

Abstract
Chromatin immunoprecipitation (ChIP) technique allows detection of proteins that bind to chromatin. While this technique has been applied extensively in cell-based studies, its tissue-based application remains poorly explored. We are specifically interested in examining estrogen-dependent transcriptional mechanism in respect of recruitment of estrogen receptor-alpha (ERα), a ligand-activated transcription factor, to uterine gene promoters in mice. Recent gene-array studies, utilizing ERα knock-out vs. wild-type mice, have revealed that estrogen regulates numerous uterine genes temporally and most importantly via ERα during the phase-II response, including three well characterized genes viz., lactoferrin (Ltf), progesterone receptor (Pgr) and cyclinD1 (Ccnd1). Here, utilizing systematic ChIP studies, we demonstrate endogenous recruitment of ERα to above uterine gene promoters following estradiol-17β (E2) injection in mice.
Keywords: Chromatin, Immunoprecipitation, Estrogen Receptor Alpha
Introduction
Interaction between transactivator proteins and DNA regulates many basic cellular functions, including gene transcription. Many techniques e.g. subtractive hybridization, differential display, microarray analysis of gene expression have been employed to monitor gene transcriptional changes within the context of cell or tissue biology. Understanding gene transcriptional regulation generally offers an opportunity to define molecular pathways that are associated with certain biologic responses. In this regard, chromatin immunoprecipitation (ChIP) technique has been used as a valuable tool to identify endogenous interaction of transactivators and the cis-regulatory regions of DNA, which indirectly analyzes endogenous gene regulation for expression. Furthermore, this approach has a great use to confirm protein(s) that are known to interact with gene previously characterized by other means, e.g. mutational analysis of the promoter. In respect of ChIP studies, it has been known that formaldehyde-mediated cross-linking between cellular proteins and DNAs, and successful reversal of interaction following immunoprecipitation are essential for successful application of this technique in any systems. The application of this technique has been instrumental in the analysis of endogenous binding of transactivators within genomes (1), although it is still at its infancy stage and should be carried-out more extensively during the post human genome-sequencing era to define molecular basis of diseases. However, ChIP studies in general are primarily concentrated on cell-based studies, although its tissue-based application provides better understanding in respect of physiological and pathophysiological responses to a complex system.
Estrogen regulates cellular growth and differentiation functions in diverse target tissues, both within and outside of the reproductive system (2). Estrogen primarily controls gene transcription via interaction with two nuclear receptors ERα (Esr1) and ERα (Esr2) that belong to a family of ligand-activated transcription factors (3). In uterine biology, estrogen elicits a biphasic response that is well recognized by the phase-I (water imbibition and macromolecular uptake) and phase-II (DNA synthesis and cell proliferation) actions, generally occur by 6 and 24 h, respectively (4). It has previously been shown that ERα is a major regulator of phase-II responses, in conjunction with the onset of genomic responses (2, 5), although phase-I responses occur with or without involving ERα (6-8). Furthermore, ERα has been shown to be extremely low in mouse uterus (9) and not involved in uterine biology (10).
According to the classical model of estrogen action, ligand first binds to the receptor and allows its dimerization and subsequent recruitment of the ligand-receptor complex to a palindromic sequence, known as estrogen responsive element (ERE) within a target gene promoter, for gene transcription (11). Although a perfect palindromic sequence was identified as AGGTCA(nnn)TGACCT, most estrogen responsive genes have imperfect palindromes or do not have recognizable ERE (12). The promoters that lack ERE-like sequences can be responsive via interaction with other DNA binding factor mediates ER association in an indirect manner (13). In this regard, a number of studies have shown that ERs can regulate transcription without binding directly to DNA but activated through interaction with AP-1 or SP1 protein complexes (2, 14). Lactoferrin (Ltf), cyclinD1 (Ccnd1) and progesterone receptor (Pgr) are well characterized estrogen-responsive uterine genes in mice (15,16). Previous studies (17-19) have established that endogenous regulation of estrogen-dependent ERα-mediated transcription of Ltf and Pgr genes utilizes a defined half consensus ERE located proximal to the transcription start site, while that of Ccnd1 occurs via AP-1 site, through an indirect association with Fos/Jun proteins, located proximal to the transcription start site. Delineation of ERα mediated interaction with distinct region of the promoter for estrogen-responsive genes in vivo in target tissues has been limited. In this regard, as presented here, a systematic approach for ChIP studies has clearly identified estrogen-dependent recruitment of endogenous ERα to above gene-specific promoter regions in the mouse uterus, suggesting this technique may be useful to study in vivo evaluation of interaction for chromatin-associated proteins in diverse tissues. In addition, although ChIP technique has been used to study uterine gene specific recruitment of protein for other studies (20), this is the first report that has a detailed protocol which should facilitate researchers in this field to carry out similar ChIP experiments.
