

Abstract
Development of new therapeutic agents for colon cancer is highly desirable. To this end, we screened a chemical library for new anticancer agents and identified a synthetic compound, 5-(2,4-dihydroxybenzylidene)-2-(phenylimino)-1,3-thiazolidin (DBPT), that kills cancer cells more effectively than it kills normal human fibroblasts. The molecular mechanism of the antitumor action of DBPT was further analyzed in three human colorectal cancer cell lines. DBPT effectively inhibited the growth of colorectal cancer cells, independent of p53 and P-glycoprotein status, whereas normal fibroblasts were unaffected at the same 50% inhibitory concentration. Over time, DLD-1 cancer cells treated with DBPT underwent apoptosis. The general caspase inhibitor z-VAD-fmk partially blocked DBPT-induced apoptosis in a dose-dependent manner. DBPT-induced apoptosis, including cytochrome c release and caspase activation, was abrogated when JNK activation was blocked with either a specific JNK inhibitor or a dominant-negative JNK1 gene. However, constitutive JNK activation alone did not replicate the effects of DBPT in DLD-1 cells, and excessive JNK activation by adenovirus encoding MKK7 had little influence on DBPT-induced apoptosis. Our results suggested that DBPT induces apoptosis in colorectal cancer cell lines through caspase-dependent and caspase-independent pathways and that JNK activation was crucial for DBPT-induced apoptosis. DBPT and its analogs might be useful as anticancer agents.
Keywords: JNK, apoptosis, colon cancer, anticancer agent, caspase
Abbreviations used in this paper: DBPT, 5-(2; 4-dihydroxybenzylidene)-2-(phenylimino)-1, 3-thiazolidin; PARP, poly(ADP-ribose) polymerase; ERK, extracellular signal-regulated kinase; HA, hemagglutinin; IC, inhibitory concentration; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; NHFB, normal human fibroblast; P-gp, P-glycoprotein; PI, propidium iodide; XTT, 2, 3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide inner salt
Introduction
Use of chemotherapeutic agents has shown to improve the outcomes of patients with advanced colorectal cancer (1, 2). Moreover, numerous new antineoplastic agents have been shown to have anticancer activity, either alone or in combination with other anticancer agents (3–5). However, for most cancer patients, currently available therapies have been only temporarily successful because they frequently lead to resistance or unacceptable levels of toxicity. Consequently, colorectal cancer is one of the leading causes of cancer-related death worldwide (6). It is therefore important to develop alternative therapeutic agents with improved efficacy and tolerability. Thus, agents that induce apoptosis selectively in cancer cells are highly desirable.
c-Jun N-terminal kinase (JNK) is an important mediator of apoptotic signaling (7). It is a member of the mitogen-activated protein kinase (MAPK) family, which also includes extracellular signal-regulated kinase (ERK) and p38 (8). JNK has at least 10 isoforms that are encoded by three genes, JNK1, JNK2, and JNK3 (9). JNK1 and JNK2 are expressed in various tissues and play crucial roles in many cellular events, including growth control, development, and apoptosis (10), whereas JNK3 is expressed only in neuronal tissue and is essential for stress-induced neuronal cell death (11). JNK activation is required for induction of apoptosis by a number of stress stimuli such as ultraviolet radiation, growth factor withdrawal, inflammatory cytokines, and chemotherapeutic agents (12–15). The exact molecular mechanism by which JNK mediates apoptotic signals remains unclear, although several recent reports have shown that JNK activation is required for stress-induced release of mitochondrial cytochrome c or Smac/DIABLO and for apoptosis mediated by the mitochondrial caspase-9 pathway (16–19).
To develop new anticancer agents that are cytotoxic to cancer cells but not normal cells, we screened a chemical library from Chembridge, Inc. (San Diego, CA) and identified a synthetic compound, 5-(2,4-dihydroxybenzylidene)-2-(phenylimino)-1,3-thiazolidin (DBPT), that kills cancer cells more effectively than it kills normal human fibroblasts (NHFBs). The molecular mechanism of DBPT’s cytotoxic effect was further characterized on human colorectal cancer cells. We found that DBPT induced JNK-mediated apoptosis in colon cancer cells through caspase-dependent and caspase-independent pathways. Our results also suggest that DBPT and its analogs might be useful as anticancer agents.
Materials and Methods
Cells and Culture Conditions
The human colon cancer cell lines DLD-1, LoVo and HCT116 (p53 wild-type and p53−/−; generously provided by Dr. Bert Vogelstein, The Johns Hopkins University, Baltimore, MD) (20) were routinely cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 100 units/ml penicillin, and 100 mg/ml streptomycin. NHFBs were maintained in Dulbecco’s modified Eagle medium with the same supplements. All cells were maintained in the presence of 5% CO2 at 37° C.
Chemicals and Antibodies
A chemical library with 10,000 compounds, DBPT, and DBPT analog MAPT (2-[(4-methylphenyl)amino]-5-(phenylmethylene)-4(5H)-thiazolone) were obtained from ChemBridge (San Diego, CA). The chemical structures of DBPT and MAPT are shown in Figure 1. The compounds were dissolved in dimethyl sulfoxide to a concentration of 10 mM and stored at 4° C as a master stock solution. The JNK-specific inhibitor SP600125, the ERK inhibitor PD98059, and the p38 inhibitor SB202190 were purchased from Calbiochem (La Jolla, CA), dissolved in dimethyl sulfoxide, stored at −20° C, and protected from light. The general caspase inhibitor z-VAD-fmk was obtained from R&D Systems (Minneapolis, MN). Antibodies to the following proteins were used for western blot analysis: caspase-3 and P-glycoprotein (P-gp) (Santa Cruz Biotechnology, Santa Cruz, CA); caspase-8 (MBL International, Woburn, MA); COX-4, poly(ADP-ribose) polymerase (PARP) and cytochrome c (BD PharMingen, San Diego, CA); JNK, phosphorylated JNK (p-JNK), ERK, phosphorylated ERK (p-ERK), p38, phosphorylated p38 (p-p38), phosphorylated c-Jun (p-c-Jun), and caspase-9 (Cell Signaling, Beverly, MA); and hemagglutinin (HA) and β-actin (Sigma, St. Louis, MO).
