein.
GFP was cloned from Aequorea victoria. Excitation of the wild-type GFP by light with a wavelength of 398 nm can induce the emission of bright green fluorescence, peaking at 508 nm. Through continuous efforts to perform protein mutation of GFP, scientists have developed various kinds of AFP with different excitation and emission wavelengths, enhanced brightness, and improved pH resistance compared to the original wide-type, such as enhanced GFP (EGFP), cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) (Fig. 2a) [12]. The original YFP exhibited several drawbacks when imaging in vivo [13]. Later generations of YFP mutants, including “Citrine” [14] and “Venus” [15], exhibit low pH (5.7) resistance, halide insensitivity, greatly improved photostability, brighter fluorescence, and faster maturation. The original generation of CFP mutants also displayed several spectroscopic disadvantages. By using similar methods to YFP mutagenesis, a CFP mutant named “Cerulean” [16] was obtained, with an improved quantum yield, a higher extinction coefficient and a longer fluorescence lifetime. Another new color GFP mutant, cyan-green fluorescent protein (CGFP) [17], whose excitation and emission wavelengths are intermediate between those of CFP and EGFP, was also found by using this tactic. In addition, “PA-GFP,” a GFP mutant, has 100 times greater fluorescence than the original and remains stable for days [18]. These fluorescent proteins were termed molecular or optical highlighters, and represent perhaps the most promising tools for investigating protein lifetimes, transport, turnover rates and so on.
Fig. 2a–c.
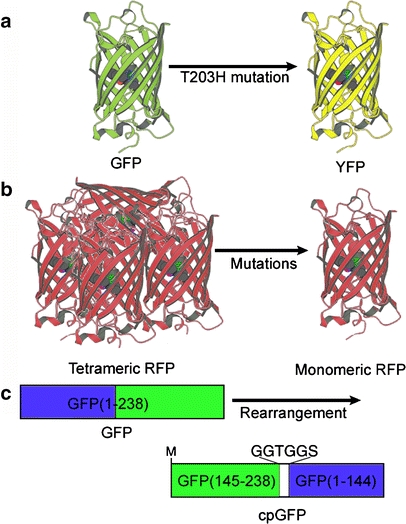
Various FP mutants. a Introduction of the mutation of Thr203His in GFP results in significantly red-shifted maximum excitation and emission wavelengths; this mutant is named YFP. b By using mutagenesis, the original tetrameric DsRed is reconstructed into the monomeric DsRed variant. c Interchanging the amino and carboxyl portions of GFP and rejoining them with a short spacer generates cpGFP
Other kinds of fluorescent protein
Long-wavelength fluorescence can provide greater tissue penetration and better spectral separation from cellular autofluorescence. Therefore, red fluorescent proteins (RFP) are very useful in applications related to multicolor protein-tracking and the construction of sensors. The first RFP with a red emission wavelength was cloned from Discosoma genus, named “DsRed” [19]. Its structure is similar to GFP, but it is tetrameric. By using mutagenesis, different derivations exhibiting several advantages have been generated, such as the fluorescent timer of DsRed which has fluorescence that changes from green to red over time [20], the monomeric DsRed, named “mRFP1” [21] (Fig. 2b), as well as DsRed2 [22], T1 [23], and mCherry [24], which have chromophores that are brighter, more soluble, and mature faster than the original generation. Another orange-emitting fluorescent protein, which has higher pH-resistance and is more effective for imaging, was discovered in Fungia concinn [25]. In addition, “Kaede” [26], which is cloned from Trachyphyllia geoffroyi, can convert from green to red fluorescence and brightens 2,000-fold under UV illumination.
New rearrangement variants of fluorescent protein
In previous studies, fluorescent proteins were treated as an indivisible entity, usually appended to the amino or carboxyl terminus of the targeted protein. However, several rearrangements of GFPs in which the amino and carboxyl portions were interchanged and rejoined with a short spacer still remained fluorescent (Fig. 2c). These circular permutations exhibit several advantages, such as altered pKa values, orientations of the chromophore with respect to a fusion partner, and greater absorbance of the exciting energy [27, 28].
Nongenetic fluorescent probes
However, there are some disadvantages of using FPs to perform OMI of the whole-body. Given the available wavelengths of excitation and emission and the power of the FP, optical imaging with a depth of penetration of approximately 1–2 mm limits their use to C. elegans, Drosophila or surface structures in small animals. To solve these problems, applying QDs (quantum dots) to OMI of the whole-body could allow us to visualize biological events in deeper tissues with better image quality.
Compared with conventional organic dyes and fluorescent proteins, semiconductor nanocrystals—quantum dots—exhibit a narrow, tunable, symmetric emission spectrum and better photochemical stability. Also, their wavelengths of maximum excitation and emission shift to shorter wavelengths with decreasing size [29, 30]. QDs present several great advantages for whole-body OMI, such as their very large molar extinction coefficients and their very bright emission, which mean that the emission can be visualized in big animals [31]; the ability to perform simultaneous multiple color imaging of targets of interest [32]; and long excited state lifetimes that provide a way to separate the QD fluorescence from background fluorescence [33]. Because QDs are capped with a monolayer of organic ligands and are hydrophobic, they cannot be used for visualization in vivo. Goldman et al. [34] reported that QDs capped with fusion proteins of a specific antibody, protein G and leucine zipper adaptor protein could be used to image the target protein in vivo (Fig. 3). Since then, bioconjugated QDs have provided new approaches for the ultrasensitive and multiple-color imaging of targeted molecules in vivo. In addition, integrating QDs with paramagnetic substances results in new multimodality imaging probes that cuple the deep imaging capability of magnetic resonance imaging with ultrasensitive OMI [35]; these would be very powerful probes if used in clinical research. More recently, a new QD conjugate was found to emit long waves in the absence of external excitation light through bioluminous resonance energy transfer [36]. Compared with existing quantum dots, self-luminous quantum dots provide great sensitivity when imaging small animals, giving high signal-to-noise ratios.
Fig. 3.

Method of conjugating QDs to target proteins. The pG-zb acts as a molecular adaptor, connecting the QDs with the target protein through interactions of its protein G portion with a specific antibody as well as interactions of its positively charged tail with QDs capped with a negatively charged dihydrolipoic acid surface
OMI techniques
General imaging microscopy
Laser scanning confocal microscopy (LSCM) (Fig. 4a) and wide-field microscopy (WFM) are the tools most widely used for in vivo OMI. The major difference between them is that LSCM only collects the fluorescence emission of in-focus light, whereas WFM collects all signals, including out-of-focus light [37]. LSCM can produce superior images of multiple-cell samples, with photobleaching. However, LSCM cannot perform multiple-color imaging because it only observes in-focus light [38]. WFM is among the most sensitive of all such methods, permits minimal exposure of the sample, uniform illumination, an unlimited choice of excitation wavelengths, and is relatively simple [37]. One of the disadvantages of WFM is that disturbances are seen in observations of thicker specimens, where the out-of-focus signals become substantial [38].
Fig. 4a–f.
