

Abstract
Translocation of cymtDNA into the nuclear genome, also referred to as numt, has been reported in many species, including several closely related to the domestic cat (Felis catus). We describe the recent transposition of 12,536 bp of the 17 kb mitochondrial genome into the nucleus of the common ancestor of the five Panthera genus species: tiger, P. tigris; snow leopard, P. uncia; jaguar, P. onca; leopard, P. pardus; and lion, P. leo. This nuclear integration, representing 74% of the mitochondrial genome, is one of the largest to be reported in eukaryotes. The Panthera genus numt differs from the numt previously described in the Felis genus in: (1) chromosomal location (F2 – telomeric region vs. D2 – centromeric region), (2) gene make up (from the ND5 to the ATP8 vs. from the CR to the COII), (3) size (12.5 kb vs. 7.9 kb), and (4) structure (single monomer vs. tandemly repeated in Felis). These distinctions indicate that the origin of this large numt fragment in the nuclear genome of the Panthera species is an independent insertion from that of the domestic cat lineage, which has been further supported by phylogenetic analyses. The tiger cymtDNA shared around 90% sequence identity with the homologous numt sequence, suggesting an origin for the Panthera numt at around 3.5 million years ago, prior to the radiation of the five extant Panthera species.
Keywords: big cats, mitochondrial DNA, nuclear insertion, numt, Panthera genus, pseudogene, tiger
Abbreviations: ATP8, ATP synthase subunit 8; bp, base pairs; Cyt b, cytochrome b; COI, cytochrome c oxidase subunit I; COII, cytochrome c oxidase subunit II; cymtDNA, cytoplasmic mitochondrial DNA; CR, control region; kb, kilobase(s); FISH, fluorescence in situ hybridization; MYA, million years ago; mtDNA, mitochondrial DNA; ND1, NADH dehydrogenase subunit 1; ND2, NADH dehydrogenase subunit 2; ND5, NADH dehydrogenase subunit 5; ND6, NADH dehydrogenase subunit 6; PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism; 16S, 16S ribosomal RNA; 12S, 12S ribosomal RNA
1. Introduction
Nuclear DNA sequences that are homologous to the mitochondrial genome, often referred to as numts (pronounced “new-mights”, Lopez et al., 1994), have been reported in numerous organisms, including more than 60 animal and plant species (reviewed in Bensasson et al., 2001). Most of the described incidences of numt are of short fragments of less than 600 bp with varying degrees of similarity with cymtDNA (Zhang and Hewitt, 1996a; Herrnstadt et al., 1999) and the process of integration has been often associated with non-homologous recombination (e.g., Roth et al., 1985; Henze and Martin, 2001). In humans, the genome sequence database has provided a broad view of the extent of mtDNA transfer, has facilitated the identification of transfer mechanisms, and has illuminated the evolutionary dynamics of numts (Mourier et al., 2001; Tourmen et al., 2002; Woischnik and Moraes, 2002; Hazkani-Covo et al., 2003; Mishmar et al., 2004; Ricchetti et al., 2004). The incorporation of mtDNAs sequences into the human nuclear genome has probably been a continuous evolutionary process, with, by some estimates, at least 612 integrations (Woischnik and Moraes, 2002). However, the incidence of novel numt insertions may be lower, since mtDNA-like sequences may also result from duplication after insertion into the nucleus (Tourmen et al., 2002; Bensasson et al., 2003; Hazkani-Covo et al., 2003). Most human numt segments encompass less than 5% of the mtDNA, and in only three instances exceed 70% of mtDNA.
Whole genome sequences of other mammals will continue to elucidate the evolutionary dynamics of numts outside of humans (Pereira and Baker, 2004; Richly and Leister, 2004). However, full genome drafts of other mammals will be limited primarily to model organisms of biomedical, taxonomic or phylogenetic interest (O’Brien et al., 2001). Therefore detailed characterizations of numts among closely related species will be necessary to provide additional insights into the characteristics of mitochondrial pseudogenes, including the study of their evolutionary histories and their distribution and abundance across species (Bensasson et al., 2001; Pons and Vogler, 2005).
There have been two documented cases of numt that have been reported in the Felidae family. The first consisted of the translocation of 7.9 kb of the mitochondrial genome into the domestic cat (Felis catus) nuclear genome (Lopez et al., 1994). This large segment is tandemly repeated 38–76 times on cat chromosome D2. The second case of numt in the Felidae family was first described in Panthera genus species based on mtDNA RFLP data (Johnson et al., 1996) and later by sequence analysis (Cracraft et al., 1998). Here we characterize the structure and evolutionary history of the Panthera numt fragment by (i) determining its chromosomal location in all the Panthera genus species (tiger, P. tigris; snow leopard, P. uncia; jaguar, P. onca; leopard, P. pardus; and lion, P. leo), (ii) comparing large portions of the numt and cymt sequences in one Panthera species (the tiger), and (iii) employing phylogenetic and coalescence analyses to assess the evolutionary history of these numt and cymt segments in species of the genus Panthera.
2. Materials and Methods
2.1. DNA isolation, amplification, cloning and sequencing
To facilitate the characterization of the Panthera numt, three distinct DNA fractions [total (t), nuclear (n), and cytoplasmic mitochondrial (cymt)] were purified from 1.5g of frozen liver from tiger (Pti065), snow leopard (Pun086), jaguar (Pon011), leopard (Ppa021), and lion (Ple181). The tDNA (mixture of nDNA and cymtDNA) was extracted from tissue according to standard procedures (Sambrook et al., 1989; Lopez et al., 1994). The nDNA fraction was purified using sucrose gradient DNA extraction methods (Bernatchez and Dodson, 1990) and the cymtDNA was purified using the Wizard Miniprep kit (Promega, Beckman et al., 1993). Four regions of the mtDNA genome were amplified in each of the fractions: (i) a portion between the ND5 gene and the CR (primers ND5F-U/CRR-U), (ii) the CR segment (primers CRF-U/CRR-U), (iii) a portion from 16S to ND2 (primers 16SF-U/ND2R-U), and (iv) the segment from ND2 to ATP8 (primers ND2F-U/ATP8R-U) (fig. 1; table S1). RFLP analysis was performed on these segments using several restriction enzymes (BamHI, HindIII, EcoRI, XhoI, etc) to test for differences in banding patterns between cymt and numt. The PCR products for the CR and 16S-ND2 segments, which exhibited different size lengths in the nDNA and cymtDNA fractions, were cloned and sequenced to unambiguously distinguish the cymt and numt products. PCR products were purified using Microcon PCR (Amicon). Cloning was carried out using Zero Blunt TOPO PCR Cloning kit (Invitrogen). The smallest CR PCR products were purified after agarose gel electrophoresis and subcloned using pCR-Blunt II-TOPO cloning vector (Invitrogen). Positive clones of cymt and numt were confirmed by comparison with the RFLP patterns. The clones were sequenced using Bigdye Terminators Cycle Sequencing Kits (PE Applied Biosystems) and run on an ABI-377 automated sequencer. Based on cymt/numt mismatches, a series of numt- and cymt-specific primers were designed for long-range PCR, allowing a more extensive sequencing and analysis of the tiger cymt and numt (table S1). Numt and cymt strand-specific primers were designed in highly variable sections or for variable sites using the virtual PCR program Amplify-2.53 (Engels, 1997). Additionally, cymt and numt portions of the 16S, ND1 and ND2 genes were amplified, cloned and sequenced for all the five Panthera species.
Fig. 1.