Figure 1.
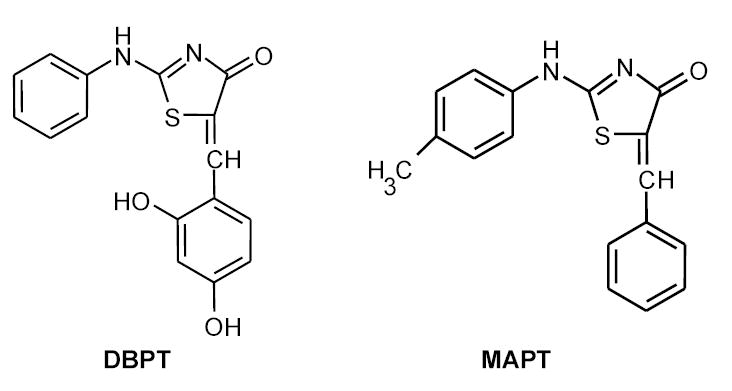
Chemical structure of 5-(2,4-dihydroxybenzylidene)-2-(phenylimino)-1,3-thiazolidin (DBPT) and its analog 2-[(4-methylphenyl)amino]-5-(phenylmethylene)- 4(5H)-thiazolone (MAPT).
Cell Proliferation Assay
The antiproliferative effects of DBPT on various cancer cell lines and NHFBs were examined using cell proliferation assays. Cells were seeded in 96-well flat-bottomed plates (5–10 × 103 in 100 μl of culture medium per well) and treated the next day with the indicated concentrations of compounds. An equal volume of dimethyl sulfoxide was used as a control. Cell viability was determined 72 h later by XTT assay using a Cell Proliferation Kit II (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer’s protocol. The experiments were performed at least three times for each cell line. The concentrations of DBPT that inhibited absorbance by 50% and 80% (IC50 and IC80, respectively) were calculated by using the CurveExpert Version 1.3 program.
Apoptosis Assay
For detection of apoptosis, fixed cells were suspended in phosphate-buffered saline containing 10 μg/ml propidium iodide (PI) (Roche Diagnostics, Indianapolis, IN) and 10 μg/ml RNase A (Sigma-Aldrich) at 37°C for 30 min. Cell-cycle analysis was performed using an epics Profile II flow cytometer (Beckman Coulter, Fullerton, CA) with MultiCycle software (Phoenix Flow Systems, San Diego, CA). Accumulation of sub-G1 cells, a known indicator of DNA fragmentation and apoptosis, was used to quantify apoptosis. The percentage of cells undergoing apoptosis was also determined by annexin V staining by using the Annexin V-FITC Apoptosis Detection Kit (PharMingen). Stained cells were analyzed using the flow cytometer and software described above. All experiments were repeated at least three times.
Plasmid Transfection and Adenovirus Vector Transduction
The plasmids pLNCX-3X HA-p46JNK1alpha (dnJNK1; encodes an HA-tagged, dominant-negative JNK1 mutant) and pLNCX-3X HA-p54JNK2alpha (dnJNK2; encodes an HA-tagged, dominant-negative JNK2 mutant) (21) were provided by Dr. L. E. Heasley (University of Colorado Health Sciences Center, Denver, CO). Plasmid transfection was performed using FuGENE6 reagent (Roche Diagnostics), and cells were selected for growth in the presence of 500 μg/ml G418. The adenoviral vector AdMKK7DN, which encodes a dominant-negative, constitutively active MKK7 mutant, was provided by Dr. Yibin Wang (University of California, Los Angeles, CA) (22). The vector encoding green fluorescence protein (AdGFP) has been reported previously (23).
Western Blot Analysis
For preparation of whole-cell extracts, cells were washed twice in cold phosphate-buffered saline, collected, and lysed in lysis buffer (62.5 mM Tris [pH 6.8], 2% SDS, and 10% glycerol) containing 1 × proteinase-inhibitor cocktail (Roche Diagnostics). The lysates were spun at 14,000 × g in a microcentrifuge at 4° C for 10 min., and the resulting supernatants were used as whole-cell extracts. For preparation of cytosolic and mitochondrial extracts, cells were washed twice in cold phosphate-buffered saline, collected, and lysed in lysis buffer from the ApoAlert Cell Fractionation Kit (Clontech, Palo Alto, CA). Cytosolic and mitochondrial extracts were isolated according to the manufacturer’s protocol. Protein concentrations were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL). Equal amounts (30–50 μg) of proteins were used for immunoblotting as described previously (24).
Statistical Analysis
Differences among the treatment groups were assessed by analysis of variance using StatSoft statistical software (Tulsa, OK). P< 0.05 was considered significant.
Results
DBPT Inhibited Proliferation of Human Colon Cancer Cells
In a primary screen of 10,000 compounds, we tested cytotoxic effects of these compounds on human colon cancer cell lines DLD1 and LoVo, human lung cancer cell lines H1299 and H460 and normal human fibroblasts. Cells seeded in 96 well plates were treated with each compound at a final concentration of about 5 μg/ml. Cells treated with solvent (DMSO) were used as controls. Cytotoxic effects were then determined by observation under microscope and by cell viability assay. This screening led us to identify DBPT as an agent that had cytotoxic effects in the four cancer cell lines tested and but not in NHFBs. We then treated human colon cancer cells with DBPT at various concentrations for 72 h and examined its effect on cell viability using XTT assays. DBPT effectively inhibited the growth of DLD-1 and HCT116 cells with IC50 values ranging from 1.6 to 5.9 μM (Table 1). This cytotoxic effect is p53-independent because DBPT inhibited the growth of both wild-type and p53-deficient HCT116 cells.
Table 1.