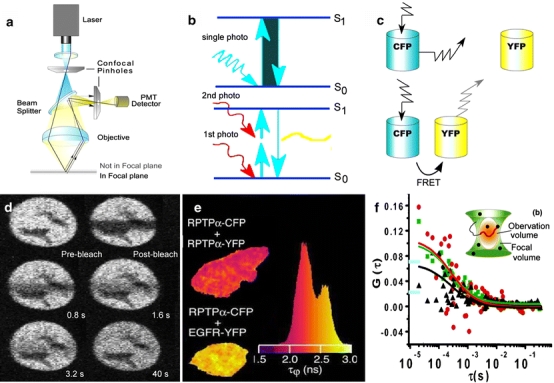
Fluorescent OMI approaches. a LSCM only collects in-focus emitted light. b The principle of multiple photon excitation is based on the use of pulsed long excitation wavelengths to excite fluorescence. c FRET occurs between a donor and an acceptor that are in molecular proximity if the emission spectrum of the donor overlaps the excitation spectrum of the acceptor. d FRAP can reveal the mobility of FP-labeling proteins. These images illustrate the change in fluorescence of cells expressed with YFP-hGR before and after photobleaching. Reproduced from [51] with permission. e FLIM can measure the time-dependent emission intensity. The histogram represents the fluorescence lifetime distributions for the donor in the presence of interactions (red) or not (yellow). Reproduced from [57] with permission. f FCS can monitor the fluorescence signals emitted from the ROI. The cross-correlation curve (black) indicates a higher level of dimer or oligomer formation in the R1- and R5-expressing cells. Reproduced from [59] with permission
Furthermore, multiple-photon laser scanning microscopy (MPLSM) has also been used in OMI [39]. The principle of MPLSM is that pulsed long excitation wavelength light, such as that provided by an infrared (IR) laser beam, is used to excite a molecule at the focal plane by multiple photons, causing fluorescence (Fig. 4b). The advantages of MPLSM are high tissue penetration ability (>30 μm), low photobleaching or photodamage, and a high signal/noise ratio (due to low autofluorescence) [40].
Second-harmonic imaging microscopy (SHIM)
SHIM is based on a nonlinear optical effect called second harmonic generation (SHG), commonly called frequency doubling. This phenomenon requires that intense laser light passes through a highly polarizable material with noncentrosymmetric molecular organization [41]. Biological macromolecules often assemble into large, ordered noncentrosymmetric structures and have highly polarizability. Therefore, high-resolution SHIM imaging can visualize living cells or tissues. This method provides several benefits: the targeted molecules are not excited, photobleaching does not occur, and it can detect information related to pathology. In addition, a combination of SHIM and MPLSM could make a powerful optical imaging technique.
Optical coherence tomography (OCT)
The principle of OCT imaging is analogous to that of ultrasound B-mode imaging, except that OCT uses light rather than acoustic waves [42]. OCT can provide cross-sectional imaging of structures below the tissue surface, analogous to histopathology. By using a state-of-the-art laser as the light source, ultrahigh-resolution imaging with axial resolutions as fine as 1–2 μm can be achieved [43]. However, the penetration depth of the technique in most tissues is limited to approximately 2–3 mm [44]. Several features of OCT imaging make it well-suited to imaging-based diagnostics and surgical guidance, such as its high resolution, its ability to quantitatively assess, and its ability to image some tissue functions.
Fluorescent molecular imaging approaches
Fluorescence resonance energy transfer (FRET)
FRET is a quantum mechanical phenomenon that occurs between a fluorescence donor and a fluorescence acceptor with a favorable dipole–dipole orientation that are in molecular proximity to each other, provided the emission spectrum of the donor overlaps the excitation spectrum of the acceptor (Fig. 4c) [45]. Therefore, imaging based on FRET can determine the proximity (within the nanometer range) between labeled biomolecules in living cells [46, 47]. FRET-based microscopy includes intensity-based detection methods and fluorescence decay kinetics-based detection methods [46]. Similar to general fluorescence imaging microscopy, FRET also suffers from various drawbacks, like autofluorescence, detector noise, optical noise and photobleaching. In addition, spectral bleed-through (SBT), or cross-talk, is a major problem in FRET [48]. FRET is currently widely used in studies of protein colocalization, conformational changes, protein interactions and signal transduction [47, 49].
Fluorescence recovery after photobleaching (FRAP)
FRAP involves observing the rate of recovery of fluorescence resulting from the movement of a fluorescent marker into an area which contains the same marker that has been rendered nonfluorescent via an intense photobleaching pulse of laser light (Fig. 4d). FRAP has proved to be a powerful method for measuring the mobilities of target molecules in various membranes, cytoplasms and nuclei [50]. For example, the mobility of the hormone receptor in the nucleus of a living cell [51], the mobility in subcellular compartments and the dynamics of vesicles inside the synapses of cultured hippocampal neurons [52] have been studied using this approach.
Fluorescence lifetime imaging microscopy (FLIM)
FLIM is a technique by which the mean fluorescence lifetime of a chromophore is measured at each spatially resolvable element of a microscope image [53]. The fluorescence lifetime is an inherent property of a chromophore that is sensitive to environmental and physical processes. Therefore, FLIM can detect interesting physical processes that can influence the excited state of a probe in vivo [54]. In addition, the mean lifetime of a donor is shifted to shorter lifetimes in the presence of an acceptor in FRET (Fig. 4e), so FLIM can detect changes in FRET [55], although this may not be valid in complex cellular systems that consist of several different lifetime components [56]. For example, using a FRET–FLIM method, the regulation of the activity of receptor protein tyrosine kinase (RPTK) through its dimerization was demonstrated in living cells [57]. The main drawbacks of FLIM are that it has relatively low sensitivity and requires expensive instrumentation.
Fluorescence correlation spectroscopy (FCS)
FCS is an approach that has emerged only recently. It is a high-sensitivity photon-counting technique that permits fluctuations in the fluorescence signal due to changes in the fluorescence quantum yield to be measured (Fig. 4f) [58]. Therefore, FCS can directly measure the mobilities of biomolecules, and it can monitor the average association and dissociation of target labeled with fluorescence probe—which vary with interaction kinetics, complex composition and complex size—in vivo without photobleaching. For example, FCS can be used to detect the clustering of somatostatin receptors [59] and the binding affinities of glucocorticoids (GC) in different subcellular compartments [60].
Applications of OMI
OMI for biomolecules
Originally, OMI of biomolecule function was performed on dead fixed cells or tissue sections in vitro. The first application of fluorescent detection in situ emerged in 1980, when RNA that was directly labeled with fluorophore was used as a probe for specific DNA sequences [61]. However, this method is not sensitive enough. Another indirect detection method allows signal to be significantly increased by binding secondary reporter to the hybridization probes that bind to targeted DNA [62] and mRNA [63]. Recently, Santangelo et al. [64] indicated that coinjection of two beacons based on FRET could be used to map the localization of a specific hybridization in living cells. Another application of FISH is immunocytochemistry (IC), which is used to detect target protein in specimens in vitro as antigen by means of antibodies labeled with fluorescence probe [65]. The technique of FISH is still being developed, with more specific fine-tuning of sensitivity and multiplicity necessary, after which its applications should gradually move from being in vitro to in vivo [66].
In addition, Fourier transform infrared (FT–IR) spectroscopy allows the simultaneous detection of nearly all organic molecules in a single spectrum and hence is ideally suited for the investigation of complex metabolic pathways. However, this advantage is only useful when powerful mathematical methods are available for the analysis of the highly complex spectra produced. Glycolytic intermediates In yeast extract were identified with FT–IR spectroscopy. The results were in good agreement with known phase relationships in oscillatory glycolysis [67]. In another experiment, glycogen, protein, lipid and nucleic acid concentrations were monitored in fish liver [68] using this method.