Schematic diagram of the relative positions of Panthera numt. The scale bar in Kb correspond to the domestic cat (Fca - Felis catus) mtDNA complete sequence (Lopez et al. 1996) aligned with the Panthera numt described in this study. The Fca numt is represented for comparison. Protein-coding genes and rRNAs are indicated in boxes. Individual capital letters correspond to the 17 tRNAs. The arrows and numbers over the CR and 16S represent gaps between the cymt and numt sequences in the tiger. Fragments amplified from cymt or numt portions are represented by lines and arrow lines with primer names labeled at the 5′ and 3′ ends and primer sequences (table 1). (A) A 2.6 kb mtDNA probe was generated by PCR and used for FISH mapping to locate the numt in the Panthera species. (B) Four segments were amplified using universal primers from three DNA fractions (tDNA, nDNA, and cymtDNA) of Panthera species and examined by RFLP (see Figure 2). Two segments (CR, 16S-ND2) were cloned and sequenced subsequently to separate cymt and numt. (C) Cymt and numt tiger sequences were obtained separately using a combination of universal and strand-specific primers designed based upon cymt/numt gaps in the CR and 16S regions (table 1).
2.2. Cytogenetic inference of the Panthera numt location: FISH mapping
The location of numt in the nuclear genome of all the Panthera species was determined by FISH. A 2.6 kb mtDNA PCR probe (fig. 1), generated from the purified cymtDNA fraction, was labeled with biotin-11 dUTP (Sigma) by nick translation (Brigati et al., 1983) in the five Panthera species, as well as the domestic cat. The final probe size was verified on a 1.2 % gel with appropriate markers. Metaphase spreads were prepared by standard cytogenetic techniques (Modi et al., 1987). FISH was performed as described in (Lichter et al., 1990). Briefly the metaphase spreads were denatured in 70% formamide 2XSSC in an 80 ºC oven for 90 s and dehydrated in cold ethanol series, 70%–90%–100%, for 3 to 5 min in each step. 400 ng of labeled probe and 10 ug of salmon sperm carrier DNA were resuspended in 50% formamide-10% dextran sulfate-2XSSC and denatured for 10 min at 75 ºC. The denatured probe cocktail was layered on the denatured metaphase chromosomes. Following 48 h of incubation at 37 ºC, post-hybridization washes, and treatment with blocking solution, the hybridized biotin labeled probe was detected by fluorescein isothioscyinate (FITC) conjugated avidin DCS (5mg/ml-Vector labs). Fluorescence signals were captured as gray scale images using a Zeiss Axioskop epi-fluorescence microscope equipped with a cooled CCD (charged coupled device) camera (Photomentics CE 200 A) and the Oncor imaging system. Gray-scale images were computer enhanced, pseudocolored, and merged using Oncor Image software. Images of reverse DAPI banded chromosomes were merged with the FITC detected signals allowing for direct visualization of localization, chromosome identification and cytogenetic loci assignment.
2.3. Sequence analyses
Sequences were inspected using SEQUENCHER (Gene Codes Co.), aligned using Clustal-X (Thompson et al., 1997), and further checked by eye. Initial sequence comparisons and measures of variability were performed using MEGA (Kumar et al., 2001). Transition/transversion ratios (Ts/Tv) and the parameter of the gamma distribution of rate variation among sites method of (Yang and Kumar, 1996) were estimated using PAMP (included in the package PAML 2.0; Yang, 1997). tRNA structure was predicted using the mfold web server (Zuker, 2003). Phylogenetic analyses of the Panthera cymt and numt sequences were performed in PAUP* 4.0b2a (Swofford, 2001) using three approaches: (i) minimum evolution (ME) heuristic search, using a Kimura two-parameter model and the neighbor-joining tree-building algorithm (Saitou and Nei, 1987) followed by branch-swapping; (ii) maximum parsimony (MP), with an exhaustive search; and (iii) maximum likelihood (ML), incorporating a gamma-corrected HKY85 model with parameters estimated from the data set. Reliability of nodes defined by the phylogenetic trees was assessed using 100 bootstrap replications (Felsenstein, 1985; Hillis and Bull, 1993) in the ME and MP analyses, and with the quartet puzzling method in the ML analysis (PUZZLE 4.0; Strimmer and von Haeseler, 1996). The molecular dating for the Panthera numt origin was estimated from the overall genetic distance between tiger numt and cymt, applying the equation of Li et al. (1981) whereby the fraction of sequence divergence is: δ = (μ1 + μ2) t, where μ1 = 2.5 × 10−8 substitutions/sites/year for cymtDNA (Hasegawa et al., 1985; Lopez et al., 1997) and μ2 = 4.7 × 10−9 substitutions/sites/year for nuclear pseudogene distance (Li et al., 1981; Lopez et al., 1997) and t is the time elapsed.
3. Results
3.1. Recognition of the genes involved in the Panthera numt
A detection strategy was devised to identify and isolate potential numt fragments based on differences in banding patterns from four distinct PCR products [(ND5-CR), (CR), (16S-ND2) and (ND2-ATP8); (fig. 1)] and RFLP’s banding patterns from three DNA fractions (tDNA, nDNA, and cymtDNA isolated from liver tissue; see Material and Methods) (fig. 2). The CR-PCR products from the tDNA fraction in all the Panthera species showed two codominant bands of around 1.7 kb and 1.5 Kb, compared with a single band from the purified cymtDNA and nDNA fraction (1.7 and 1.5 Kb, respectively) (fig. 2A). We determined by band pattern and sequence analysis that the 1.7 Kb fragment was cymt and that the 1.5 Kb fragment was the numt copy. Numt PCR products were identified also from the three other regions, (ND5-CR), (16S-ND2; fig. 2B) and (ND2-ATP8) based on different RFLP patterns of Hind III and Bam HI digestion among the three DNA fractions. These combined results suggested that the Panthera numt encompasses a region within the ND5 to the ATP8 gene, including eight protein coding genes, two rRNA genes, 17 tRNA genes, and the non-coding CR (fig. 1).
Fig. 2.
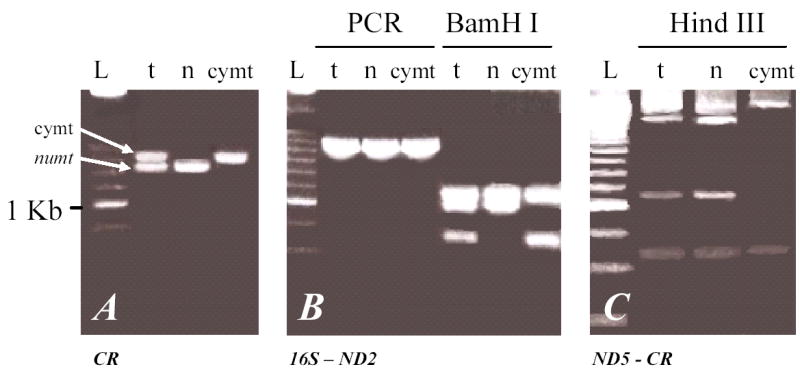
Differences in the banding patterns from PCR products amplified from total (t), nuclear (n), and cytosolic mitocondrial (cymt) Panthera DNA fractions from two of the four segments surveyed that showed presence of numt copies. The two segments represented in this figure were chosen for depiction due to the clear distinction of numt sequences caused by the large deletions in CR and 16S. The banding patterns observed were similar in all Panthera species and thus only a single species profile is represented. (A) Control region fragment. (B) Region between 16S and ND2 gene followed by restriction enzyme digestion of BamH I. Lane L represents DNA size ladder 250 (BRL, i.e. the brightest band is 1.0 kb and each step represents 250 bp).