IC50 and IC80 for DBPT in human colorectal cancer cell lines and NHFBs
|
DBPT concentration |
|||
|---|---|---|---|
| Cell line | P-gp status | IC50(μM) | IC80(μM) |
| DLD-1 | +++ | 1.6 ± 0.3 | 2.7 ± 0.3 |
| HCT116 (p53+) | ± | 4.9 ± 0.3 | ND |
| HCT116 (p53−) | ± | 5.9 ± 0.3 | ND |
| NHFB | ND | 22.9 ± 7.2 | ND |
Cells were grown in the presence of DBPT for 72 h, and cell viability was determined by XTT assay. P-gp levels were evaluated by western blot analysis. p53+, wild type; p53−, p53 deficient; ND, not determined. Each value in the DBPT columns represents the mean ± SD of three independent experiments.
In addition, we measured the expression level of P-gp, a product of the human multidrug resistance MDR1 gene (25), in three colon cancer cell lines (Table 1). The amount of P-gp in DLD-1 cells was markedly higher than in HCT116 cells, but the sensitivity of DLD-1 cells to DBPT was also much higher than that of HCT116 cells, indicating that the activity of DBPT in colon cancer cells was not affected by P-gp status.
We also evaluated the dose-response effect of DBPT in NHFB cells (Table 1). The IC50 in NHFB cells was 4 to 14 times higher than in cancer cells, indicating that DBPT may selectively induce cytotoxic effects in cancer cells. Interestingly, MAPT, a DBPT analog with similar chemical structure (Fig. 1), did not induce any cytotoxic effects in any of the cells tested, even at 31 μM, the highest concentration tested (data not shown).
DBPT Induced Apoptosis and Caspase Activation in Colon Cancer Cells
To elucidate the mechanisms by which DBPT induces cytotoxicity, we used flow cytometry to analyze apoptosis in DLD-1 and HCT116 cells after DBPT treatment (Fig. 2A). DBPT induced a marked, time-dependent increase in the percentage of annexin V-positive DLD-1 and HCT116 cells, with peak values of 65.2% and 44.5% 48h after treatment with 3 μM and 5 μM DBPT, respectively. To further examine the ability of DBPT to induce apoptosis, we treated DLD-1 cells with 3 μM DBPT for 6, 12, 24, or 48 h and evaluated caspase activation by western blot analysis (Fig. 2B). Cleavage of caspase-3, caspase-8, caspase-9, and PARP was easily detectable after 48 h.
Figure 2.
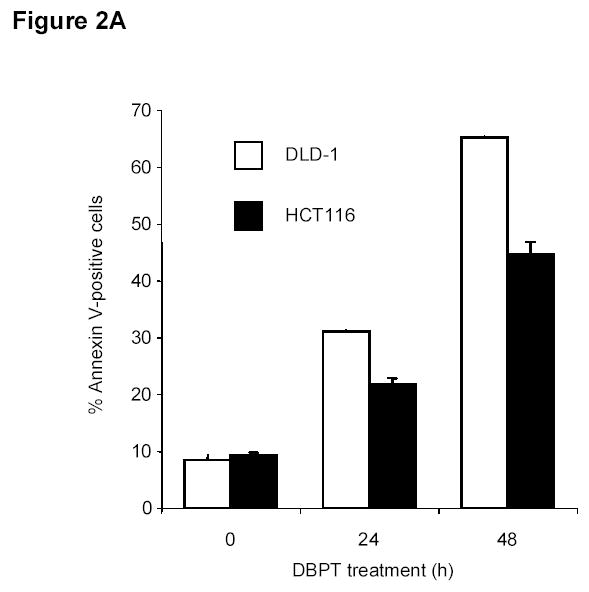
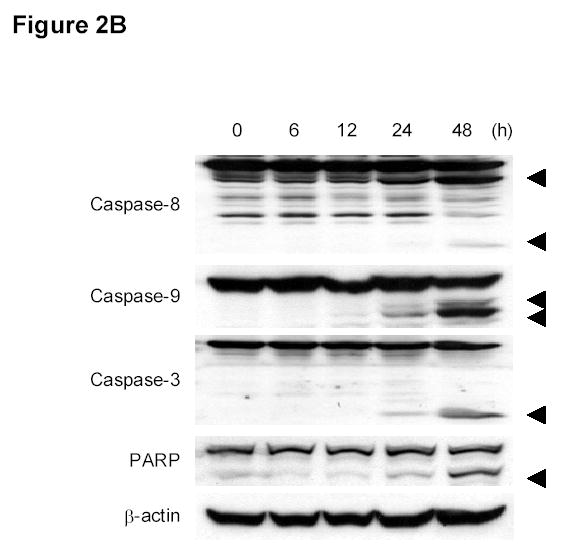
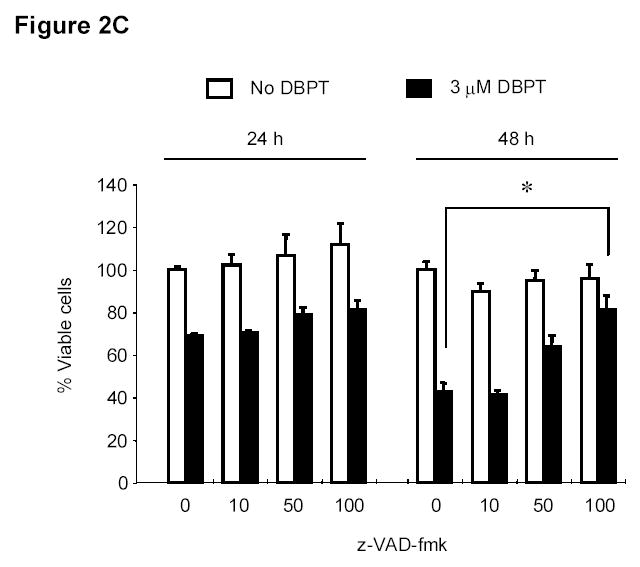
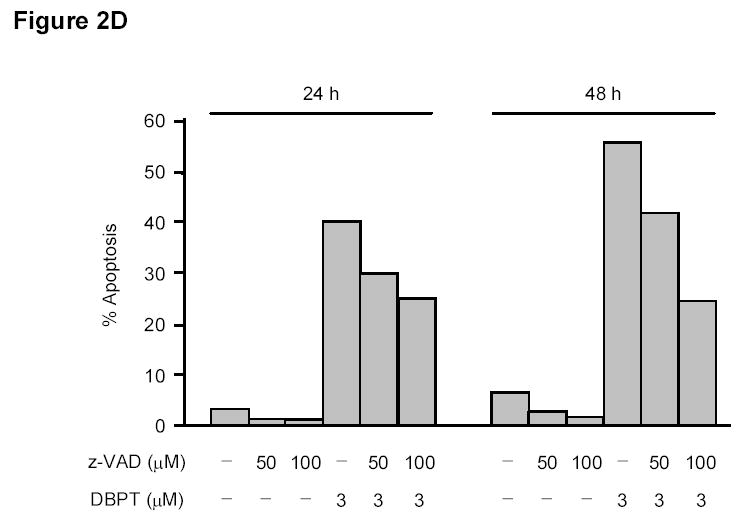
DBPT induced apoptosis in colon cancer cells by activating caspases. A, DLD-1 and wild-type HCT116 cells were treated with 3 and 5 μM DBPT, respectively, for the indicated times and stained with fluorescein isothiocyanate-labeled annexin V and PI. Stained cells were analyzed by flow cytometry to determine the apoptotic ratio. Data represent mean ± SD of three independent experiments. B, DLD-1 cells were treated with 3 μM DBPT for the indicated time periods, and whole-cell extracts were analyzed by western blotting. Activation of caspases and PARP were detected 24 and 48 h after DBPT treatment. Arrowheads represent cleavage proteins. β-actin was used as a loading control. C, DLD-1 cells were pretreated with 10, 50, or 100 μM z-VAD-fmk for 30 min and treated with DBPT for 24 or 48 h. Cell growth inhibition was determined by XTT assay. Data represent mean ± SD of three independent experiments. *, P< 0.01 compared with DBPT treatment alone. D, DLD-1 cells were pretreated with 50 or 100 μM z-VAD-fmk for 30 min and treated with DBPT for 24 or 48 h. The percentage of cells in sub-G1 phase was determined by flow cytometry.