Single-molecule OMI is a very young field that holds great promise. It is not a technique, but a way of thinking that can allow us to detect individual molecular interactions, protein dynamics and signaling transduction pathways in living cells, which is difficult and sometimes impossible to achieve using conventional techniques [46]. For example, Sonnleitner et al. observed that the voltage-gated ion channel does not directly open or close under the conditions of single-molecular interaction using this approach [69]. Karymov et al. reported a method that could be used to directly follow junction branch migration in the holiday junction at the single-molecule level, and they detected that branch migration was a stepwise random process where the overall kinetics were dependent on the Mg2+ concentration [70].
Using OMI to monitor physiological processes in living cells
Monitoring gene expression and RNA localization
The fusion of gene promoter and fluorescent protein cDNA can be used to noninvasively analyze the expression pattern of a target gene, which is a very common approach used to study gene expression in vivo and is widely used in life science research. To minimize the accumulation of the background fluorescence that is generated by leaky, undegraded level (i.e. from previously expressed fluorescent protein that has not yet degraded), several chimeric EGFPs with shorter fluorescence half-lives were constructed by fusing protein [71, 72]. However, this approach leads to lower sensitivity, and cannot reveal low levels of expression. To overcome these drawbacks, a new approach that is similar to the amplification of an enzymatic reporter was applied, which was then used in “enhancer-trap” strategies [73, 74]. The principle behind this is that the fluorescence of FP, revealing targeted gene expression in different tissues, is magnified through a transcript factor, which allows weak signals to be seen.
In addition, DNA and RNA sequences and other molecules could be visualized indirectly by labeling their binding proteins with FP in vivo. For example, by using RNA binding protein MS2 labeled with GFP, the mobility of targeted RNA could be visualized in vivo [75]. In another study, a similar method was used to observe gene expression correlated with a change in chromatin structure under the progress of transcription in real time [76]. Compared with FISH in vivo, the disadvantage of this method is the introduction of a protein complex of considerable size.
Monitoring protein and subcellular organelle dynamics
FP can also be used to monitor the behavior of the targeted protein, such as its appearance, degradation, localization, translocation and interaction in vivo. For example, by using a fusion protein of GR and FP, the dynamics of the exchange of GR with its binding sequence array can be monitored [77]. In addition, visualization of the dynamics of the GFP-GR fusion protein in vivo in more physiological conditions was achieved by using GFP-GR knock-in mice [78]. Another interesting application involves identifying the localization of an unknown protein on a large scale, a method termed the “protein trap,” in which the imaging of cells containing the fusion protein of FP and a cDNA library was used to screen for target protein at the localization of interest [79].
Fusion protein that targets a given subcellular organelle is often used to study the dynamics of subcellular organelles. For example, Shaw et al. [80] revealed the dynamics of migration across the cell cortex of individual cortical microtubules in Arabidopsis by using tubulin fused to FP. In a similar study, this method was used to measure the rates of tubulin polymer growth, shortening and transition [81]. Furthermore, new subcellular structures can also be discovered by visualizing fusion FP, such as the novel discrete area in the nucleus, in which phytochrome species were induced to accumulated by light [82].
Monitoring general protein–protein interactions
Detecting the proximity of two biomolecules based on FRET is an approach widely used to observe protein interactions in real time in vivo. Most commonly, CFP and YFP are respectively fused to each of their putative interaction proteins, which are coexpressed in one living cell so that FRET between two fused FPs of interaction can be detected (Fig. 5a). A variety of protein interactions in different cells have been successfully visualized so far using intermolecular FRET, such as the interaction between phytochrome B and cryptochrome 2 [83], the rearrangement of G-protein subunits [84], and the oligomeric states of the ligands B7-1 and B7-2 [85]. Protein–protein interactions can also be imaged via protein complementation assays. The putative interaction proteins are respectively fused to two complementary fragments of one fluorescent protein, and then interaction of the proteins can reinvoke the fluorescence [46, 86], in a technique which is analogous to yeast two-hybrid assays (Fig. 6a). Using this method, Hu et al. simultaneously visualized several interactions among bZIP and Rel family transcription factors in one cell [87, 88].
Fig. 5a–d.

General designs of FRET-based fluorescent probes. a An intermolecular probe consists of two interacting proteins that are labeled with CFP and YFP, respectively, which interact and result in FRET. b An intramolecular probe consists of CFP and YFP fused together with a cleavable linker or protein, which can be cleaved by proteolysis and disrupt FRET. c An intramolecular probe consists of sandwiching two domains between CFP and YFP, which can interact after phosphorylation or binding to calcium, resulting in a change in FRET. d An intramolecular probe consists of CFP, YFP and a protein/domain, which permits conformational change by binding to another biomolecule, leading to a change in FRET
Fig. 6a–d.
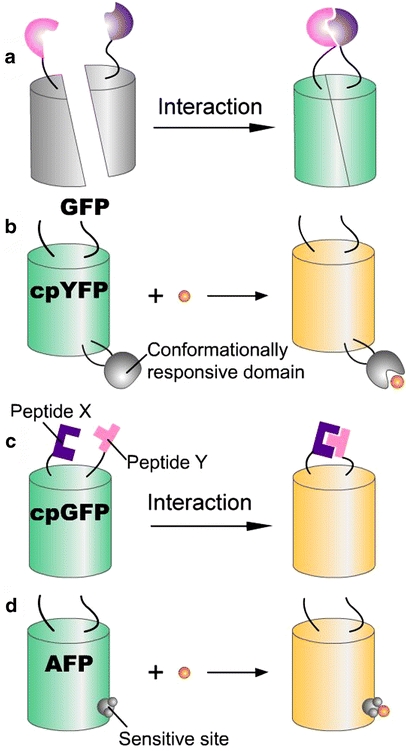
Single FP-based fluorescent probes. a The probe consists of the fusion of two interacting proteins to two complementary fragments of one FP, respectively, which can interact and reinvoke the fluorescence. b Insertion of a conformationlly responsive domain/protein into cpYFP can lead to a change of fluorescence when its conformation is changed. c The probe consists of the fusion of two interacting proteins/domains to the amino and carboxyl termini of cpGFP, which can interact and change the cpGFP fluorescence. d By using mutagenesis, AFP can be engineered to be directly sensitive to a small molecule, such as Cl−, H+
Monitoring protease and kinase activity
The first reporter of proteolysis consists of BFP and GFP fusing together with a protease-sensitive linker, which exhibits the FRET phenomenon. Proteolysis can disrupt the FRET by separating the donor and acceptor fluorescent proteins [89]. In recent studies, fusion proteins of CFP, YFP and a specific recognition sequence were used to measure the different caspase proteolytic activities during different apoptosis pathways (Fig. 5b) [90–92]. In another study, YFP-Bid-CFP fusion protein was used to visualize the activation of Bid protein by proteolytic cleavage, and translocation of the cleaved Bid to mitochondria was observed directly [93].
Reporters for the activity of tyrosine kinases and serine/threonine kinases have been made by sandwiching a substrate peptide for the kinase of interest and a phosphoaminoacid-binding domain, such as Src-homology-2 (SH2) or 14-3-3 protein, between two FPs (Fig. 5c). Phosphorylation of the substrate peptide induces the formation of an intramolecular complex with the phosphoaminoacid-binding domain, which leads to a change in the FRET. Meanwhile, these indicators also report the opposite phosphatase activity. This generic concept has so far been adapted to create probes for the phosphorylation of the kinase of interest in different signal transduction pathways, including phosphorylation of protein kinase A (PKA) [94], activation of serine/threonine kinase Akt [95], and activation of Src kinase on the cell membrane [96].