Materials and Methods
Animal
Adult CD1 (Charles Rivers laboratory, Raleigh, NC) mice were housed in our institutional animal care facility according to NIH and institutional guidelines for laboratory animals. Adult ovariectomized mice (8-10 weeks old) were rested for 12 days to reduce levels of endogenous ovarian steroid hormones. They were given a single subcutaneous injection (0.1ml/mouse) with sesame seed oil (as a control) or estradiol-17β (E2, 100ng/mouse) dissolved in oil and necropsied after 24 h. Previous studies have implicated that estrogen induces expression of numerous genes via ERα during this time in the mouse uterus (5).
Tissue collection and formaldehyde fixation
Uterine tissues were removed after opening the peritoneal cavity and placed on saline solution soaked tissue paper for further cleaning from the adipose tissues. Previous studies have shown that formaldehyde effectively crosslinks protein to DNA, RNA and protein (20). For tissue fixation, we slit uteri longitudinally through the lumen and cut into small pieces (3-4 mm in length) using the scalpel blade. Tissues were then suspended in 1% formaldehyde (EMD Chemicals Inc., Gibbstown, NJ; Cat# FX0418-1) solution (200 μl/mouse) for 10 min at room temperature. In general, longer incubation causes permanent cross-linking which may be difficult to reverse for successful PCR analysis (20). Following an optimum cross-linking, the reaction was terminated by the addition of glycine to a final concentration 0.125 M (using 28.6 μl of 1 M glycine in 200 μl total volume). Tissues were then collected by centrifugation at 5000g for 5 min at 4°C and washed twice with ice-cold PBS.
Cell rupture and isolation of protein bound DNA
Washed tissue pellet from the above mentioned step was suspended in 200 μl ice cold lysis buffer (Tissue-PE LB; Genotech, St. Louis, MO) containing 1X protease arrest (Cat# 786-108; Genotech) in 1.7 ml eppendorf tube. Tissues were then homogenized in presence of 0.5 gm acid washed glass beads (Sigma, Cat# G1277), using strong vortex (Fisher Vortex Genie2) at high speed (with a setting 8) for 40 min at 4°C. Tissue lysate was then transferred to a new tube after making a small hole at the bottom with 18-gauge needle. Following the transfer, a short spin was done to remove cell debris or beads. At this stage, the supernatant contains desirable chromatin complex that can be kept frozen in liquid nitrogen.
Optimum DNA fragmentation
While gene specific primers are used to analyze chromatin DNA, its size specific fragmentation (usually within ≈ 500 bp) is necessary prior to chromatin immunoprecipitation in order to avoid amplification of unwanted DNA (20), and this can be achieved by optimum sonication. Optimum size fragmentation can enrich distinct region of DNA following immunoprecipitation; large fragments will lead misleading results by pooling distal region. In the current study, the optimum sonication was indeed achieved based on our analysis using 5 pulses, each for 10 sec at maximum speed (setting the power at maximum, with tune set at 3) using Micro-ultrasonic cell disrupter (Kontes, Vineland, NJ). During sonication, samples were kept in ice for at least 1 min between pulses to eliminate unnecessary warming. Finally, soluble fragmented chromatin samples were collected after pulse spin and kept frozen in liquid nitrogen for future analysis.