We next investigated whether inhibition of caspase activation affects DBPT-induced apoptosis. DLD-1 cells were pretreated with the general caspase inhibitor z-VAD-fmk at 10, 50, or 100 μM for 30 min and then treated with DBPT for another 24 to 48 h. The cells were analyzed for viability by XTT assay and for apoptosis by flow cytometry (Fig. 2C and D). Pretreatment with z-VAD-fmk blocked DBPT-mediated cell growth inhibition in a dose-dependent manner. The most significant difference in cell viability was observed between cells treated with DBPT alone (42.6%) and those with 100 μM z-VAD-fmk plus DBPT for 48h (81.3%) (P< 0.01, Figure 2C). Consistently, pretreatment with z-VAD-fmk markedly diminished the DBPT mediated apoptotic (Sub-G1) cells (Fig. 2D). These results indicate that DBPT-induced apoptosis in DLD-1 cells was related to caspase activation, although z-VAD-fmk did not completely block apoptosis.
DBPT Activated MAPK Signaling
Many cellular stresses and stimuli induce apoptosis and modulate MAPK signaling pathways (26, 27). However, the role of these pathways in cell death is not completely understood. We examined the activation of the MAPKs JNK, ERK, and p38 after DBPT treatment (Fig. 3). Lysates of DBPT-treated cells were analyzed by western blotting for MAPK phosphorylation, a hallmark of MAPK activation. Control cells were treated with MAPT. Treatment of DLD-1 cells with DBPT increased the amounts of phosphorylated JNK and p38, which were easily detectable at 24 and 48 h after the treatment. In contrast, treatment with MAPT did not result in detectable activation of JNK and p38. Phosphorylated ERK was slightly decreased at the early time points (6 – 24 h) but increased at 48 h after DBPT treatment. Phosphorylated ERK in MAPT treated cells were only temporarily reduced (at 24 h). There was no obvious difference in phosphorylated ERK levels in cells 48 h after treatment with either DBPT or MAPT.
Figure 3.
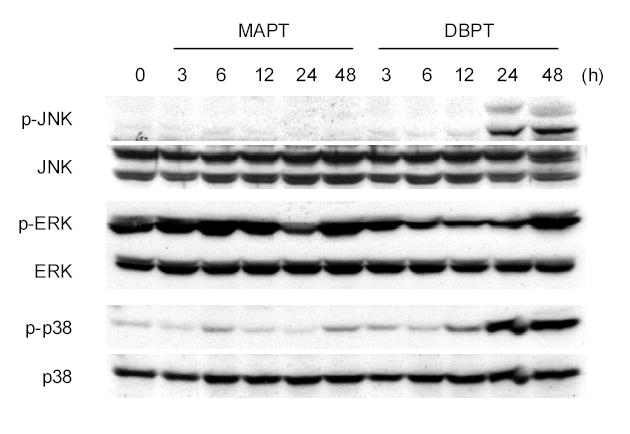
DBPT induces MAPK activity. DLD-1 cells were treated with 3 μM DBPT or MAPT for the indicated times. Whole-cell lysates were analyzed for total and active (phosphorylated) JNK, ERK, and p38 by western blot analysis.
JNK Inhibitor SP600125 Blocked DBPT-Induced Apoptosis
To further investigate the role of MAPK activation in DBPT-induced apoptosis, we treated DLD-1 cells with DBPT in the presence or absence of the p38 inhibitor SB202190 (28, 29), ERK-specific inhibitor PD98059, or JNK inhibitor SP600125 (30) and analyzed the sub-G1 population by flow cytometry. Western blot analysis showed that pretreatment with SP600125 and PD98059 diminished DBPT-induced activation of JNK and ERK, respectively. SB202190, an inhibitor that is specific for the α and β isoforms of p38 and does not inhibit p38 phosphorylation but inhibits p38 kinase activity, had little effect on p38 phosphorylation as previously reported by others (28, 29). Interestingly, pretreatment with JNK inhibitor SP600125 completely blocked DBPT-induced apoptosis (Fig. 4B). The percentages of apoptotic cells after treatment with 50 μM SP600125, 3 μM DBPT, or both were 7%, 38%, and 4%, respectively. The difference between DBPT alone and DBPT plus SP600125 was significant (P< 0.001). In contrast, the p38 inhibitor SB202190 and ERK inhibitor PD98059 had either no effects or only moderate effects on DBPT-induced apoptosis. Apoptotic cells after treatment with DBPT alone, DBPT plus SB202190, and DBPT plus PD98059 were 38%, 42% and 27%, respectively.