Monitoring changes in calcium
Genetically encoded fluorescent indicators for calcium without cofactors that can target to specific intracellular locations were first constructed by Miyawaki et al. [97]. This indicator was termed “cameleon.” The cameleon consists of the tandem fusion of CFP, calmodulin, the calmodulin-binding peptide M13 and YFP. Binding of Ca2+ allows calmodulin to wrap around the M13 domain, which increases the FRET between flanking FPs (Fig. 5c). Replacing YFP with cpYFP [28], Venus [98] or EYFP [99] led to a new generation of cameleon that exhibited better spatial and temporal resolution as well as better environmental resistance. In order to measure high concentrations of Ca2+, the CaM–M13 interface of cameleon was re-engineered to reduce its affinity to Ca2+, which improved the imaging of Ca2+ in the endoplasmic reticulum [100].
Other indicators for calcium that are not based on FRET have also been reported. cpGFP or cpYFP can tolerate the insertion of another entire protein, and conformational change of the fusion cpFP can change its fluorescence. Therefore, the insertion of calmodulin into cpYFP (called “camgaroos”) (Fig. 6b) [14, 27] and the tandem fusion protein of M13, cpGFP and calmodulin (termed “G-CaMP”) (Fig. 6c) [101] have both been used as calcium indicators because a conformational change occurs in the fusion cpFPs upon calcium binding.
Monitoring changes in cyclic nucleotides
The first fluorescent sensor of cAMP consists of cAMP-dependent protein kinase (PKA) in which the catalytic subunit and the regulatory subunit are labeled with a fluorescein and a rhodamine, respectively. Binding of cAMP to the labeled regulatory subunit leads to the dissociation of the labeled catalytic subunit, which disrupts the FRET between the two fluorescent dyes [102]. Later, this system was developed into a genetic cAMP indicator with BFP and GFP replacing the fluorescent dyes [103]. The most recent cAMP indicator was constructed by sandwiching a full length Epac1 between CFP and YFP (Fig. 5d) [104].
A genetically encoded indicator for another cyclic nucleotide, cGMP, has been reported by Sato et al. [105]. This indicator consists of a tandem fusion of BFP, nondimerizing mutants of cGMP-dependent protein kinase Iα (PKG Iα) and GFP. An increase in FRET between the two FPs can be detected upon the cGMP-induced conformational change of PKG Iα. Another sensor, termed “cygnet-1” [106], was constructed to have high selectivity for cGMP over cAMP. Here, the truncation of PKG Iα consisted of residues 1–77 rather than 1–47 reported in a previous study [105], and it exhibited decreased FRET fluorescence upon the binding of cGMP.
Monitoring changes in transmembrane voltage
Measuring electrical activity in living cells with high spatial and temporal resolution is a fundamental problem in studies of excited cell information processing. To address this problem, Siegel and Isacoff [107] first constructed a novel fluorescent probe that could be used to measure transmembrane voltage in vivo, termed the fluorescent shaker or “FlaSh”, in which the modified GFP was inserted into a voltage-sensitive K+ channel so that voltage-dependent rearrangements in the K+ channel fusion protein could induce changes in the GFP fluorescence. A similar voltage sensor was generated by inserting GFP into another channel, the rat μI skeletal muscle voltage-gated Na+ channel, and was named “SPARC” [108]. In other studies, Sakai et al. [109] developed a new voltage-sensitive indicator based on FRET, in which changes in membrane voltage can lead to changes in FRET.
Monitoring changes in pH in living cells
The chromophore of a FP is surrounded by a hydrogen-bonding network within the β-barrel, which means that the FP is pH-sensitive because of direct interactions between its hydrogen-bonding network and external protons [12]. The majority of GFP, YFP and their circularly permuted constructions described in previous parts of this review exhibit sensitivity to acid (Fig. 6d), whereas the original AFP—Renilla reniformis GFP—and DsRed are insensitive to acid. Therefore, acid-sensitive variants can serve as ideal donors in pH indicators based on FRET, and DsRed is the most desirable acceptor [110]. Another pH sensor has been generated by using a new group of pH-sensitive GFP variants, termed “deGFPs,” which exhibit rapid changes in emission from green to blue as the pH decreases, and these are suitable for ratiometric measurements in vivo [111, 112].
Monitoring signal transduction from cell to cell
Scientists have also monitored intercellular changes in biomolecules and physiological events, which have been visualized in single cells in real time in a cell population. For example, Bedner et al. monitored the intercellular permeability to cAMP of six different gap junction channels by using sensors that are highly sensitive to cAMP concentration [113]. The change in calcium concentration from cell to cell was also monitored via calcium dyes [114]. In addition to these biomicromolecules, the propagation of intercellular apoptotic or survival events in monolayer cells mediated by the gap junction channel were also studied through OMI [115].
Using OMI to investigate tissue structure and function
It has recently been shown that SHIM can be used to directly detect several structural protein arrays in tissues, such as collagen arrays in mouse [116], neurons and muscular structures of the pharynx in C. elegans [117] without the need for fluorescent labeling. SHIM also readily retrieves more detailed molecular information than that obtained through the application of fluorescent labeling. Therefore, SHIM can be used to study or diagnose several diseases that are related to the assembly polarity of microtubule complexes in native brain tissue [118]. In addition, SHIM has proven to be crucial to neuroscience investigations in thick tissue preparations. For example, the fast neuronal membrane potential transient in mammalian brain slices labeled with FM4-64 dye was investigated by this method [119]. By labeling with another dye, the action potential was recorded with high temporal and spatial resolution on soma and neurite membranes [120].
OCT also has the ability to perform in situ, real-time imaging of tissue pathology, and it can be used to guide excisional biopsy in order to reduce false negatives caused by sampling errors. In particular, OCT enables the internal architectural morphology of the retina to be visualized noninvasively, and it can be used to diagnose and monitor retinal diseases, which cannot be achieved through any other methods [121]. The development of high-speed OCT imaging combined with small fiber-optic probes has enabled in vivo endoscopic imaging, such as the visualization of the oral cavity [122], the larynx and the bladder [123]. In addition, OCT can perform functional imaging of tissues, such as brain activity [124], inflammatory/neoplastic morphologic changes [125]. Raman spectroscopy (RS) is another powerful diagnostic tool that enables tissue identification and classification, as demonstrated by measurements of the brain tissue of a six-month-old pig by fiber-optic probes [126] and analysis of the molecular composition of human bronchial tissue structures [127]. Also, a combination of confocal RS and LSCM has been employed to obtain detailed information about the subsurface structures in the skin with high spatial resolution in a completely noninvasive manner [128].
OMI of organisms
Employing fluorescent protein
When performing whole-body OMI of organisms, the utilization of genetically encoded fluorescence probes has enabled scientists to decipher spatial and temporal changes in biological events inside complex organisms [129]. Over the past few decades, FPs have been used to generate various fluorescent probes that have been used to image physiological and biochemical events in the monolayer cells. Extracting information from whole-body imaging is more relevant to real physiological conditions, but up to now, only a few fluorescent indicators have been used in complex organisms to visualize physiological activity in vivo.
To study the feeding behavior of C. elegans, Kerr et al. [130] introduced a calcium indicator based on cameleon to C. elegans, which allowed them to measure the Ca2+ signals evoked in pharyngeal muscles and individual neurons under the stimulation of feeding in vivo (Fig. 7a,b). Similar visualization of cameleon localized in other tissues was used to study the functions of serotonin and G proteins [131], the role of G proteins in signal transduction [132], and the ASH neuron response [133] in C. elegans. The spatial and temporal representation of odorant-evoked Ca2+ signals in the Drosophila brain was explored using cameleon [134] and G-CaMP (Fig. 7c–f) [135]. Calcium imaging was also used in other animals, such as zebrafish (Fig. 7g–i) [136] and mouse [137, 138]. In addition, an indicator with pH sensitivity was used to visualize spatial patterns of defined neuronal activity in the mouse [139]. More recently, the proteolytic activity of calpain was visualized by introducing fluorescent indicator into living mouse muscle, which gave the first 3-D imaging of FRET in vivo [140].