Immunoprecipitation
The fragmented chromatin samples were first diluted to a total volume (0.5 ml) using 20 mM HEPES and then subjected to immunoprecipitation using ERα specific rabbit polyclonal antibody (Cat# sc542; Santa Cruz Biotechnology, Inc., CA) bound with Protein A Sepharose [(1μg antibody conjugated with 3 mg Protein A Sepharose beads (Amersham Biosciences Inc., Cat# 170780-01)] or normal rabbit serum (used as IgG control) bound with Protein A Sepharose (in a same ratio as that of primary antibody), for 1 h at RT followed by 2 h at 4°C. Protein A Sepharose beads were collected after centrifugation and washed once in lysis buffer, followed by washing twice in HEPES buffer (20mM HEPES, 150 mM NaCl, 0.1 Triton-X 100, 10% glycerol). Finally, Sepharose bound proteins were eluted (2X) with 100 μl elution buffer (1% SDS and 0.1 M NaHCO3) for each time by constant shaking using a vortex at maximum setting for 15 min. The pooled eluted samples were then centrifuged at 14000g for 5 min at 4°C to remove any trace of protein A Sepharose beads and transferred to clean tubes. The collected solution (~200 μl) was mixed with RNase A (10 μg) and NaCl (adjusted to a final concentration 0.3 M), and subjected to reverse cross-linking between DNA and protein by incubation at 65°C for 5 h, followed by precipitation using 100% ethanol (2.5 volume) at -20°C for overnight. Samples were then centrifuged at 20,000g for 15 min at 4°C to collect the pellet containing both DNA and protein mixtures. The pellet was then dissolved in 100 μl of TE (pH 8.0) by incubation at 45°C for 2 h in presence of proteinase K (37.5 μg) (Fisher Scientific, Cat# BP1700-500). Finally, DNA was extracted once with phenol:chloroform:isoamyl alcohol (24:24:1 v/v), once with chloroform and precipitated with 100% ethanol. Finally, the DNA pellet was washed once with 70% ethanol, dried and dissolved in 20 μl TE buffer (pH 7.5) for further analysis by PCR.
PCR analysis
The recovered DNAs from the above were analyzed by comparative PCR (21). The PCR reaction was performed in a mixture (25 μl), containing 1.5 μl DNA; reaction buffer (10 mM Tris-HCl, pH 8.3; 2.5 mM MgCl2 and 50 mM KCl); 200 μM each of dATP, dTTP, dGTP and dCTP; 0.5 μM each of the respective primer pair (shown in Table 1) and 0.75 units Ampli Taq™ DNA polymerase (Perkin-Elmer Cetus, Norwalk, CT). The reaction was carried out in a Peltier Thermal Cycler (PTC-200; MJ Research) using a cycling parameter as follows: first cycle at 94°C for 2 min, followed by an appropriate cycle number for amplification (as shown in Table 1) using the denaturation step at 94°C for 30 sec, annealing step at a specific annealing temperature (see Table 1 for a set of primer pair) for 30 sec and extension reaction step at 72°C for 20 sec. The amplified DNAs were analyzed by 2% agarose gel. In standard ChIP procedures, while highly abundant repeated regions of the chromatin appear to precipitate non specifically (22), however, it is generally not a problem because of gene specific primers are used to amplify regions around the putative transcription factor binding sites on the promoter of a specific gene.
Results and Discussion
Systematic analysis of formaldehyde-mediated fixation and its reversibility during chromatin immunoprecipitation of uterine extracts using ERα antibody
As depicted in Fig. 1, formaldehyde-mediated fixation of ERα to gene-specific promoters, size specific chromatin fragmentation, and reversal of the protein-chromatin complex following immunoprecipitation play key regulatory steps for successful application of chromatin immunoprcipitation assays.
Fig. 1. Schematic representation of chromatin immunoprecipitation protocols for uterine analysis of ERα recruitment to gene specific promoters.
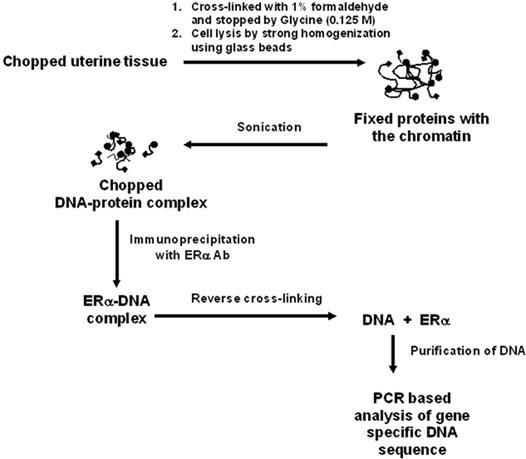
To examine estrogen-dependent association between ERα and uterine gene promoters, uterine tissues were collected after injections of E2 or vehicle (as control) for 24 h in adult ovariectomized mice. As expected, Western blot analysis clearly revealed that estrogen was capable of inducing ERα levels in uterine extracts as compared to control (Fig. 2A).