Figure 4.
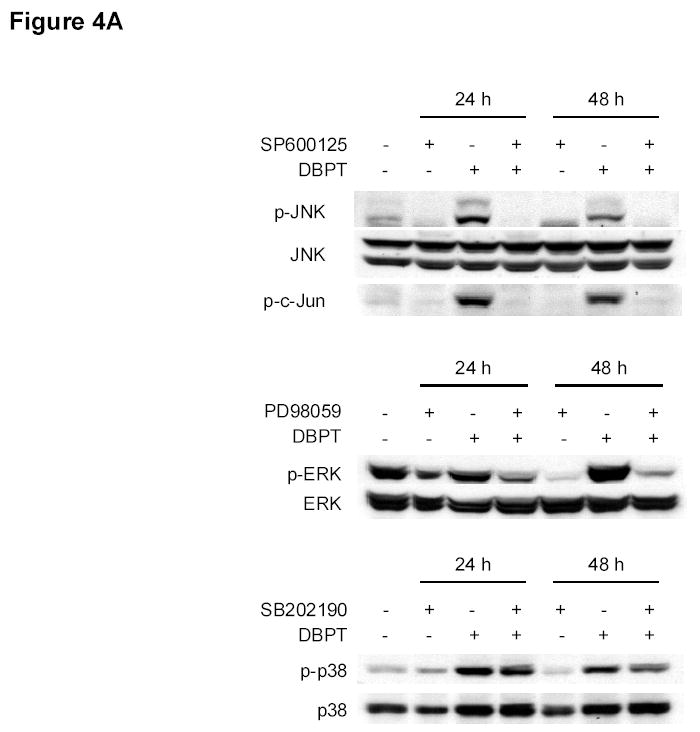
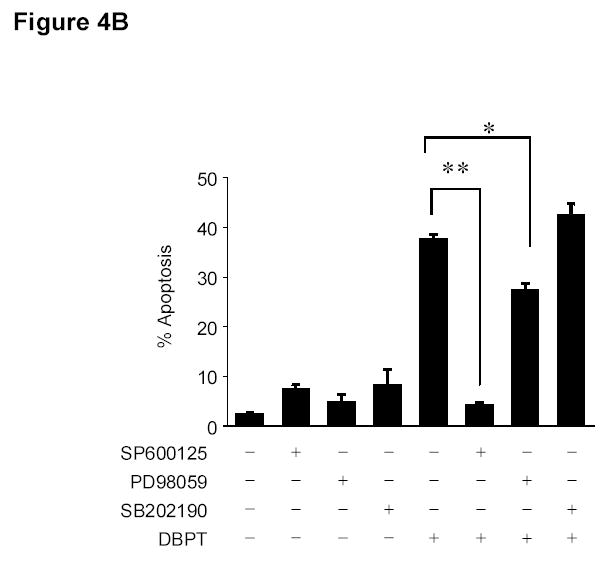
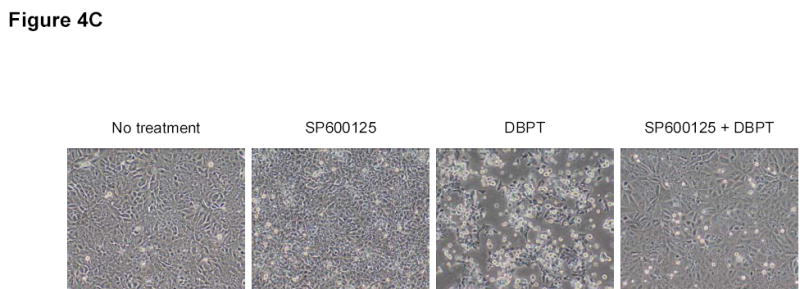
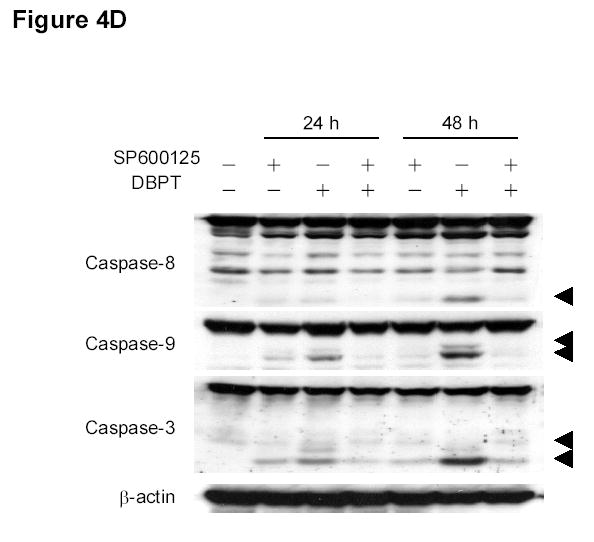
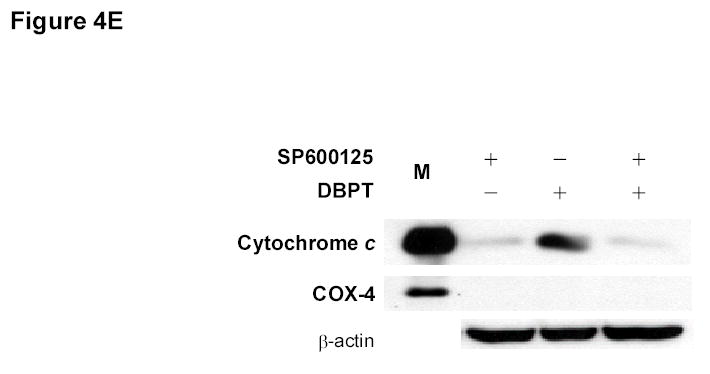
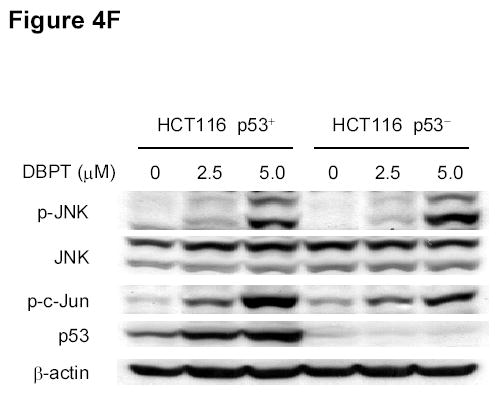
Effect of MAPK inhibitors on DBPT-induced apoptosis. A, DLD-1 cells were treated with 3 μM DBPT in the presence or absence of 50 μM PD98059 (ERK inhibitor), 40 μM SB202190 (p38 inhibitor), or 50 μM SP600125 (JNK inhibitor) for 24 or 48 h. Whole-cell lysates were analyzed for JNK, c-Jun, ERK, and p38 activation by immunoblotting. B, Apoptotic cells after treatment as described in A. Data represent mean ± SD from three independent experiments performed in triplicate. *, P< 0.01, **, P< 0.001 among the indicated groups. C, phase-contrast photomicrographs of DLD-1 cells treated with 50 μM SP600125, 3 μM DBPT or both for 24 h. Magnification, × 100. D, Dection of caspase activation by immunoblotting. DLD-1 cells were treated with DBPT in the presence or absence of SP600125 for 24 or 48 h. Whole-cell lysates were analyzed for caspase activation. β-actin was used as a loading control. Arrowheads indicate cleavage proteins. E, DLD-1 cells were treated with DBPT in the presence or absence of SP600125 for 24 h. Cytosolic fractions were analyzed by immunoblotting with anti-cytochrome c. Mitochondrial fractions (M) were used as a positive control. COX-4 and β-actin were used as a loading controls for mitochondrial and cytosolic fractions. F, wild-type (p53+) and p53-deficient (p53−) HCT116 cells were treated with 2.5 or 5 μM DBPT for 24 h. Cell extracts were analyzed for the indicated proteins by western blotting.
The suppression of DBPT-induced apoptosis by the JNK inhibitor was also obvious when observed under microscope. DLD-1 cells treated with DBPT alone showed the typical morphological changes of cytopathies, and became rounded and detached from the plate (Fig. 4C). Those changes were abrogated by the presence of SP600125. SP600125 pretreatment also reduced DBPT-mediated cleavage of caspase-8, caspase-9, and caspase-3 (Fig. 4D). Additionally, DBPT-mediated release of cytochrome c from mitochondria was substantially attenuated in the presence of SP600125 (Fig. 4E). These results show that DBPT-mediated caspase activation and cytochrome c release were JNK dependent and that JNK activation had a key role in DBPT-mediated apoptosis.