Fig. 7a–k.

Whole-body OMI using FPs and QDs. Imaging of Ca2+ signals in pharyngeal muscles under the conditions of noncontraction (a) and contraction (b) (red color indicates higher calcium) in transgenic C. elegans expressing cameleon. Reproduced from [130] with permission. c–f Ca2+ signals evoked by different odors in Drosophila brain expressing G-CaMP. Reproduced from [135] with permission. Imaging of a three-day-old transgenic fish (g) carrying the cameleon and its RB neurons (h) by confocal optical section, and the change in the fluorescence ratio (i) (representing the calcium concentration) in an RB neuron under electrical stimulation of the skin. Reproduced from [136] with permission. j Simultaneous multicolor imaging in a mouse injected with QD-encoded microbeads; k QD imaging of a prostate tumor in the mouse. Reproduced from [33] with permission
Employing QDs
Due to several advantages of QDs over FP, QDs have been used to image large animals in vivo, which holds great promise for clinical applications, especially in the imaging of tumors. Akerman et al. [141] report the application of QDs coated with specific targeting peptides to the imaging of different tissues in tumor in vitro, but this QD probe cannot be detected in living animals. Gao et al. [33] report on a new class of multifunctional QDs that were encapsulated with an ABC triblock copolymer and then linked with a tumor-targeting antibody. Using subcutaneous injection or systemic injection of these QDs into the mouse, simultaneous multicolor fluorescence imaging of the prostate tumor with efficient background removal and precise delineation of weak spectral signatures based on wavelength resolution was achieved (Fig. 7j,k). More recent research showed that the utilization of QDs in combination with GFP labeling can differentiate tumor vessels from both perivascular cells and the matrix, and QDs linked to bone marrow-derived precursor cells can visualize tumor vasculature [142]. These OMI techniques, as well as their applications at different biological levels, are summarized in Table 1.
Table 1.
OMI in systems biology
| Techniques | Physical processes | Resolution | Sensitivity | Penetration | Observation methods | Probes | Indicator localization | References |
|---|---|---|---|---|---|---|---|---|
| WFM LSCM MPLSM | RNA transcription and expression | High | High | High | DFI, FRET | FPs | In vitro, cells, tissues, organism | [63, 64, 71–76] |
| Protein localization and dynamics | DFI | FPs | In vitro, cells, tissues, organism | [51, 52, 65, 77–79] | ||||
| Subcellular organelle dynamics | DFI, FRAP | FPs | Cells | [80–82] | ||||
| Protein interaction | FRET, FLIM, FCS | FPs, cpFPs | Cells, mouse | [46, 47, 49, 57, 59, 60, 83–88, 140] | ||||
| Protease and kinase activity | FRET | FPs | [89–96] | |||||
| Calcium | DFI, FRET | FPs, cpFPs | Cells, C. elegans, fish, mouse, Drosophila | [14, 27, 28, 97–101, 130–138] | ||||
| cAMP, cGMP | FRET | FPs | Cells | [102–106] | ||||
| Electrical activity | DFI, FRET | FPs | Cells | [107–109] | ||||
| pH | DFI, FRET | FPs | Cells, mouse | [110–112, 139] | ||||
| Tumor | DFI | QDs | Mouse | [33, 141, 142] | ||||
| SHIM | Biomacromolecules | Low | High | Low | NI | – | Mouse, C. elegans, brain tissue | [116–118] |
| Membrane potential | NI | Dyes | Cells, brain tissue | [119, 120] | ||||
| OCT | Morphology of tissue | High | Low | High | EI | – | Human | [122, 123] |
| Activity and disease of organ | EI | – | Human | [124, 125] | ||||
| RS | Composition of structure | Low | High | Low | NI | – | Brain tissues, bronchial tissue | [126, 127] |
| FT-IR | Kinetics of reaction intermediates | Low | High | Low | NI | – | Yeast extract, fish liver | [67, 68] |
Abbreviations: DFI, Direct fluorescent imaging; NI, nonlinear imaging; EI, light-echo imaging
Future development
As described above, OMI is a feasible analytical technique for monitoring the dynamics of biological events in vivo and in real time anywhere from single-molecule to whole-body level, which makes it well-suited to systems biology measurements. Its noninvasive, ultrasensitive, high-resolution and real time nature has resulted in the widespread use of OMI. However, its disadvantages are also very apparent, such as its high cost, its low throughput, and its limited detectable depth, which is a particular hindrance in whole-body analysis. It should be pointed out that OMI is still far from fulfilling all of the requirements of systems biology (as concluded in our previous paper [8])—in terms of sensitivity, selectivity or specificity, linear range of quantitation, throughput, robustness, flexibility and cost—that are definitely required in genomics, transcriptomics, proteomics, and metabolomic profiling and dynamics, although it it clear that OMI is a promising method for these types of bioanalysis.
In the near future, several routes to improving OMI for systems biology need to be followed. Firstly, it is crucial to develop better molecular probes for OMI; in other words probes that have improved biocompatibilities and high quantum yields, are easy-to-tag, display multiple colors and have better penetration capabilities. The importance of the molecular probe to OMI cannot be overstressed. Current endeavors in QD development are heading in a good direction but this field is still in its infancy [143]. However, this doesn’t mean that the development of small organic dyestuffs and biogenetic FP is less important: on the contrary, applications of these two kinds of molecular probes will still constitute most OMI studies [144]. Secondly, the integration of several modalities or approaches should be emphasized, since it makes OMI much more informative. Thirdly, efforts to improve OMI instrumentation are also important, since they will result in better time and space resolution and better sensitivity. For example, the 4Pi technique has improved the spatial resolution of OMI down to the mid-nanometer level, which is over the diffraction limit. The rapid progress currently being achieved in nanotechnology and nano/microelectromechanical systems looks set to lead to the manufacture of low-cost and high-efficiency OMI instruments [145]. Finally, the application of OMI to the life sciences is important; it is the reason behind the development of OMI. In particular, the application of OMI to systematic measurements of biological systems will be of great interest, which is also the aim of systems biology.
Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (Nos. 20405006, 30570468), the National High Technology Research and Development Program of China (863 Program: 2003AA231011), the Program for New Century Excellent Talents in University (NCET), and the Program for Distinguished Young Scientists of the Hubei Province (2004ABB004).