Fig. 2. Analysis of different steps during chromatin immunoprecipitation using ERα antibody of uterine extracts.
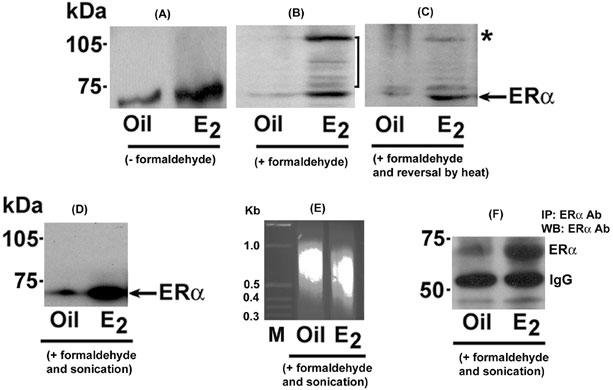
Uterine tissues were collected following injection of oil (as vehicle) or E2 (100 ng/mouse) for 24 h in ovariectomized mice. Uterine tissues extracts without cross-linking (A), following the cross-linking using 1% formaldehyde (B), following the cross-linking and reversal by heat treatment (C) or following the cross-linking and fragmentation of the chromatin by sonication (D) were analyzed by Western blotting (WB) using ERα antibody. Arrows indicates the position of un-complexed ERα band, while a vertical line in (B) represents macromolecular association of ERα. In panel C, the asterisk (*) denotes a band exhibiting reversal of ERα association. Results show that ERα-associated high molecular complex with retarded migration in SDS-PAGE gel can be reversed by prolonged heat treatment (B vs. C). Additionally, the application of sonication following fixation does not cause any detrimental effect to ERα protein for its normal detection after WB (D) and IP/WB (F), but eliminates higher molecular retarded bands as seen in (B). Furthermore, following the sonication step, DNA fragments were recovered by phenol:chloroform extraction and alcohol precipitation, and then analyzed by 2% agarose gel (E). M = 1 kb marker.
Interestingly, the treatment of 1% formaldehyde caused induction of cross-linking between ERα and cellular macromolecules, presumably with chromosomal DNA, only after E2, as judged by the presence of several ERα-immunoreactive higher molecular bands on the Western blot (Fig. 2B). For reversing the cross-linking, tissue extracts were subjected to heat treatment and analyzed by Western blotting using ERα antibody (Fig. 2C). Results show that we were indeed able to reverse the majority of cross-linking, as indicated by asterisk (*) (Fig. 2C). Furthermore, we wanted to examine whether sonication speed / duration (as described in materials and methods) achieves effective fragmentation of the chromatin without any detrimental effects on the associated protein. As shown in Fig. 2D, Western blot analysis clearly revealed the presence of intact ERα, but elimination of higher molecular bands, suggesting appropriate chromatin fragmentation was indeed achieved (Fig. 2D). Consistent to these results, further analysis of DNA fragments appeared to be in the range of ≈ 400-800bp (Fig. 2E). Moreover, ERα was indeed detected following immunoprecipitation and Western blotting (Fig. 2F), suggesting the suitability of ERα antibody is established, since the selection of an appropriate antibody in above studies is a key requirement for successful progression of ChIP experiments.
Estrogen regulates recruitment of ERα to estrogen responsive gene promoters
Previous studies have established that endogenous regulation of Ltf, Pgr and Ccnd1 genes by estrogen occurs via ERα interaction either directly or indirectly at the promoters (17-19). Based on this information, in conjunction with the available mouse genome project sequence (ENSEMBL mouse geneview), we designed the primers for the proximal region, as well as distal region located further upstream with no recognizable ERα binding sites for these genes, as shown in Table 1. β-Actin (Actb) gene specific primers were also designed from an arbitrary location within 5’-flanking region of the promoter to use as a non-specific gene target. In the present study, we demonstrated by chromatin immunoprecipitation assay that a distinct promoter region of above genes was recognized by ERα in a ligand-dependent manner in the mouse uterus (Fig. 3). More specifically, using ERα antibody, we were able to detect desired amplified products after PCR only in the proximal region, but not the distal, by E2. While, the control (oil) group did not show any such results. Furthermore, although the presence of both promoter regions (proximal and distal) for above genes before the ChIP assay was confirmed by PCR (Input), ChIP assays using pre-immune serum (IgG) did not detect any such amplified bands. Overall, these results suggest that ERα recruitment to above genes was promoter region-specific.