To determine whether DBPT-induced JNK activation was associated with p53 status, we treated wild-type and p53-deficient HCT116 cells with DBPT at two different concentrations and monitored the expression of p-JNK and p-c-Jun. DBPT activated JNK and c-Jun after 24 h in both cell lines, suggesting that DBPT-mediated JNK activation was independent of p53 (Fig. 4F). Interestingly, treatment with DBPT also led to the accumulation of p53 in HCT116 (p53 +/+) cells. Whether this accumulation of p53 also correlates with JNK activation is not clear.
Suppression of DBPT-Induced JNK Activation and Apoptosis by Dominant-Negative JNK1
To further investigate the role of JNK activation in DBPT-induced apoptosis, we stably transfected DLD-1 cells with plasmid expressing either of the dominant-negative mutant dnJNK1 or dnJNK2 (Fig. 5A), treated the cells with 3 μM DBPT, and analyzed cell viability and apoptosis. The expression of dnJNK1 in DLD-1 cells markedly blocked DBPT-induced growth inhibition after 24 h (P< 0.01) and 48 h (P< 0.001). In contrast, the expression of dnJNK2 had no effect at 24 h, but enhanced DBPT-induced cell death at 48 h when compared with the same treatment for empty vector-transfected control cells (P< 0.01) (Fig. 5B). Flow cytometric analysis of cells treated with 3 μM DBPT for 24 h showed that DBPT-mediated apoptosis was markedly reduced in two dnJNK1 transfectants (P< 0.01) but not in any dnJNK2 transfectants as compared with an empty control vector (Fig. 5C). We also examined the JNK activation in dnJNK1 or dnJNK2 transfectants treated with DBPT for 24 and 48 h. Western blot analysis showed that dnJNK1 but not dnJNK2 suppressed the DBPT-mediated JNK activation (Fig. 5D). Similarly, dnJNK1 but not dnJNK2 inhibited DBPT-mediated caspase-3 cleavage. Those results indicate that JNK1 activation is crucial for DBPT-mediated apoptosis and that different JNK isoforms (or their respective genes) may have different roles in DBPT-induced apoptosis.
Figure 5.