3.2. Chromosomal location of the Panthera numt
A 2.6 kb mtDNA probe including ND5, ND6, and CytB regions (fig. 1) was hybridized on a metaphase spread of the five Panthera genus species and the domestic cat. Strong hybridization fluorescent signals were observed on chromosome F2 at q1.1 in all the Panthera species (fig. 3A to E), but on chromosome D2 at the centromere of the domestic cat (fig. 3F), as previously described by Lopez et al. (1994).
Fig. 3.
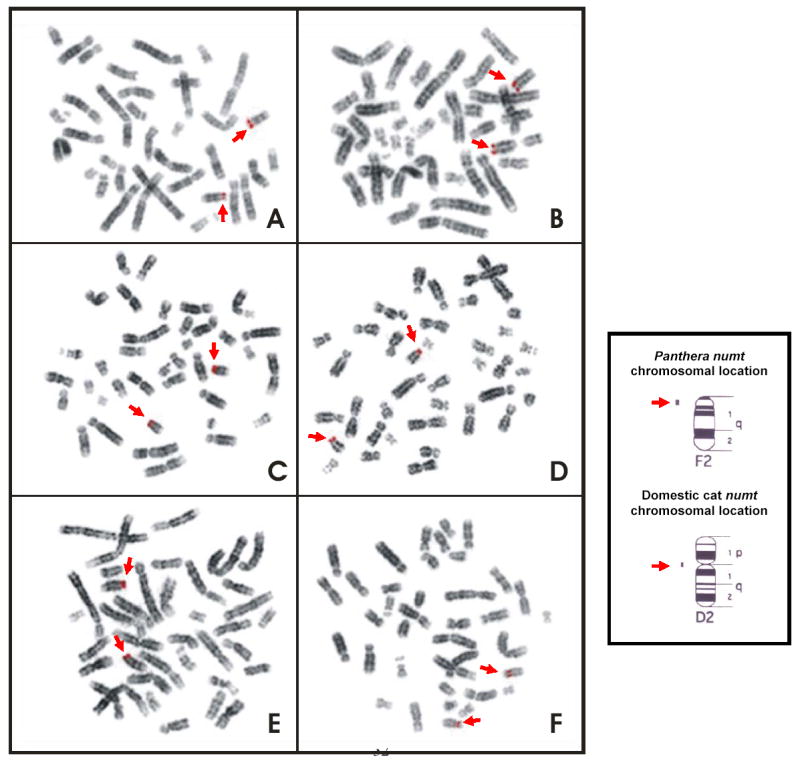
Image of fluorescent in-situ hybridization (FISH) of the metaphase chromosomes for each of the five Panthera species and the domestic cat using the probe including the partial sequences from ND5 and Cytb region (2.6 kb). (A) Tiger, P. tigris. (B) Lion, P. leo. (C) Jaguar, P. onca. (D) Leopard, P. pardus. (E) Snow leopard, P. uncia. (F) Domestic cat, F. catus. Signals revealed on the telomeric region of the chromosome F2 (F2q12) in all the Panthera species (A–E) and on the centromeric region of the chromosome D2 (D2p11) in the domestic cat (F).
3.3. Comparative sequence analyses of tiger numt and cymt
Using large deletions in CR (25 bp) and 16S (23bp) of the Panthera numt, we designed strand-specific primers for numt and cymt for long-range PCR amplification and sequencing in tiger (fig. 1). Sequences from clones and PCR products were concatenated into a fragment of 12,898 bp for cymt and 12,536 bp for numt (GenBank accession numbers DQ151550 and DQ151551) (fig. 1). The size difference between numt and cymt was caused mostly by the 340 bp gap in the RS3 region, a 23 bp gap in the HVS-1 region of CR, and a 25 bp gap of the 16S gene in numt (fig. S1). The numt sequence started in the middle of the ND5 gene position (corresponding to position Fca 12,918 in the domestic cat; Lopez et al. 1996) and almost reached the end of ATP8 gene (position Fca 8,840). This 12,536 bp (~12.5 kb) of tiger numt included approximately 75% of the 17 kb mitochondrial genome, as described in the domestic cat (Lopez et al., 1996) The tiger numt contains a truncated ND5 gene (1533 bp), and complete ND6 (527 bp), Cyt b (1143 bp), 12S (960 bp), 16S (1545 bp), ND1 (958 bp), ND2 (1044 bp), COI (1550 bp), and COII genes (684 bp), a truncated ATP8 gene (183 bp), a CR sequence (1,181 bp) with a large deletion (340 bp) removing most of the RS-3 with the d(CA)-rich 8-bp [ACACACGT] motif, and full sequences for 17 interspersed tRNAs (fig. S1; table S1).
Fig S1.





Alignment of tiger cymt and numt sequences. (A) Sequences from the eight protein coding genes. (B) Sequences from the two rRNA genes. (C) Sequences from the seventeen tRNA genes. (D) Sequences from the control region. Stop codons were marked as * and gaps were marked as -. Replication directions are represented as H for heavy chain (5′–3′) and as L for light chain (3′–5′) after each gene. Overlapped sequences between ND5 and ND6 were underlined.
3.4. Numt and cymt sequence characterization in tiger
The nucleotide composition of tiger numt and cymt sequences were similar, 32.31% A, 26.04% C, 15.13% G, and 26.38% T in numt compared with 32.34% A, 26.19% C, 15.10% G, and 26.32% T in cymt. Numt and cymt shared three different types of genes (rRNA, tRNA, and protein coding) plus the CR (fig. S1). Markedly different patterns of sequence variation were observed between different numt and cymt genes, with sequence similarities ranging from 82% in ATP8 to 100% in three tRNA (table 2). Sequence variation between numt and cymt was due to both base-pair substitutions (n = 803) and indels (n = 523 bp). Most of the mutational changes between numt and cymt were transitions (710/803 = 88%) with the highest proportion of transitional changes occurring in the protein coding genes (5635/611 = 92%) and the lowest in RNAs (83/103 = 81%). Transitions from T to C were more common than from A to G. To infer whether these genes retained function, sequences from the protein coding genes of cymt and numt were translated into amino acid using the mitochondrial and universal genetic codes, respectively. All cymt protein coding gene sequences could be translated into amino acid sequences, but in the numt sequences 32 extra stop codons were observed (fig. S1A and S2). The variable sites between cymt and numt in protein-coding genes were not distributed evenly (fig. S1A), suggesting that conserved segments may lie within the functional domains of the mtDNA proteins, which are more prone to evolutionary constrains. Likewise, in 12S there were 26 variable sites in the first half from positions 1 to 530 bp and no variable sites from positions 531 to 1,027. In the 1,575 bp fragment of 16S, 74 of 82 (90%) variable sites occurred in the first 520 bp (1–520 bp) and the third 500 bp (1,040–1,575 bp) compared with only 8 variable sites (less than 10%) in the middle, (from 521 to 1,039 bp) (fig. S1B). Seventeen tRNA genes were sequenced in both cymt and numt (fig. S1C). Three tRNA genes (tRNA-Gln, -Pro, and -Val) had identical sequences in both cymt and numt. The number of variable sites in the other tRNA genes ranged from one in tRNA-met to 12 in tRNA-Phe. Average percentage sequence similarity between cymt and numt in tRNA genes was 95% and in rRNA 95.5% (table 2). Lower sequence similarity was observed for the protein coding genes (90.9%) and the CR (91%; excluding the 186 bp gap of RS3 region).
Table 2.