Fig. 3. Analysis of recruitment of ERα to estrogen-responsive Ltf, Pgr and Ccnd1 gene promoters.
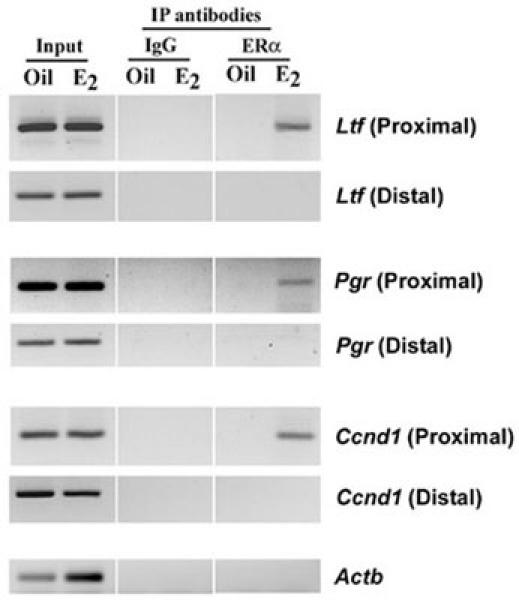
ChIP analysis was performed using ERα antibody or normal serum IgG (as control) as described in materials and methods. The presence of the promoter DNA before immunoprecipitation was confirmed by PCR (Input). PCR products were resolved in 2% agarose gels containing ethidium bromide.
Although we used whole uterus for our analysis, previous studies have already established that Ltf and Ccnd1 genes are regulated by estrogen in uterine epithelial cells, while that of Pgr detected in subluminal stroma (15, 16). To address the issue if association of ERα to a distinct promoter region could be correlated with ERα dependent gene transcription, we performed RT-PCR analysis in parallel for above genes using RNAs isolated from E2- and oil-treated uterine tissues. Our analysis of expression of mRNAs for Ltf, Pgr and Ccnd1 genes was based on the relative changes as measured by comparative RT-PCR, using a lower PCR cycle number to achieve linear amplification for the genes of interest. The PCR cycle numbers used for our analyses were 25, 28, 30 and 22 for Ltf, Pgr, Ccnd1 and ribosomal protein (rpl7) genes, respectively. Rpl7 was used as a constitutive gene.
As shown in Figure 4, our results showed that Ltf, Pgr and Ccnd1 gene specific mRNAs were indeed inducible after the injection of E2, as compared to the control. Overall, these results suggest that estrogen dependent recruitment of ERα to distinct promoter regions could be correlated with estrogen-dependent gene transcription in the mouse uterus.
Fig. 4. Analysis of mRNA expression for Ltf, Pgr and Ccnd1 genes.
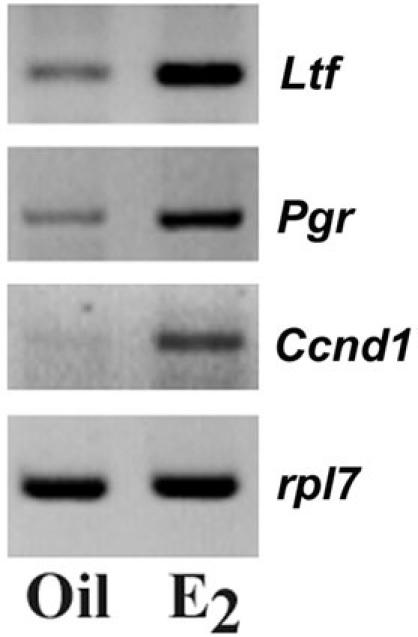
RT-PCR analysis of Ltf, Pgr and Ccnd1 genes in the uterus. Total RNA was extracted and analyzed by comparative RT-PCR (21). Ribosomal protein L7 (rpl7) was used as a constitutive gene. Amplified DNA bands were visualized by ethidium bromide staining.