Abbreviations
- AFP
Aequorea fluorescent protein
- CFP
Cyan fluorescent protein
- cpGFP
Circularly permuted green fluorescent protein
- cpYFP
Circularly permuted yellow fluorescent protein
- EGFP
Enhanced green fluorescent protein
- EYFP
Enhanced yellow fluorescent protein
- FCS
Fluorescence correlation spectroscopy
- FISH
Fluorescence in situ hybridization
- FLIM
Fluorescence lifetime imaging microscopy
- FP
Fluorescent protein
- FRAP
Fluorescence recovery after photobleaching
- FRET
Fluorescence resonance energy transfer
- FSM
Fluorescent speckle microscopy
- GFP
Green fluorescent protein
- LSCM
Laser scanning confocal microscopy
- MPLSM
Multiple-photon laser scanning microscopy
- OMI
Optical molecular imaging
- QDs
Quantum dots
- RS
Raman spectroscopy
- ROI
Regions of interest
- SBT
Spectral bleed-through
- WFM
Wide-field microscopy
- YFP
Yellow fluorescent protein
References
- 1.Kitano H (ed) (2001) Foundations of systems biology. MIT Press, Cambridge, MA
- 2.Liu ET (2005) Cell 121:505–506 [DOI] [PubMed]
- 3.Palsson B (2000) Nat Biotechnol 18:1147–1150 [DOI] [PubMed]
- 4.Hood L, Perlmutter RM (2004) Nat Biotechnol 22:1215–1217 [DOI] [PubMed]
- 5.Kitano H (2002) Science 295:1662–1664 [DOI] [PubMed]
- 6.Weiner N (1948) Cybernetics or control and communication in the animal and the machine. MIT Press, Cambridge, MA
- 7.Corttingham K (2005) Anal Chem 77:197A [DOI] [PubMed]
- 8.Liu BF, Xu B, Zhang G, Du W, Luo Q (2006) J Chromatogr A 1106:19–29 [DOI] [PubMed]
- 9.Stephens DJ, Allan VJ (2003) Science 300:82–86 [DOI] [PubMed]
- 10.Farkas DL (2003) Nat Biotechnol 21:1269–1271 [DOI] [PubMed]
- 11.Farkas D (2003) Nat Biotechnol 21:1269–1271 [DOI] [PubMed]
- 12.Tsien RY (1998) Annu Rev Biochem 67:509–544 [DOI] [PubMed]
- 13.Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ (1996) Science 273:1392–1395 [DOI] [PubMed]
- 14.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY (2001) J Biol Chem 276:29188–29194 [DOI] [PubMed]
- 15.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) Nat Biotechnol 20:87–90 [DOI] [PubMed]
- 16.Rizzo MA, Springer GH, Granada B, Piston DW (2004) Nat Biotechnol 22:445–449 [DOI] [PubMed]
- 17.Sawano A, Miyawaki A (2000) Nucleic Acids Res 28:e78 [DOI] [PMC free article] [PubMed]
- 18.Patterson GH, Lippincott-Schwartz J (2002) Science 297:1873–1877 [DOI] [PubMed]
- 19.Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA (1999) Nat Biotechnol 17:969–973 [DOI] [PubMed]
- 20.Terskikh A, Fradkov A, Ermakova G, Zaraisky A, Tan P, Kajava AV, Zhao X, Lukyanov S, Matz M, Kim S, Weissman I, Siebert P (2000) Science 290:1585–1588 [DOI] [PubMed]
- 21.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY (2002) Proc Natl Acad Sci USA 99:7877–7882 [DOI] [PMC free article] [PubMed]
- 22.Terskikh AV, Fradkov AF, Zaraisky AG, Kajava AV, Angres B (2002) J Biol Chem 277:7633–7636 [DOI] [PubMed]
- 23.Bevis BJ, Glick BS (2002) Nat Biotechnol 20:83–87 [DOI] [PubMed]
- 24.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY (2004) Nat Biotechnol 22:1567–1572 [DOI] [PubMed]
- 25.Karasawa S, Araki T, Nagai T, Mizuno H, Miyawaki A (2004) Biochem J 381:307–312 [DOI] [PMC free article] [PubMed]
- 26.Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A (2002) Proc Natl Acad Sci USA 99:12651–12656 [DOI] [PMC free article] [PubMed]
- 27.Baird GS, Zacharias DA, Tsien RY (1999) Proc Natl Acad Sci USA 96:11241–11246 [DOI] [PMC free article] [PubMed]
- 28.Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A (2004) Proc Natl Acad Sci USA 101:10554–10559 [DOI] [PMC free article] [PubMed]
- 29.Murray CB, Kagan CR, Bawendi MG (2000) Annu Rev Mater Sci 30:545–610
- 30.Green M, O’Brien P (1999) J Chem Soc Chem Commun 22:2235–2241
- 31.Leatherdale CA, Woo WK, Mikulec FV, Bawendi MG (2002) J Phys Chem B 106:7619–7622
- 32.Gao XH, Nie SM (2003) Trends Biotechnol 21:371–373 [DOI] [PubMed]
- 33.Jakobs S, Subramaniam V, Schonle A, Jovin TM, Hell SW (2000) FEBS Lett 479:131–135 [DOI] [PubMed]
- 34.Goldman ER, Anderson GP, Tran PT, Mattoussi H, Charles PT, Mauro JM (2002) Anal Chem 74:841–847 [DOI] [PubMed]
- 35.Wang DS, He JB, Rosenzweig N (2004) Nano Lett 4:409–413
- 36.So MK, Xu C, Loening AM, Gambhir SS, Rao J (2006) Nat Biotechnol 24:339–343 [DOI] [PubMed]
- 37.Gerlich D, Ellenberg J (2003) Nat Cell Biol Suppl S14–S19 [PubMed]
- 38.Swedlow JR, Platani M (2002) Cell Struct Funct 27:335–341 [DOI] [PubMed]
- 39.Denk W, Strickler JH, Webb WW (1990) Science 248:73–76 [DOI] [PubMed]
- 40.Lansford R, Bearman G, Fraser SE (2001) J Biomed Opt 6:311–318 [DOI] [PubMed]
- 41.Campagnola PJ, Loew LM (2003) Nat Biotechnol 21:1356–1360 [DOI] [PubMed]
- 42.Fujimoto JG (2003) Nat Biotechnol 21:1361–1367 [DOI] [PubMed]
- 43.Drexler W, Morgner U, Kartner FX, Pitris C, Boppart SA, Li XD, Ippen EP, Fujimoto JG (1999) Opt Lett 24:1221–1223 [DOI] [PubMed]
- 44.Schmitt JM, Knüttel A, Yadlowsky M, Eckhaus MA (1994) Phys Med Biol 39:1705–1720 [DOI] [PubMed]
- 45.Sekar RB, Periasamy A (2003) J Cell Biol 160:629–633 [DOI] [PMC free article] [PubMed]
- 46.Zhang J, Campbell RE, Ting AY, Tsien RY (2002) Nat Rev Mol Cell Biol 3:906–918 [DOI] [PubMed]
- 47.Miyawakib A (2003) Dev Cell 4:295–305 [DOI] [PubMed]
- 48.Berney C, Danuser G (2003) Biophys J 84:3992–4010 [DOI] [PMC free article] [PubMed]
- 49.Herman B, Krishnan RV, Centonze VE (2004) Methods Mol Biol 261:351–370 [DOI] [PubMed]