Characterization of the size, similarity, and nucleotide substitution patterns from pairwise comparison of tiger cymt (12.8 kb) and numt (12.5 kb) sequences. Stop codons within numt were determined after frame shift or indels.
| Pattern of substitutions
|
Pattern of gaps in numt |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | Changes between cymt and numt (bp)
|
Ts
|
Tv
|
||||||||||||
| Segments | cymt | numt | Subst. | Gaps | A-G | T-C | G-C | A-T | T-G | A-C | Ts/Tv ratio | Insertions (bp) | (bp) Deletions | Number of stop codons within numt | Percent differences of nucleotide |
| Protein Coding Genes | |||||||||||||||
| ND5 | 1530 | 1533 | 124 | 11 | 35 | 76 | 4 | 3 | 1 | 5 | 8,5 | 7(1bpx3+2bpx2) | 4(1bpx4) | 1 | 8,8 |
| ND6 | 528 | 527 | 38 | 5 | 14 | 19 | - | 1 | 3 | 1 | 6,6 | 2(1bpx2) | 3(1bpx1+2bpx1) | - | 8,1 |
| ND1 | 957 | 958 | 71 | 5 | 15 | 52 | 1 | 1 | 1 | 1 | 16,8 | 3(1bpx4) | 2(1bpx2) | 3 | 7,0 |
| ND2 | 1044 | 1044 | 84 | 4 | 26 | 46 | 4 | 4 | - | 4 | 6,0 | 2(1bpx2) | 2(1bpx2) | - | 8,4 |
| CytB | 1140 | 1143 | 94 | 15 | 29 | 59 | 1 | 2 | 1 | 2 | 14,7 | 9(1bpx9) | 6(1bpx6) | - | 10,5 |
| COI | 1545 | 1550 | 124 | 19 | 48 | 65 | 1 | 2 | 6 | 2 | 10,3 | 12(1bpx9+3bpx1) | 7(1bpx7) | 23 | 9,3 |
| COII | 684 | 684 | 62 | 8 | 27 | 33 | - | - | 1 | 1 | 30,0 | 4(1bpx2+2bpx1) | 4(1bpx4) | - | 10,2 |
| ATP8 | 182 | 183 | 23 | 9 | 10 | 9 | 1 | 2 | 1 | 1 | 3,8 | 5(1bpx3+2bpx1) | 4(1bpx2+2bpx1) | 5 | 17,6 |
| Total | 7610 | 7622 | 620 | 76 | 204 | 359 | 12 | 15 | 14 | 17 | 9,7 | 44 | 32 | 32 | 9,1 |
| rRNAs | |||||||||||||||
| 12S | 957 | 956 | 19 | 9 | 7 | 9 | - | - | - | 3 | 5,3 | 4(1bpx4) | 5(1bpx5) | - | 2,9 |
| 16S | 1575 | 1545 | 42 | 44 | 13 | 22 | 1 | 4 | - | 1 | 5,8 | 7(1bpx7) | 37(6bpx1+1bpx6+25bpx1) | - | 5,5 |
| Total | 2532 | 2501 | 61 | 53 | 20 | 31 | 1 | 4 | - | 4 | 5,7 | 11 | 42 | - | 4,5 |
| tRNAs | |||||||||||||||
| tRNA-Glu | 71 | 70 | - | 1 | - | - | - | - | - | - | - | - | 1(1bpx1) | - | 1,4 |
| tRNA-Thr | 70 | 70 | 4 | - | 1 | 2 | - | 1 | - | - | 3 | - | - | - | 5,7 |
| tRNA-Pro | 66 | 66 | - | - | - | - | - | - | - | - | - | - | - | - | 0 |
| tRNA-Phe | 71 | 75 | 6 | 6 | 3 | 1 | 1 | - | 1 | - | 2 | 5(2bpx1+1bpx3) | 1(1bpx1) | - | 16,9 |
| tRNA-Val | 68 | 68 | - | - | - | - | - | - | - | - | - | - | - | 0 | |
| tRNA-Leu | 75 | 75 | 4 | - | - | 2 | - | 1 | 1 | - | 1 | - | - | - | 5,3 |
| tRNA-Ile | 69 | 69 | 2 | - | 2 | - | - | - | - | - | 2 | - | - | - | 2,9 |
| tRNA-Gln | 74 | 74 | - | - | - | - | - | - | - | - | - | - | - | - | 0 |
| tRNA-Met | 69 | 69 | 1 | - | - | 1 | - | - | - | - | 1 | - | - | - | 1,4 |
| tRNA-Trp | 69 | 69 | 3 | 4 | - | 1 | - | 1 | 1 | - | 0,5 | 2(1bpx2) | 2(1bpx2) | - | 10,1 |
| tRNA-Ala | 69 | 69 | 3 | - | 1 | 2 | - | - | - | - | 3 | - | - | - | 4,3 |
| tRNA-Asn | 73 | 73 | 3 | - | 1 | 1 | - | - | 1 | - | 2 | - | - | - | 4,1 |
| tRNA-Cys | 66 | 66 | 2 | - | 1 | - | - | 1 | - | - | 1 | - | - | - | 3,0 |
| tRNA-Tyr | 68 | 66 | 4 | 4 | - | 2 | 1 | - | - | 1 | 1 | 1(1bpx1) | 3(1bpx3) | - | 11,8 |
| tRNA-Ser | 70 | 70 | 3 | - | 1 | 2 | - | - | - | - | 3 | - | - | - | 4,3 |
| tRNA-Asp | 69 | 70 | 4 | 1 | 2 | 2 | - | - | - | - | 4 | 1(1bpx1) | - | - | 7,2 |
| tRNA-Lys | 69 | 69 | 4 | - | 1 | 3 | - | - | - | - | 4 | - | - | - | 5,8 |
| Total | 1186 | 1188 | 43 | 16 | 13 | 19 | 2 | 4 | 4 | 1 | 2,9 | 9 | 7 | - | 5,0 |
| Control region | 1539 | 1181 | 79 | 378 | 25 | 39 | 2 | 6 | 2 | 5 | 4,3 | 10(1bpx7+3bpx1) | 368(1bpx5+23bpx1+340bpx1) | - | 30 |
| Total | 12867 | 12492 | 803 | 523 | 262 | 448 | 17 | 29 | 20 | 27 | 7,6 | 74 | 449 | 32 | 10,3 |
Fig S2.

Alignment of the amino acid sequences of three protein coding genes in tiger cymt and numt. Stop codons represented by *. The gaps are represented as -.
3.5. Phylogenetic relationships of the Panthera numts
The phylogenetic relationships of the cymt and numt sequences in the five Panthera species was investigated using concatenated sequences (1,206 bp) from three mitochondrial genes, 16S (403 bp), ND1 (502 bp), and ND2 (301 bp) (fig. 4A). The cymt/numt specific-amplification of such genes was facilitated by the 23 bp deletion of the 16S Panthera numt. Two distinct monophyletic clusters, with very strong bootstrap support, defined cymt and numt sequences (results were identical considering ME, MP or ML analyses, or each of the single gene sequences analyses). Little internal structure among Panthera species was observed in either cymt and numt sequences. Cymt sequences showed a five fold faster rate of divergence (average pairwise distance = 0.066 ± 0.006) compared to numts (0.013 ± 0.002) (see also fig. 4A), similar to the pattern observed in Felis numt (Lopez et al., 1994). Additionally, the phylogenetic relationships between the domestic cat numt (Lopez et al. 1997) and the tiger numt (this study) clearly suggest that the two classes of numts within Felidae are distinct synapomorphies (fig. 4B).
Fig. 4.