Efficiency of primary antibody in chromatin immunoprecipitation studies is an important step of this procedure. In our experience, we have noted that the background noise occasionally results due to non-specific pull down fraction by antibody. Under the circumstances, specific steps were taken to minimize this unwanted situation and may include longer antibody incubation time (overnight at 4°C) followed by more stringent wash (twice in lysis buffer and three times in HEPES buffer). It should be noted here that negative results may not reveal the fact that the given protein is not associated with the chromosomal site, the lack of cross-linking may also contribute to that factor. Standard ChIP experiments may provide quantitative information about the relative levels for association of a given protein to the DNA (23), however, there are some limitations in which two proteins co-occupy a given DNA sequence that may not be revealed by this method. Moreover, as we examined only one probable ERα recruitment site for each of the genes, the recruitment through other probable sites within the genomic sequence may be necessary to consider for global gene regulation in the context of tissue biology. Nonetheless, appropriate use of this method will no doubt provide a genetic footprint in respect of the association of transcription regulatory factors to the chromatin in different functional states.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants (ES07814 and HD37830) to SKD. The authors have no conflicts of interest to declare related to this publication.
Abbreviations
- ChIP
chromatin immunoprecipitation
- ERα
estrogen receptor-alpha
- Ltf
lactoferrin
- Pgr
progesterone receptor
- Ccnd1
cyclin D1
- IP
immunoprecipitation
Appendix
Protocols
Formaldehyde fixation
Collect uterine tissues of ovariectomized mice following injections of E2 or oil by 24 h.
Treat one whole uterus, after cutting into small pieces (3-4 mm) in 200 μl 1% formaldehyde for 10min at room temperature.
Stop the reaction after addition of 28.6 μl glycine (1 M).
Centrifuge at 5000g for 5min at 4°C.
Wash twice with ice cold PBS (each time with centrifugation removing supernatant).
Cell rupture and sonication
Collect washed tissue pellet in 200 μl lysis buffer containing 1X protease arrest.
Add 0.5 g acid washed glass beads.
Vortex at high speed for 40 min, with frequent incubation on ice in every 2-3 minutes.
Collect cell lysate into a new tube.
Do a rapid short spin and collect the supernatant.
Sonicate with 5 pluses of 10 sec each at maximum speed. Keep the samples on ice between the pluses.
Spin to remove cell debris and collect supernatant.
Immunoprecipitation
Weigh Protein A Sepharose (Amersham Biosciences Inc., Cat#170780-01), 3 mg/sample. Add 1 ml of 20 mM HEPES.
Centrifuge for 5 min using high speed at 4°C. Discard supernatant without disturbing Sepharose.
Add 1 ml of 20 mM HEPES and vortex well.
Repeat steps 14 and 15.
Add 1 μg antibody (or normal rabbit serum as IgG control) per 3 mg Protein A Sepharose. Mix continuously using a rotator at RT for 1h.
Centrifuge and wash protein A Sepharose / antibody complex three times using 1ml HEPES buffer.
Mix the complex in 50 μl of 50 mM HEPES and keep at 4°C until use.
Dilute the whole supernatant from step 12 to 0.5 ml using 20 mM HEPES, prior to immunoprecipitation step.
Incubate each sample with 3 mg antibody-conjugated protein A Sepharose for 1 h at RT, followed by 2 h at 4°C with rotation.
Centrifuge at high speed for 5 min and collect the Sepharose complex.
Wash the complex three times with cold lysis buffer, followed by HEPES buffer using 1 ml each.
Collect the pellet and elute with elution buffer twice, each time 100 μl.
Centrifuge at 14000g for 5 min at 4°C.
To this 200 μl recovered volume, add 1 μl RNase A (10 mg/ml) and 12 μl 5 M NaCl.
Heat at 65°C for 5 h.
Mix with 2.5 volume of 100% ethanol for precipitation overnight.
Centrifuge at 20,000g for 15 min at 4°C to collect the pellet.
Add 100 μl TE (pH 8.0) to dissolve the pellet along with 25 μl 5X proteinase K buffer and 1.5 μl proteinase K (25 mg/ml) and incubate at 45°C for 2 h.