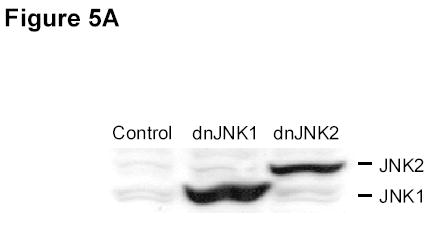
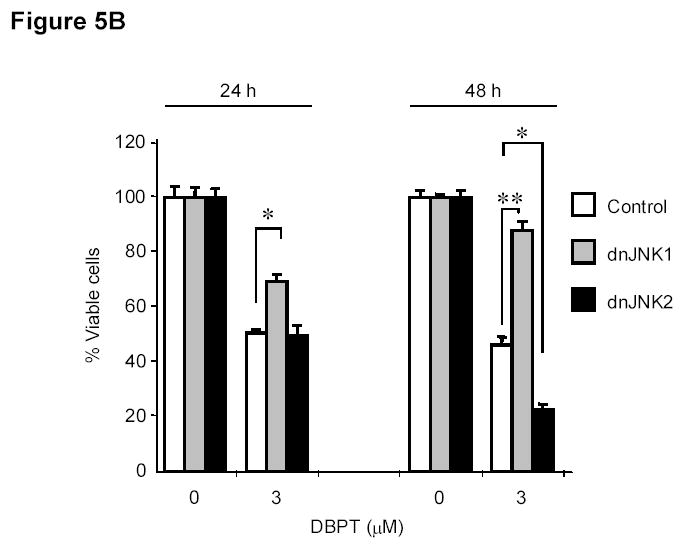
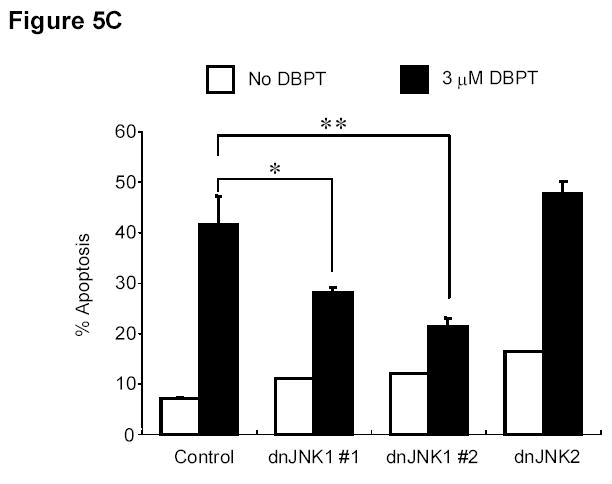
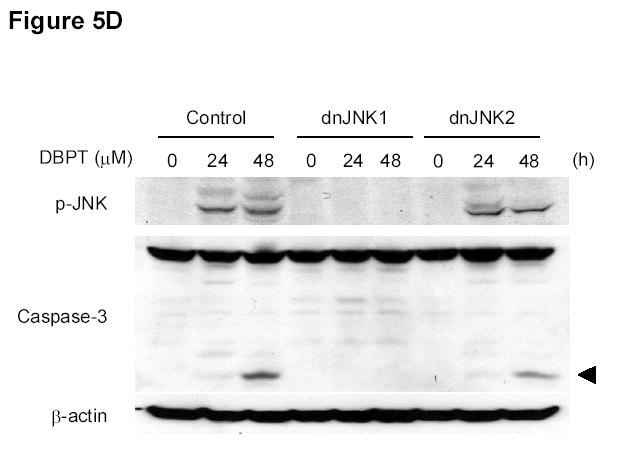
JNK1 activation has a crucial role in DBPT-induced apoptosis. A, DLD-1 cells transfected with dnJNK1 or dnJNK2 plasmids were analyzed by western blot with anti-HA antibodies. DLD-1 cells stably transfected with dnJNK1, dnJNK2 or an empty vector (control) were treated with 3 μM DBPT for 24 or 48 h. Cell viability was determined by XTT assay (B), and apoptotic ratio was determined by flow cytometry (C). Data represent mean ± SD of three independent experiments. *, P< 0.01, **, P< 0.001 among the indicated groups. D, DLD-1 cells stably transfected with dnJNK1 or dnJNK2 were treated with 3 μM DBPT for 24 and 48 h. Whole-cell lysates were analyzed for indicated antibodies by western blot analysis.
Constitutive JNK Activation Alone Did Not Replicate the Effects of DBPT
To determine whether ectopic JNK activation can itself affect cell proliferation, we treated DLD-1 cells with an expression vector encoding MKK7, a kinase upstream of JNK. Treatment with the AdMKK7DN vector activated JNK and c-Jun, but not ERK and p38, after 24 and 48 h (Fig. 6A). An XTT assay revealed that AdMKK7DN did not inhibit the growth of DLD-1 cells after 24 and 48 h compared with the control vector AdGFP (Fig. 6B). To investigate whether constitutive JNK activation by AdMKK7DN will have effect on DBPT-induced apoptosis, DLD-1 cells were treated with DBPT in the presence or absence of AdMKK7DN and apoptosis was determined by flow cytometry assay. The results showed that activation of JNK by AdMKK7DN moderately enhanced DBPT-induced apoptosis at 24 h (P< 0.01). However, this effect was not observed at 48 h (Fig. 6C), indicating that excessive activation of JNK had only minor impact on DBPT-induced apoptosis.
Figure 6.