(A) Phylogenetic minimum evolution tree (Kimura two-parameter) of the five Panthera species cymts and numts (1,206 bp concatenated sequences of the 16S, ND1, and ND2). The taxon abbreviation is as follow: Pti – tiger, Pun – snow leopard, Pon – jaguar, Ppa – leopard, Ple – lion, and Nne – Clouded leopard (Neofelis nebulosa). The rooting of the tree was obtained with the slowest evolving mtDNA fragment (16S) to avoid long-branch attraction caused by the high rate of divergence of mtDNA. (B) Phylogenetic minimum evolution tree (Kimura two-parameter) illustrating the relationship between the domestic cat numt (Lopez et al. 1996) and the tiger numt (this study) (7,683 bp alignment). The taxon abbreviation is as follow: Fca – domestic cat, Aju – cheetah (Acinonyx jubatus), Pti – tiger, and Cfa – dog (Canis familiaris). GenBank accession numbers are as follow: Fca-cymt (U20753); Fca-numt (U20754); Aju-cymt (NC_005212); and Cfa-cymt (NC_002008). Bootstrap support values were identical in the ME, MP and ML analyses. Bootstrap values are placed at each branchpoint for the minimum evolution/maximum parsimony/maximum likelihood phylogenetic analyses, respectively (ME/MP/ML).
4. Discussion
4.1. Origin of the Panthera numt
An independent origin of the Panthera numt from that of the domestic cat (Lopez et al., 1994) is strongly supported by its distinct chromosomal location, size, contents, and structure. The numt location in all the Panthera species was mapped by FISH on chromosome F2 (fig. 3A to E). However, the signal using the same probe on the domestic cat produced a signal on chromosome D2 (fig. 3F), as previously described (Lopez et al., 1994). The tiger numt, is considerable larger than domestic cat’s, with a single unit of 12.5 kb that includes genes from middle of ND5 to part of ATP 8 subunit (fig. 1). By contrast, the domestic cat numt has a unit of 7.9 kb (with genes from middle of CR to COII) that is tandemly repeated with 38 to 76 copies, having an overall integrated size of 300 to 600 kb (Lopez et al., 1994). To test for a tandem arrangement in tiger numt, we performed inverse PCR with several different primer sets. However, because we did not observe any PCR products, this suggests that the Panthera numt is not tandemly repeated and is most-likely a single segment on the chromosome F2.
The phylogenetic analysis performed on cymt and numt sequences from the five extant Panthera species strongly supports a single origin for all these numts along the branch leading to the most-recent common ancestor of the genus (fig. 4A) and that the domestic cat numt and the tiger numt lineages are distinct synapomorphies within the Cat family (fig. 4B). Using an overall genetic distance of 10.3 % between tiger numt and cymt (table 2), we estimate that numt and cymt began to diverge around 3.45 MYA, which would be consistent with the known evolutionary history of the Panthera lineage. Analyses of nuclear and mtDNA sequences across all felid species suggests that a common ancestor of the five species of roaring cats diverged from the clouded leopard 5.96 MYA and began to speciate into unique evolutionary lineages 3.47 MYA (O’Brien, 1996; Johnson and O’Brien, 1997; Johnson et al., submitted). Overall, our results support the occurrence within the Felidae family of two independent translocations of cytoplasmic mtDNA into the nuclear genome: one in the Panthera genus (around 3 MYA) and the other in the domestic cat lineage (around 1.8 MYA; Lopez et al. 1994).
4.2. Numt as a pseudogene: evolution and functional implications
Once mtDNA fragments become incorporated into the nuclear genome, they immediately are exposed to different modes of evolution, which will influence the divergence patterns between the two sequences (Lopez et al., 1994; Lopez et al., 1996; Lopez et al., 1997). These include lower mutation rates due to nuclear DNA repair, a distinct genetic code, and the possibility of recombination. In addition, numts apparently evolve without the functional selective constraints as their mitochondrial counterparts (Gellissen et al., 1983; Perna and Kocher, 1996). The tiger cymt showed a high bias in transitions over transversions, a well-recognized characteristic of mtDNA (Brown et al., 1982) that was not observed for the numt sequence (nDNA). The phylogenetic analyses depict the more-rapid rate of cymt divergence among Panthera. This is caused by the higher mutation rate of mtDNA, particularly for protein-coding genes (Lopez et al., 1997).
Genes within the tiger numt fragment have several characteristics that would preclude these sequences from producing functional gene products. First, in the protein coding genes of numt, there are often several termination codons or frame shift mutations in all possible open reading frames (table 2; fig. S1A), many of which were caused by differences in the genetic codes between the nucleus and mitochondria (Anderson et al., 1981; Brown, 1985). Second, the numt 16S has a large deletion (23 bp), which would appear to disrupt the normal secondary structure (fig. S1B). Third, two regulatory elements (CSB 2 and 3) of the CR that are involved in transcriptional promotion catalyzed by mitochondrial RNA polymerase and trans-activating factors do not function in nuclear genes (Schinkel and Tabak, 1989). The numt CR also lacks most of the repetitive segment three (RS-3), which is involved in mtDNA replication and transcription (fig. S1D). The importance of mtDNA CR in the nuclear genome is at least in part dependent on the presence of promoter regions and functional sequences, because as far as is known, the CR is only functional with promoter and several protein-binding sites (Chang and Clayton, 1985). Due to the large deletions of the hypervariable segment one (HVS-1) and RS-3, the numt CR sequence is presumably not functional. Fourth, all of the cymt tRNA sequences formed typical cloverleaf shapes of class 1 tRNAs (Lewin, 1994). However, some numt tRNAs, like for example, tRNA-Thr and tRNA-Tyr, formed imperfect shapes due to several unpaired free-bases that likely cause loss of function (fig. 5). The differing degrees of similarity among tiger cymt and numt genes, specifically the highly conserved rRNAs or invariant tRNA genes contrasted with the more-divergent protein-coding genes and the CR (table 2; fig. S1C and S1D), highlight the differential rates of nucleotide substitution among mitochondrial genes relatively to its homologues numt molecular “fossils”. In the mammalian mitochondria, the average nucleotide divergence is much lower in rRNA genes relative to protein-coding genes or the CR (Lopez et al., 1997).
Fig. 5.
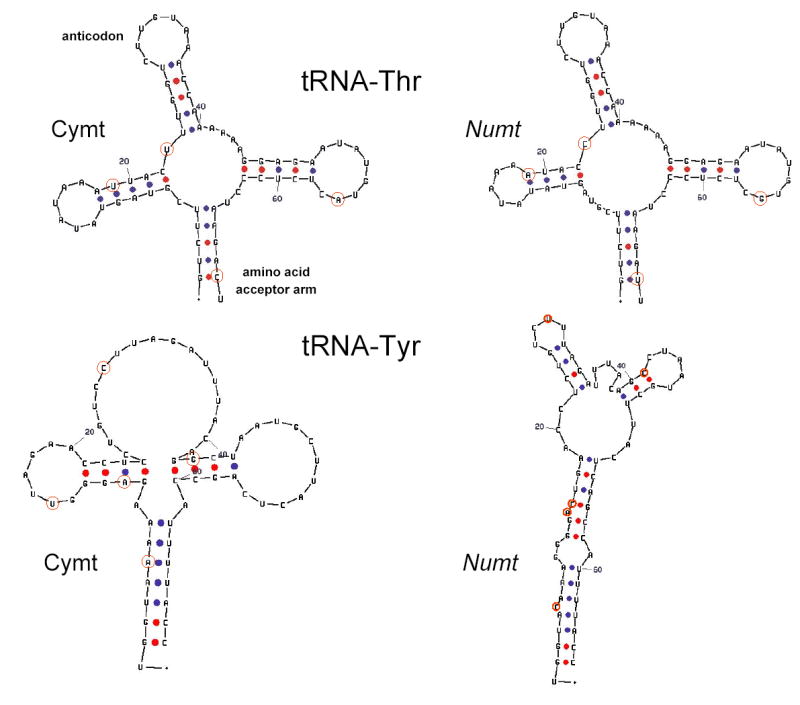
Proposed secondary structure for tRNA-Thr and tRNA-Tyr based on DNA sequence data from the tiger cymt and numt. Dots represent Watson-Crick bonds. Red circle indicate that the nucleotide is variable between cymt and numt. Numbers represent the direction of the sequences from 5′ to 3′.