Extract DNA by strong vortex after mixing in equal volume with phenol: chloroform: isoamyl alcohol (24:24:1).
Centrifuge at 20,000g for 15 min at RT.
Collect ≈ 100 μl aqueous layer into a new tube, and mix again by strong vortex after addition of equal volume of chloroform:isoamyl alcohol (24:1).
Centrifuge at 20,000g for 5 min at RT.
Collect the aqueous layer.
Mix with 2.5 volume of 100% ethanol, precipitate overnight at -20°C and centrifuge as in step 32.
Collect the pellet, wash with 70% ethanol, dry and dissolve in 20 μl TE (pH 7.5).
Follow PCR for gene specific sequence amplification. PCR reaction mixture (25 μl), contains 1.5 μl DNA; 1X reaction buffer (10 mM Tris-HCl, pH 8.3; 2.5 mM MgCl2 and 50 mM KCl); 200 μM each of dATP, dTTP, dGTP and dCTP; 0.5 μM each primer and 0.75 units Ampli Taq™ DNA polymerase (Perkin-Elmer Cetus, Norwalk, CT). PCR cycling conditions were as follows: 1st cycle: 94°C for 2 min; Subsequent cycles (see Table 1 for the total number of cycles using a set of primer-pair) for amplification were as follows: 94°C for 30 sec, a specific annealing temperature for the primer set (see Table 1) for 30 sec and 72°C for 20 sec. Analyze PCR amplified products using agarose gel (2%) in the presence of ethidium bromide.
Table 1.
Primers used for PCR amplification of DNAs after chromatin immunoprecipitation.
| Gene | ID# | Sequence Region | Primer Sequence | Size (bp) |
| Ltf | ENSMUSG00000032496 | -469 to -273 (proximal) | 5’-TCTAGGCTGACTCCGCTCTC-3’(s) 5’-TAGAGGTGGGACATGGGGTA-3’ (as) |
197 |
| -4252 to -4076 (distal) | 5’-CATGTGCATGTATGTGAGATGAA-3’ (s) 5’-ATCCCCTGTCAGTGCCTTC-3’ (as) |
177 | ||
| Pgr | ENSMUSG00000031870 | -268 to -87 (proximal) | 5’-AGGACAGGAGCTGACCAAGA-3’ (s) 5’-AGTCATGACGACCCAAGCTC-3’ (as) |
182 |
| -2158 to -1937 (distal) | 5’-ACTGTCCAGAATGCCTCCAC-3’ (s) 5’-ATCACCAGGGAGGTGCTACA-3’ (as) |
222 | ||
| Ccnd1 | ENSMUSG00000070348 | -1162 to -984 (proximal) | 5’-AGGTGGAGAAACACCACCAC-3’ (s) 5’-CGGTTTGCCCAAGAAAAATA-3’ (as) |
179 |
| -4424 to -4126 (distal) | 5’-TCAGCAAGCACTCTGACCTG-3’ (s) 5’-CGGATCTCTTGTCAGGAAGC-3’ (as) |
229 | ||
| Actb | ENSMUSG00000029580 | -2831 to -2598 | 5’-GTCGTACCACTGGCATTGTG-3’ (s) 5’-CTCTCAGCTATGGTGGTGAA-3’ (as) |
233 |
References
- Hearnes JM, Mays DJ, Schavolt KL, Tang L, Jiang X, Pietenpol JA. Chromatin immunoprecipitation-based screen to identify functional genomic binding sites for sequence-specific transactivators. Mol Cell Biol. 2005;25:10148–10158. doi: 10.1128/MCB.25.22.10148-10158.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endo Rev. 1999;20(3):358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Ann Rev Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- Ray S, Hou X, Zhou H-E, Wang H, Das SK. Bip is a m olecular link between the phase-I and phase-II estrogenic responses in uterus. Mol Endocrinol. 2006;20(8):1825–1837. doi: 10.1210/me.2006-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, Korach KS. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol Endocrinol. 2003;17:2070–2083. doi: 10.1210/me.2003-0146. [DOI] [PubMed] [Google Scholar]
- Das SK, Taylor JA, Korach KS, Paria BC, Dey SK, Lubahn DB. Estrogenic responses in estrogen receptor-α deficient mice reveal a novel estrogen signaling pathway. Proc Natl Acad Sci USA. 1997;94:12786–12791. doi: 10.1073/pnas.94.24.12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Tan J, Raja S, Halder J, Paria BC, Dey SK. Estrogen targets genes involved in protein processing, calcium homeostasis and Wnt signaling in the mouse uterus independent of estrogen receptor-α and - β. J Biol Chem. 2000;275:28834–28842. doi: 10.1074/jbc.M003827200. [DOI] [PubMed] [Google Scholar]