- 50.Phair RD, Misteli T (2001) Nat Rev Mol Cell Biol 2:898–907 [DOI] [PubMed]
- 51.Schaaf MJ, Cidlowski JA (2003) Mol Cell Biol 23:1922–1934 [DOI] [PMC free article] [PubMed]
- 52.Shtrahman M, Yeung C, Nauen DW, Bi GQ, Wu XL (2005) Biophys J 89:3615–3627 [DOI] [PMC free article] [PubMed]
- 53.Bastiaens PI, Squire A (1999) Trends Cell Biol 9:48–52 [DOI] [PubMed]
- 54.Dong CY, French T, So PT, Buehler C, Berland KM, Gratton E (2003) Methods Cell Biol 72:431–464 [DOI] [PubMed]
- 55.Wouters FS, Verveer PJ, Bastiaens PI (2001) Trends Cell Biol 11:203–211 [DOI] [PubMed]
- 56.Subramaniam V, Hanley QS, Clayton AH, Jovin TM (2003) Methods Enzymol 360:178–201 [DOI] [PubMed]
- 57.Tertoolen LG, Blanchetot C, Jiang G, Overvoorde J, Gadella Jr TW, Hunter T, den Hertog J (2001) BMC Cell Biol 2:8 [DOI] [PMC free article] [PubMed]
- 58.Berland KM (2004) Meth Mol Biol 261:383–398 [DOI] [PubMed]
- 59.Patel RC, Kumar U, Lamb DC, Eid JS, Rocheville M, Grant M, Rani A, Hazlett T, Patel SC, Gratton E, Patel YC (2002) Proc Natl Acad Sci USA 99:3294–3299 [DOI] [PMC free article] [PubMed]
- 60.Maier C, Runzler D, Schindelar J, Grabner G, Waldhausl W, Kohler G, Luger A (2005) J Cell Sci 118:3353–3361 [DOI] [PubMed]
- 61.Bauman JG, Wiegant J, Borst P, van Duijn P (1980) Exp Cell Res 128:485–490 [DOI] [PubMed]
- 62.Manuelidis L, Langer-Safer PR, Ward DC (1982) J Cell Biol 95:619–625 [DOI] [PMC free article] [PubMed]
- 63.Singer RH, Ward DC (1982) Proc Natl Acad Sci USA 79:7331–7335 [DOI] [PMC free article] [PubMed]
- 64.Santangelo PJ, Nix B, Tsourkas A, Bao G (2004) Nucleic Acids Res 32:e57 [DOI] [PMC free article] [PubMed]
- 65.Brandtzaeg P (1998) J Immunol Methods 216:49–67 [DOI] [PubMed]
- 66.Tanke HJ, Dirks RW, Raap T (2005) Curr Opin Biotech 16:49–54 [DOI] [PubMed]
- 67.Thomas M, LAAszlAA Z, Petro K, Andrea M, Stefan CM (2006) BioSystems 83:188–194 16236430
- 68.Cakmak G, Togan I, Severcan F (2006) Aquat Toxicol 77:53–63 [DOI] [PubMed]
- 69.Sonnleitner A, Mannuzzu LM, Terakawa S, Isacoff EY (2002) Proc Natl Acad Sci USA 99:12759–12764 [DOI] [PMC free article] [PubMed]
- 70.Karymov M, Daniel D, Sankey OF, Lyubchenko YL (2005) Proc Natl Acad Sci USA 102:8186–8191 [DOI] [PMC free article] [PubMed]
- 71.Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, Kain SR (1998) J Biol Chem 273:34970–34975 [DOI] [PubMed]
- 72.Chiang CF, Okou DT, Griffin TB, Verret CR, Williams MN (2001) Arch Biochem Biophys 394:229–235 [DOI] [PubMed]
- 73.Phelps CB, Brand AH (1998) Methods 14:367–379 [DOI] [PubMed]
- 74.McGuire SE, Roman G, Davis RL (2004) Trends Genet 20:384–391 [DOI] [PubMed]
- 75.Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM (1998) Mol Cell 2:437–445 [DOI] [PubMed]
- 76.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, Spector DL (2004) Cell 116:683–698 [DOI] [PMC free article] [PubMed]
- 77.Stavreva DA, Muller WG, Hager GL, Smith CL, McNally JG (2004) Mol Cell Biol 24:2682–2697 [DOI] [PMC free article] [PubMed]
- 78.Usuku T, Nishi M, Morimoto M, Brewer JA, Muglia LJ, Sugimoto T, Kawata M (2005) Neuroscience 135:1119–1128 [DOI] [PubMed]
- 79.Gonzalez C, Bejarano LA (2000) Trends Cell Biol 10:162–165 [DOI] [PubMed]
- 80.Shaw SL, Kamyar R, Ehrhardt DW (2003) Science 300:1715–1718 [DOI] [PubMed]
- 81.Dhonukshe P, Gadella TW Jr (2003) Plant Cell 15:597–611 [DOI] [PMC free article] [PubMed]
- 82.Kircher S, Gil P, Kozma-Bognar L, Fejes E, Speth V, Husselstein-Muller T, Bauer D, Adam E, Schafer E, Nagy F (2002) Plant Cell 14:1541–1555 [DOI] [PMC free article] [PubMed]
- 83.Mas P, Devlin PF, Panda S, Kay SA (2000) Nature 408:207–211 [DOI] [PubMed]
- 84.Bunemann M, Frank M, Lohse MJ (2003) Proc Natl Acad Sci USA 100:16077–16082 [DOI] [PMC free article] [PubMed]
- 85.Bhatia S, Edidin M, Almo SC, Nathenson SG (2005) Proc Natl Acad Sci USA 102:15569–15574 [DOI] [PMC free article] [PubMed]
- 86.Michnick SW, Remy I, Campbell-Valois FX, Vallee-Belisle A, Pelletier JN (2000) Methods Enzymol 328:208–230 [DOI] [PubMed]
- 87.Hu CD, Chinenov Y, Kerppola TK (2002) Mol Cell 9:789–798 [DOI] [PubMed]
- 88.Hu CD, Kerppola TK (2003) Nat Biotechnol 21:539–545 [DOI] [PMC free article] [PubMed]
- 89.Heim R, Tsien RY (1996) Curr Biol 6:178–182 [DOI] [PubMed]
- 90.Suzuki M, Ito Y, Sakata I, Sakai T, Husimi Y, Douglas KT (2005) Biochem Biophys Res Commun 330:454–460 [DOI] [PubMed]
- 91.Mahajan NP, Harrison-Shostak DC, Michaux J, Herman B (1999) Chem Biol 6:401–409 [DOI] [PubMed]
- 92.Rehm M, Dussmann H, Janicke RU, Tavare JM, Kogel D, Prehn JH (2002) J Biol Chem 277:24506–24514 [DOI] [PubMed]
- 93.Onuki R, Nagasaki A, Kawasaki H, Baba T, Uyeda TQ, Taira K (2002) Proc Natl Acad Sci USA 99:14716–14721 [DOI] [PMC free article] [PubMed]
- 94.Zhang J, Ma Y, Taylor SS, Tsien RY (2001) Proc Natl Acad Sci USA 98:14997–15002 [DOI] [PMC free article] [PubMed]
- 95.Ananthanarayanan B, Ni Q, Zhang J (2005) Proc Natl Acad Sci USA 102:15081–15086 [DOI] [PMC free article] [PubMed]
- 96.Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S (2005) Nature 434:1040–1045 [DOI] [PubMed]
- 97.Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY (1997) Nature 388:882–887 [DOI] [PubMed]
- 98.Evanko DS, Haydon PG (2005) Cell Calcium 37:341–348 [DOI] [PubMed]
- 99.Truong K, Sawano A, Mizuno H, Hama H, Tong KI, Mal TK, Miyawaki A, Ikura M (2001) Nat Struct Biol 8:1069–1073 [DOI] [PubMed]