The maintenance in the function of genes translocated from organelle to nucleus occurred numerous times in evolutionary history, contributing to the compact and economical mitochondrial genomes observe today (Perna and Kocher, 1996). The mammalian mitochondrial genome of 15,000–17,000 bp and thirty-seven coding genes contrasts with the hundreds of nuclear genes that have function in the mitochondria, such as nuclear-encoded members of the citric acid cycle, cytochrome chain, and oxidative phosphorylation pathways. As with numt, these nuclear genes, following the Serial Endosymbiosis Theory (Margulis, 1970; Yang et al., 1985), are thought to have originated from the transfer of mtDNA genes to the nucleus, with subsequent duplication and divergence. A reduction in the accumulation of deleterious mutations is a prime benefit for cymt genes that are subsequently located in the nuclear genome (where DNA repair is more efficient). However, functional gene transfers have been documented almost exclusively in plants (e.g., Adams et al., 2002) and green algae (e.g., Perez-Martinez et al., 2000; Funes et al., 2002), suggesting that in animals, where the mitochondrial genetic code differs from the standard code (Wolstenholme, 1992), most numts are non functional upon arrival.
4.3. The mtDNA as a reliable molecular marker
The maternal inheritance, cellular abundance, and lack of recombination of the mtDNA have allowed biologists to phylogenetically study many metazoan animal. However, mitochondrial-like DNA sequences in the nuclear genome of many organisms, and their amplification or coamplification during PCR is a recognized complication (Perna and Kocher, 1996; Zhang and Hewitt, 1996a). Because nuclear insertions are paralogs of the authentic mitochondrial sequences, they will confound phylogenetic and population genetic analyses when inadvertently included, especially when using more slowly evolving segments (Arctander, 1995; Collura and Stewart, 1995; Vanderkuyl et al., 1995; Zhang and Hewitt, 1996b). Mitochondrial-like sequences in the nuclear genome can negate the advantages of mtDNA as a molecular marker in population studies. The occurrence of numt, as with sequence heteroplasmy, necessitates more-complicated data collection and analysis and in some species, like gorillas that have a large variety of numt sequences bearing high similarity to cymtDNA, can make analysis of mtDNA impractical (Thalmann et al., 2004).
One implication is that explicit measures need to be taken to authenticate mtDNA sequences generated. Previously reported mtDNA tiger sequences have been incorrectly labeled (fig. S3). In some cases, the reported gene sequences were mixed sequences of cymt and numt (Masuda et al., 1994; Ledje and Arnason, 1996). In another case, sequences were preferentially collected from nuclear copies (Johnson and O’Brien, 1997). The full sequence for both tiger cymt and numt is presented here, providing a valuable contribution for research in felids. Such data has greatly facilitated the validation of the matrilineal genealogy of current tiger subspecies (Luo et al., 2004) and certainly will be highly useful for research on the other closely related Panthera species. Refined accurate population genetic inferences will represent an effective contribution for the conservation and the management of these endangered cat species.
Fig S3.

Comparison of mtDNA sequence errors in tiger. Cymt and numt sequences generated in this study were compared with previous reported tiger mtDNA sequences. (A) 12S gene sequences from (Ledje and Arnason, 1996). (B) 12S gene sequences from Masuda et al. (1996). (C) 16S gene sequences from Johnson and O’Brien (1997). In some cases, the reported gene sequences were mixed sequences of cymt and numt (A and B) while in another case, sequences were preferentially collected from nuclear copies (C).
The relative scarcity of numts described in Felidae species to date contrasts with the high frequency of numts observed in primates, particularly in humans, as revealed by the human genome database (e.g., Mourier et al., 2001; Tourmen et al., 2002; Woischnik and Moraes, 2002). The prevalence of reported numts varies widely among metazoans (reviewed in Bensasson et al., 2001), with human and plant genomes harboring the largest numt repertoires (Richly and Leister, 2004). The cat genome project, which was recently included in the Large-Scale Sequencing Research Network, will facilitate more detailed evaluation of the dynamics and extent of numt insertions in this Felidae species.
Table 1.
Primers used to amplify the Panthera cymt and numt portions surveyed in this study.
| Primer Name | Sequence | Specificity |
|---|---|---|
| ND5F-U | 5′-GTGCAACTCCAAATAAAAG-3′ | Panthera sp. |
| CytBR-U | 5′-ATTAATAATTTTGATAAGGGGGTGCGAT-3′ | Panthera sp. |
| CRF-U | 5′-TCAAAGCTTACACCAGTCTTGTAAACC-3′ | universal |
| CRR-U | 5′-TAACTGCAGAAGGCTAGGACCAAACCT-3′ | universal |
| 16SF-U | 5′-ACGACGGCCAGTGTGCAAAGGTAGCATAATCA-3′ | Panthera sp. |
| ND2R-U | 5′-CAACCCGTTAACCTCGGGTACTCAGAAGT-3′ | Panthera sp. |
| ND2F-U | 5′-ACTTCTGAGTACCCGAGGTTAACGGGTTG-3′ | Panthera sp. |
| ATP8R-U | 5′-GCTATGACCGGCGAATAGATTTTCGTTCA-3′ | universal |
| CRF-N | 5′-ACTCCCACAACACAGACGCACAGT-3′ | P. tigrisN |
| CRF-C | 5′-CGTTAATACAGAACACACAACACG-3′ | P. tigris C |
| CRR-N | 5′-CATTGTGCGTTTGTGTTATGGG-3′ | P. tigris N |
| CRR-C | 5′-CGTGTTGTGTGTTCTGTAT-3′ | P. tigris C |
| 16SF-N | 5′-CGTTTGTTCAACGACTACCGG-3′ | P. tigris N |
| 16SF-C | 5′-CAAAGTCCTACGTGATCTG-3′ | P. tigris C |
| 16SR-N | 5′-CGTGGACTACTCCGGTAATCG-3′ | P. tigris N |
| 16SR-C | 5′-CAGAACTCAGATCACGTAG-3′ | P. tigris C |
The meanings of the abbreviations are as follows; U - Panthera species specific or universal primer, N - numt specific, C - cymt specific, F - forward, R - reverse. The source of universal primers is Kocher et al. (1989), Johnson et al. (1998), or designed from this study, and N, C primers were designed for this study using clones from CR and 16S-ND2 gene regions.
Acknowledgments
A. Antunes was supported in part by a Postdoctoral grant (SFRH/BPD/5700/2001) from the Portuguese Foundation for the Science and Technology (Fundação para a Ciência e a Tecnologia). This publication has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Comments made by two anonymous referees improved a previous version of this manuscript.