- Hou X, Tan Y, Li M, Dey SK, Das SK. Canonical Wnt Signaling Is Critical to Estrogen Mediated Uterine Growth. Mol Endocrinol. 2004;18:3035–3049. doi: 10.1210/me.2004-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Paria BC, Dey SK, Das SK. Differential uterine expression of estrogen and progesterone receptors correlates with uterine preparation for implantation and decidualization in the mouse. Endocrinology. 1999;140:5310–5321. doi: 10.1210/endo.140.11.7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc Natl Acad Sci USA. 1998;95:15677–15682. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC. A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res. 1988;16:647–663. doi: 10.1093/nar/16.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukovich DA, Mukherjee R, Benfield PA. A novel, cell-type-specific mechanism for estrogen receptor-mediated gene activation in the absence of an estrogen-responsive element. Mol Cell Biol. 1994;14:7134–7143. doi: 10.1128/mcb.14.11.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol Endocrinol. 2004;18:1859–1875. doi: 10.1210/me.2003-0044. [DOI] [PubMed] [Google Scholar]
- Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol. 1999;13:1672–1685. doi: 10.1210/mend.13.10.0357. [DOI] [PubMed] [Google Scholar]
- Das SK, Tan J, Johnson DC, Dey SK. Differential spatiotemporal regulation of lactoferrin and progesterone receptor genes in the mouse uterus by primary estrogen, catechol estrogen, and xenoestrogen. Endocrinology. 1998;139:2905–2915. doi: 10.1210/endo.139.6.6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W, Pollard JW. Progesterone inhibits estrogen-induced cyclin D1 and cdk4 nuclear translocation, cyclin E- and cyclin A-cdk2 kinase activation, and cell proliferation in uterine epithelial cells in mice. Mol Cell Biol. 1999;19:2251–2264. doi: 10.1128/mcb.19.3.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Teng CT. Estrogen response module of the mouse lactoferrin gene contains overlapping chicken ovalbumin upstream promoter transcription factor and estrogen receptor-binding elements. Mol Endocrinol. 1992;6:355–364. doi: 10.1210/mend.6.3.1584212. [DOI] [PubMed] [Google Scholar]
- Planas-Silva MD, Shang Y, Donaher JL, Brown M, Weinberg RA. AIB1 enhances estrogen dependent induction of cyclin D1 expression. Cancer Res. 2001;61:3858–3862. [PubMed] [Google Scholar]
- Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. 2004;88:113–122. doi: 10.1016/j.jsbmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Kazi AA, Jones JM, Koos RD. Chromatin immunoprecipitation analysis of gene expression in the rat uterus in vivo: estrogen-induced recruitment of both estrogen receptor alpha and hypoxia-inducible factor 1 to the vascular endothelial growth factor promoter. Mol Endocrinol. 2005;19:2006–2019. doi: 10.1210/me.2004-0388. [DOI] [PubMed] [Google Scholar]
- Rahman MA, Li M, Li P, Wang H, Dey SK, Das SK. Hoxa-10 deficiency alters region-specific gene expression and perturbs differentiation of natural killer cells during decidualization. Dev Biol. 2006;290:105–117. doi: 10.1016/j.ydbio.2005.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem Sci. 2000;25:99–104. doi: 10.1016/S0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- Yahata T, Shao W, Endoh H, Hur J, Coser KR, Sun H, Ueda Y, Kato S, Isselbacher KJ, Brown M, Shioda T. Selective coactivation of estrogen-dependent transcription by CITED1 CBP/p300-binding protein. Genes Dev. 2001;15:2598–2612. doi: 10.1101/gad.906301. [DOI] [PMC free article] [PubMed] [Google Scholar]