- 100.Palmer AE, Jin C, Red JC, Tsien RY (2004) Proc Natl Acad Sci USA 101:17404–17409 [DOI] [PMC free article] [PubMed]
- 101.Nakai J, Ohkura M, Imoto K (2001) Nat Biotechnol 19:137–141 [DOI] [PubMed]
- 102.Adams SR, Harootunian AT, Buechler YJ, Taylor SS, Tsien RY (1991) Nature 349:694–697 [DOI] [PubMed]
- 103.Zaccolo M, Pozzan T (2002) Science 295:1711–1715 [DOI] [PubMed]
- 104.Dipilato LM, Cheng XD, Zhang J (2004) Proc Natl Acad Sci USA 101:16513–16518 [DOI] [PMC free article] [PubMed]
- 105.Sato M, Hida N, Ozawa T, Umezawa Y (2000) Anal Chem 72:5918–5924 [DOI] [PubMed]
- 106.Honda A, Adams SR, Sawyer CL, Lev-Ram V, Tsien RY, Dostmann WR (2001) Proc Natl Acad Sci USA 98:2437–2442 [DOI] [PMC free article] [PubMed]
- 107.Siegel MS, Isacoff EY (1997) Neuron 19:735–741 [DOI] [PubMed]
- 108.Ataka K, Pieribone VA (2002) Biophys J 82:509–516 [DOI] [PMC free article] [PubMed]
- 109.Sakai R, Repunte-Canonigo V, Raj CD, Knopfel T (2001) Eur J Neurosci 13:2314–2318 [DOI] [PubMed]
- 110.Miyawaki A, Mizuno H, Nagai T, Sawano A (2003) Methods Enzymol 360:202–225 [DOI] [PubMed]
- 111.Hanson GT, McAnaney TB, Park ES, Rendell ME, Yarbrough DK, Chu S, Xi L, Boxer SG, Montrose MH, Remington SJ (2002) Biochemistry 41:15477–15488 [DOI] [PubMed]
- 112.McAnaney TB, Park ES, Hanson GT, Remington SJ, Boxer SG (2002) Biochemistry 41:15489–15494 [DOI] [PubMed]
- 113.Bedner P, Niessen H, Odermatt B, Kretz M, Willecke K, Harz H (2006) J Biol Chem 281:6673–6681 [DOI] [PubMed]
- 114.Dakin K, Zhao Y, Li WH (2005) Nat Methods 2:55–62 [DOI] [PubMed]
- 115.Nodin C, Nilsson M, Blomstrand F (2005) J Neurochem 94:1111–1123 [DOI] [PubMed]
- 116.Lyubovitsky JG, Spencer JA, Krasieva TB, Andersen B, Tromberg BJ (2006) J Biomed Opt 11:14013 [DOI] [PubMed]
- 117.Filippidis G, Kouloumentas C, Voglis G, Zacharopoulou F, Papazoglou TG, Tavernarakis N (2005) J Biomed Opt 10:024015 [DOI] [PubMed]
- 118.Dombeck DA, Sacconi L, Blanchard-Desce M, Webb WW (2005) J Neurophysiol 94:3628–3636 [DOI] [PubMed]
- 119.Nuriya M, Jiang J, Nemet B, Eisenthal KB, Yuste R (2006) Proc Natl Acad Sci USA 103:786–790 [DOI] [PMC free article] [PubMed]
- 120.Dombeck DA, Blanchard-Desce M, Webb WW (2004) J Neurosci 24:999–1003 [DOI] [PMC free article] [PubMed]
- 121.van Velthoven ME, Verbraak FD, Yannuzzi LA, Rosen RB, Podoleanu AG, de Smet MD (2006) Retina 26:129–136 [DOI] [PubMed]
- 122.Feldchtein FI, Gelikonov GV, Gelikonov VM, Iksanov RR, Kuranov RV, Sergeev AM, Gladkova ND, Ourutina MN, Warren Jr JA, Reitze DH (1998) Opt Express 3:239–250 [DOI] [PubMed]
- 123.Zagaynova EV, Streltsova OS, Gladkova ND, Snopova LB, Gelikonov GV, Feldchtein FI, Morozov AN (2002) J Urol 167:1492–1496 [PubMed]
- 124.Maheswari RU, Takaoka H, Kadono H, Homma R, Tanifuji M (2003) J Neurosci Meth 124:83–92 [DOI] [PubMed]
- 125.Whiteman SC, Yang Y, van Pittius DG, Stephens M, Parmer J, Spiteri MA (2006) Clin Cancer Res 12:813–818 [DOI] [PubMed]
- 126.Santos LF, Wolthuis R, Koljenovic S, Almeida RM, Puppels GJ (2005) Anal Chem 77:6747–6752 [DOI] [PubMed]
- 127.Koljenovic S, Bakker Schut TC, van Meerbeeck JP, Maat AP, Burgers SA, Zondervan PE, Kros JM, Puppels GJ (2004) J Biomed Opt 9:1187–1197 [DOI] [PubMed]
- 128.Caspers PJ, Lucassen GW, Puppels GJ (2003) Biophys J 85:572–580 [DOI] [PMC free article] [PubMed]
- 129.Miyawaki A (2003) Curr Opin Neurobiol 13:591–596 [DOI] [PubMed]
- 130.Kerr R, Lev-Ram V, Baird G, Vincent P, Tsien RY, Schafer WR (2000) Neuron 26:583–594 [DOI] [PubMed]
- 131.Shyn SI, Kerr R, Schafer WR (2003) Curr Biol 13:1910–1915 [DOI] [PubMed]
- 132.Fukuto HS, Ferkey DM, Apicella AJ, Lans H, Sharmeen T, Chen W, Lefkowitz RJ, Jansen G, Schafer WR, Hart AC (2004) Neuron 42:581–593 [DOI] [PubMed]
- 133.Hilliard MA, Apicella AJ, Kerr R, Suzuki H, Bazzicalupo P, Schafer WR (2005) EMBO J 24:63–72 [DOI] [PMC free article] [PubMed]
- 134.Fiala A, Spall T, Diegelmann S, Eisermann B, Sachse S, Devaud JM, Buchner E, Galizia CG (2002) Curr Biol 12:1877–1884 [DOI] [PubMed]
- 135.Wang JW, Wong AM, Flores J, Vosshall LB, Axel R (2003) Cell 112:271–282 [DOI] [PubMed]
- 136.Higashijima S, Masino MA, Mandel G, Fetcho JR (2003) J Neurophysiol 90:3986–3997 [DOI] [PubMed]
- 137.Nyqvist D, Mattsson G, Kohler M, Lev-Ram V, Andersson A, Carlsson PO, Nordin A, Berggren PO, Jansson L (2005) J Endocrinol 186:333–341 [DOI] [PubMed]
- 138.Hara M, Bindokas V, Lopez JP, Kaihara K, Landa LR Jr, Harbeck M, Roe MW (2004) Am J Physiol Cell Physiol 287:C932–C938 [DOI] [PubMed]
- 139.Metzger F, Repunte-Canonigo V, Matsushita S, Akemann W, Diez-Garcia J, Ho CS, Iwasato T, Grandes P, Itohara S, Joho RH, Knopfel T (2002) Eur J Neurosci 15:40–50 [DOI] [PubMed]
- 140.Stockholm D, Bartoli M, Sillon G, Bourg N, Davoust J, Richard I (2005) J Mol Biol 346:215–222 [DOI] [PubMed]
- 141.Akerman ME, Chan WCW, Laakkonen P, Bhatia SN, Ruoslahti E (2002) Proc Natl Acad Sci USA 99:12617–12621 [DOI] [PMC free article] [PubMed]
- 142.Stroh M, Zimmer JP, Duda DG, Levchenko TS, Cohen KS, Brown EB, Scadden DT, Torchilin VP, Bawendi MG, Fukumura D, Jain RK (2005) Nat Med 11:678–682 [DOI] [PMC free article] [PubMed]
- 143.Seydel C (2003) Science 300:80–81 [DOI] [PubMed]
- 144.Lippincott-Schwartz J, Patterson GH (2003) Science 300:87–91 [DOI] [PubMed]
- 145.Hell SW (2003) Nat Biotechnol 21:1347–1355 [DOI] [PubMed]