References
- Adams KL, Qiu YL, Stoutemyer M, Palmer JD. Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc Natl Acad Sci U S A. 2002;99 (15):9905–9912. doi: 10.1073/pnas.042694899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, et al. Sequence and Organization of the Human Mitochondrial Genome. Nature. 1981;290 (5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Arctander P. Comparison of a Mitochondrial Gene and a Corresponding Nuclear Pseudogene. Proc R Soc Lond B. 1995;262 (1363):13–19. doi: 10.1098/rspb.1995.0170. [DOI] [PubMed] [Google Scholar]
- Beckman K, Smith M, Orrego C. Purification of mitochondrial DNA with Wizard Minipreps DNA Purification System. Promega Note. 1993;43:10–13. [Google Scholar]
- Bensasson D, Feldman MW, Petrov DA. Rates of DNA duplication and mitochondrial DNA insertion in the human genome. J Mol Evol. 2003;57 (3):343–354. doi: 10.1007/s00239-003-2485-7. [DOI] [PubMed] [Google Scholar]
- Bensasson D, Zhang DX, Hartl DL, Hewitt GM. Mitochondrial pseudogenes: evolution’s misplaced witnesses. Trends Ecol Evol. 2001;16 (6):314–321. doi: 10.1016/s0169-5347(01)02151-6. [DOI] [PubMed] [Google Scholar]
- Bernatchez L, Dodson JJ. Allopatric Origin of Sympatric Populations of Lake Whitefish (Coregonus-Clupeaformis) as Revealed by Mitochondrial-DNA Restriction Analysis. Evolution. 1990;44 (5):1263–1271. doi: 10.1111/j.1558-5646.1990.tb05230.x. [DOI] [PubMed] [Google Scholar]
- Brigati DJ, et al. Detection of Viral Genomes in Cultured-Cells and Paraffin-Embedded Tissue-Sections Using Biotin-Labeled Hybridization Probes. Virology. 1983;126 (1):32–50. doi: 10.1016/0042-6822(83)90460-9. [DOI] [PubMed] [Google Scholar]
- Brown W. The mitochondrial genome of animals. In: Macintyre RJ, editor. Molecular Evolutionary Genetics. Plenum Press; New York: 1985. pp. 95–130. [Google Scholar]
- Brown WM, Prager EM, Wang A, Wilson AC. Mitochondrial-DNA Sequences of Primates - Tempo and Mode of Evolution. J Mol Evol. 1982;18 (4):225–239. doi: 10.1007/BF01734101. [DOI] [PubMed] [Google Scholar]
- Chang DD, Clayton DA. Priming of Human Mitochondrial-DNA Replication Occurs at the Light-Strand Promoter. Proc Natl Acad Sci U S A. 1985;82 (2):351–355. doi: 10.1073/pnas.82.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collura RV, Stewart CB. Insertions and Duplications of Mtdna in the Nuclear Genomes of Old-World Monkeys and Hominoids. Nature. 1995;378 (6556):485–489. doi: 10.1038/378485a0. [DOI] [PubMed] [Google Scholar]
- Cracraft J, Felsenstein J, Vaughn J, Helm-Bychowski K. Sorting out tigers (Panthera tigris): mitochondrial sequences, nuclear inserts, systematics, and conservation genetics. Animal Conservation. 1998;1:139–150. [Google Scholar]
- Engels B. University of Wisconsin.; 1997. Amplify: Software for PCR, version 2.53B. [Google Scholar]
- Felsenstein J. Confidence-Limits on Phylogenies - an Approach Using the Bootstrap. Evolution. 1985;39 (4):783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Funes S, et al. The typically mitochondrial DNA-encoded ATP6 subunit of the F1F0-ATPase is encoded by a nuclear gene in Chlamydomonas reinhardtii. J Biol Chem. 2002;277 (8):6051–6058. doi: 10.1074/jbc.M109993200. [DOI] [PubMed] [Google Scholar]
- Gellissen G, Bradfield JY, White BN, Wyatt GR. Mitochondrial-DNA Sequences in the Nuclear Genome of a Locust. Nature. 1983;301 (5901):631–634. doi: 10.1038/301631a0. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22 (2):160–74. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hazkani-Covo E, Sorek R, Graur D. Evolutionary dynamics of large numts in the human genome: Rarity of independent insertions and abundance of post-insertion duplications. J Mol Evol. 2003;56 (2):169–174. doi: 10.1007/s00239-002-2390-5. [DOI] [PubMed] [Google Scholar]
- Henze K, Martin W. How do mitochondrial genes get into the nucleus? Trends Genet. 2001;17 (7):383–387. doi: 10.1016/s0168-9525(01)02312-5. [DOI] [PubMed] [Google Scholar]
- Herrnstadt C, et al. A novel mitochondrial DNA-like sequence in the human nuclear genome. Genomics. 1999;60 (1):67–77. doi: 10.1006/geno.1999.5907. [DOI] [PubMed] [Google Scholar]
- Hillis DM, Bull JJ. An Empirical-Test of Bootstrapping as a Method for Assessing Confidence in Phylogenetic Analysis. Systematic Biology. 1993;42 (2):182–192. [Google Scholar]
- Johnson WE, Dratch PA, Martenson JS, O’Brien SJ. Resolution of recent radiations within three evolutionary lineages of felidae using mitochondrial restriction fragment length polymorphism variation. J Mammal Evol. 1996;3 (2):97–120. [Google Scholar]
- Johnson WE, Eizirik E, Murphy WJ, Pecon-Slaterry J, Antunes A, Teeling AE, O’Brien SJ. The Disjunct Late Miocene Radiation of the Felidae. (Submitted) [Google Scholar]
- Johnson WE, O’Brien SJ. Phylogenetic reconstruction of the Felidae using 16S rRNA and NADH-5 mitochondrial genes. J Mol Evol. 1997;44 (Suppl 1):S98–S116. doi: 10.1007/pl00000060. [DOI] [PubMed] [Google Scholar]
- Kocher TD, et al. Dynamics of Mitochondrial-DNA Evolution in Animals - Amplification and Sequencing with Conserved Primers. Proc Natl Acad Sci U S A. 1989;86 (16):6196–6200. doi: 10.1073/pnas.86.16.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001;17 (12):1244–1245. doi: 10.1093/bioinformatics/17.12.1244. [DOI] [PubMed] [Google Scholar]
- Ledje C, Arnason U. Phylogenetic relationships within caniform carnivores based on analyses of the mitochondrial 12S rRNA gene. J Mol Evol. 1996;43 (6):641–9. doi: 10.1007/BF02202112. [DOI] [PubMed] [Google Scholar]
- Lewin B. Oxford University Press Inc.; New York: 1994. Genes V. [Google Scholar]
- Li WH, Gojobori T, Nei M. Pseudogenes as a paradigm of neutral evolution. Nature. 1981;292 (5820):237–9. doi: 10.1038/292237a0. [DOI] [PubMed] [Google Scholar]
- Lichter P, et al. High-Resolution Mapping of Human Chromosome-11 by Insitu Hybridization with Cosmid Clones. Science. 1990;247 (4938):64–69. doi: 10.1126/science.2294592. [DOI] [PubMed] [Google Scholar]
- Lopez JV, Cevario S, O’Brien SJ. Complete nucleotide sequences of the domestic cat (Felis catus) mitochondrial genome and a transposed mtDNA tandem repeat (Numt) in the nuclear genome. Genomics. 1996;33 (2):229–246. doi: 10.1006/geno.1996.0188. [DOI] [PubMed] [Google Scholar]
- Lopez JV, Culver M, Stephens JC, Johnson WE, Obrien SJ. Rates of nuclear and cytoplasmic mitochondrial DNA sequence divergence in mammals. Mol Biol Evol. 1997;14 (3):277–286. doi: 10.1093/oxfordjournals.molbev.a025763. [DOI] [PubMed] [Google Scholar]
- Lopez JV, Yuhki N, Masuda R, Modi W, O’Brien SJ. Numt, a recent transfer and Tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J Mol Evol. 1994;39 (2):174–190. doi: 10.1007/BF00163806. [DOI] [PubMed] [Google Scholar]
- Luo SJ, et al. Phylogeography and Genetic Ancestry of Tigers (Panthera tigris) PLoS Biology. 2004;2 (12):e442. doi: 10.1371/journal.pbio.0020442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulis L. Yale University Press; New Haven, CT: 1970. Origin of eukaryotic cells. [Google Scholar]
- Masuda R, Yoshida MC, Shinyashiki F, Bando G. Molecular phylogenetic status of the iriomote cat Felis iriomotensis, inferred from mitochondrial DNA sequence analysis. Zoolog Sci. 1994;11 (4):597–604. [PubMed] [Google Scholar]
- Mishmar D, Ruiz-pesini E, Brandon M, Wallace DC. Mitochondrial DNA-like sequences in the nucleus (NUMTs): Insights into our African origins and the mechanism of foreign DNA integration. Human Mutation. 2004;23 (2):125–133. doi: 10.1002/humu.10304. [DOI] [PubMed] [Google Scholar]
- Modi WS, Nash WG, Ferrari AN, O’Brien SJ. Cytogenetic Methodologies for Gene Mapping and Comparative Analyses in Mammalian Cell Culture Systems. Gene Analysis Techniques. 1987;4 (4):75–85. doi: 10.1016/0735-0651(87)90021-5. [DOI] [PubMed] [Google Scholar]
- Mourier T, Hansen AJ, Willerslev E, Arctander P. The human genome project reveals a continuous, transfer of large mitochondrial fragments to the nucleus. Mol Biol Evol. 2001;18 (9):1833–1837. doi: 10.1093/oxfordjournals.molbev.a003971. [DOI] [PubMed] [Google Scholar]
- O’Brien SJ. Molecular genetics and phylogenetics of the Felidae. In: Nowell K, Jackson P, editors. Status survey and conservation action plan: Wild cats. IUCN; 1996. pp. XXIII–XXIV. [Google Scholar]
- O’Brien SJ, Eizirik E, Murphy WJ. Genomics - On choosing mammalian genomes for sequencing. Science. 2001;292 (5525):2264–2266. doi: 10.1126/science.1059393. [DOI] [PubMed] [Google Scholar]
- Pereira SL, Baker AJ. Low number of mitochondrial pseudogenes in the chicken (Gallus gallus) nuclear genome: implications for molecular inference of population history and phylogenetics. BMC Evol Biol. 2004;4 (1):17. doi: 10.1186/1471-2148-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Martinez X, et al. Unusual location of a mitochondrial gene - Subunit III of cytochrome c oxidase is encoded in the nucleus of chlamydomonad algae. J Bio Chem. 2000;275 (39):30144–30152. doi: 10.1074/jbc.M003940200. [DOI] [PubMed] [Google Scholar]
- Perna NT, Kocher TD. Mitochondrial DNA - Molecular fossils in the nucleus. Current Biology. 1996;6 (2):128–129. doi: 10.1016/s0960-9822(02)00441-4. [DOI] [PubMed] [Google Scholar]
- Pons J, Vogler AP. Complex Pattern of Coalescence and Fast Evolution of a Mitochondrial rRNA Pseudogene in a Recent Radiation of Tiger Beetles. Mol Biol Evol. 2005;22 (4):991–1000. doi: 10.1093/molbev/msi085. [DOI] [PubMed] [Google Scholar]
- Ricchetti M, Tekaia F, Dujon B. Continued colonization of the human genome by mitochondrial DNA. PLoS Biology. 2004;2 (9):1313–1324. doi: 10.1371/journal.pbio.0020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly E, Leister D. NUMTs in sequenced eukaryotic genomes. Mol Biol Evol. 2004;21 (6):1081–1084. doi: 10.1093/molbev/msh110. [DOI] [PubMed] [Google Scholar]
- Roth DB, Porter TN, Wilson JH. Mechanisms of Nonhomologous Recombination in Mammalian-Cells. Mol Cell Biol. 1985;5 (10):2599–2607. doi: 10.1128/mcb.5.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch E, Maniatis T. Cold Spring Harbor Laboratory Press; New York, NY: 1989. Molecular Cloning: a Laboratory Manual. [Google Scholar]
- Schinkel AH, Tabak HF. Mitochondrial Rna-Polymerase - Dual Role in Transcription and Replication. Trends Genet. 1989;5 (5):149–154. doi: 10.1016/0168-9525(89)90056-5. [DOI] [PubMed] [Google Scholar]
- Strimmer K, von Haeseler A. Quartet puzzling: A quartet maximum-likelihood method for reconstructing tree topologies. Mol Biol Evol. 1996;13 (7):964–969. [Google Scholar]
- Swofford DL. Sinauer; Sunderland, MA: 2001. ‘PAUP* Phylogenetic Analysis Using Parsimony and Other Methods’ Computer Program. [Google Scholar]
- Thalmann O, Hebler J, Poinar HN, Paabo S, Vigilant L. Unreliable mtDNA data due to nuclear insertions: a cautionary tale from analysis of humans and other great apes. Mol Ecol. 2004;13 (2):321–335. doi: 10.1046/j.1365-294x.2003.02070.x. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL-X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourmen Y, et al. Structure and chromosomal distribution of human mitochondrial pseudogenes. Genomics. 2002;80 (1):71–77. doi: 10.1006/geno.2002.6798. [DOI] [PubMed] [Google Scholar]
- Vanderkuyl AC, Kuiken CL, Dekker JT, Perizonius WRK, Goudsmit J. Nuclear Counterparts of the Cytoplasmic Mitochondrial 12s Ribosomal-Rna Gene - a Problem of Ancient DNA and Molecular Phylogenies. J Mol Evol. 1995;40 (6):652–657. doi: 10.1007/BF00160513. [DOI] [PubMed] [Google Scholar]
- Woischnik M, Moraes CT. Pattern of organization of human mitochondrial pseudogenes in the nuclear genome. Genome Research. 2002;12 (6):885–893. doi: 10.1101/gr.227202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstenholme DR. Genetic novelties in mitochondrial genomes of multicellular animals. Curr Opin Genet Dev. 1992;2 (6):918–25. doi: 10.1016/s0959-437x(05)80116-9. [DOI] [PubMed] [Google Scholar]
- Yang D, Oyaizu Y, Oyaizu H, Olsen GJ, Woese CR. Mitochondrial Origins. Proc Natl Acad Sci U S A. 1985;82 (13):4443–4447. doi: 10.1073/pnas.82.13.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Computer Applications in the Biosciences. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Kumar S. Approximate methods for estimating the pattern of nucleotide substitution and the variation of substitution rates among sites. Mol Biol Evol. 1996;13 (5):650–659. doi: 10.1093/oxfordjournals.molbev.a025625. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Hewitt GM. Nuclear integrations: Challenges for mitochondrial DNA markers. Trends Ecol Evol. 1996a;11 (6):247–251. doi: 10.1016/0169-5347(96)10031-8. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Hewitt GM. Highly conserved nuclear copies of the mitochondrial control region in the desert locust Schistocerca gregaria: Some implications for population studies. Mol Ecol. 1996b;5 (2):295–300. doi: 10.1111/j.1365-294x.1996.tb00317.x. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]