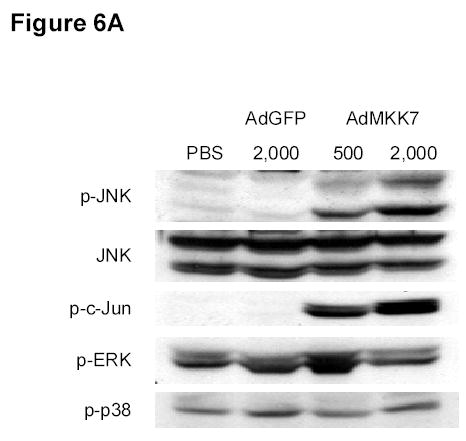
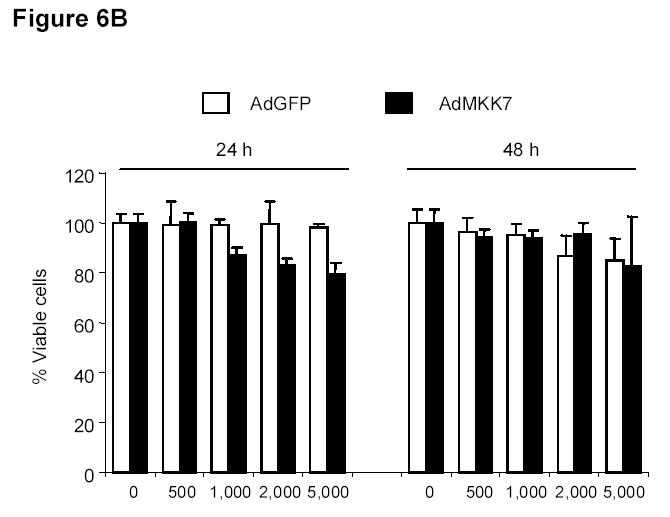
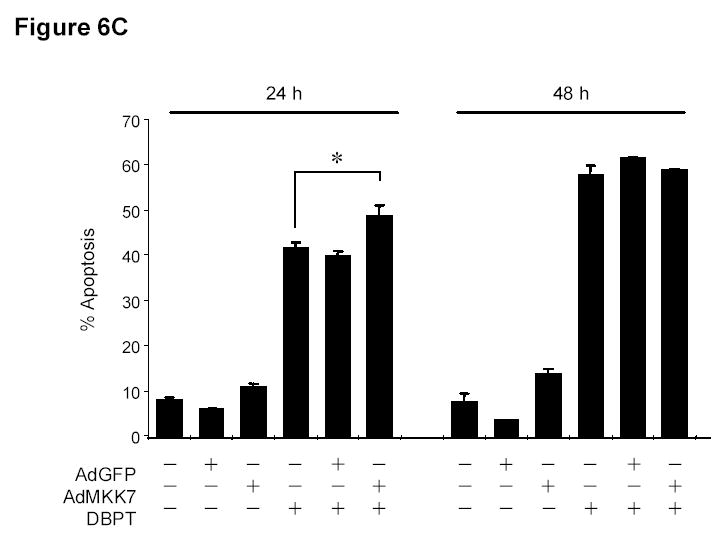
JNK activation alone has no effect on cell proliferation. A, DLD-1 cells were treated with either AdGFP or AdMKK7DN at the indicated multiplicity of infection (MOI) for 24 h, and cell lysates were subjected to western blot analysis for the indicated proteins. Lysate from cells treated phosphate-buffered saline (PBS) was used as a control. B, DLD-1 cells were treated with AdGFP or AdMKK7DN at the indicated MOI for 24 or 48 h. The effect of JNK activation on cell growth inhibition was determined by XTT assay. Data represent mean ± SD of three independent experiments. C, DLD-1 cells were pretreated with AdGFP or AdMKK7DN at 2000 MOI for 24 h and then treated with 3 μM DBPT for additional 24 and 48 h. The apoptotic ratio of stained cells with PI was determined by flow cytometry. Data represent mean ± SD from three independent experiments performed in triplicate.
Discussion
We identified a synthetic compound that effectively inhibited the growth of three human colorectal cancer cell lines in vitro. DBPT had little effect on the growth of NHFBs at the IC80 values used for colorectal cancer cells, suggesting that DBPT might selectively induce cytotoxicity in human colorectal cancer cells. Although p53 has a crucial role in cell cycle regulation and apoptosis induction in various types of cancer cells after treatment with chemotherapeutic agents (31, 32), DBPT effectively inhibited cell growth in both wild-type and p53-deficient HCT116 cells and p53-mutant DLD-1 cells, suggesting that DBPT-induced cell growth inhibition does not depend on p53 status. However, DBPT can induce accumulation of p53 in p53 wt HCT116 cells. Thus, it is still possible that p53 may have a contribution to DBPT-mediated cytotoxic effects in p53-positive cells.
In addition, our data showed that DLD-1 cells express moderate levels of P-gp, a glycoprotein that confers multi-drug resistance in some cell lines (Table 1). Consistently, previous report has shown that the level of P-gp expression in DLD-1 cells was higher than that of other cancer cell lines and were relatively resistant to paclitaxel in vitro (33). Notably, DLD-1 cells were more sensitive to DBPT than were HCT116 cells, which have no detectable P-glycoprotein. Furthermore, the growth-inhibitory effect of DBPT on P-glycoprotein-overexpressing cells was comparable to the effect on parental cells that did not express P-glycoprotein (data not shown). These results indicate that the effect of DBPT was not affected by P-glycoprotein status and that DBPT might not be a substrate of P-glycoprotein. DBPT may thus be a potent treatment for patients who do not respond to paclitaxel therapy because of P-glycoprotein overexpression.
Although most antineoplastic agents induce apoptosis in cancer cells, the mechanisms by which they do so remain unclear. In this study, we showed that caspase activation was involved in DBPT-induced apoptosis, as is the case with numerous chemotherapeutic agents (34–38). Caspase-3, caspase-8, and caspase-9 were activated by DBPT in DLD-1 cells, and the general caspase inhibitor z-VAD-fmk blocked DBPT-induced apoptosis in a dose-dependent manner. However, even treatment with up to 100 μM z-VAD-fmk did not completely block DBPT-induced apoptosis, indicating that it involved both caspase-dependent and caspase-independent pathways.
Our data also showed that JNK activation was required for DBPT-induced apoptosis. JNK has a wide variety of biological functions, including acceleration of cell proliferation and transduction of survival signals under some stress conditions (17). Nevertheless, many reports have focused on the roles of JNK in the regulation of apoptosis when cells are exposed to DNA damage, chemotherapeutic agents, or cytokines (12–14). The mechanism by which JNK becomes activated in response to apoptotic signaling is not completely understood. In this study, we found that DBPT-induced apoptosis, including cytochrome c release and caspase activation, was abrogated when cells were pretreated with either the specific JNK inhibitor SP600125 or a dominant-negative JNK1 gene. This result is consistent with previous reports that activated JNK regulates phosphorylation of mitochondrial proteins during apoptosis and induces apoptosis through a mitochondrial pathway (16, 19, 39). Interestingly, DBPT-induced cell death was slightly enhanced in dominant-negative JNK2 transfected cells, indicating different roles for JNK1 and JNK2 in DBPT-induced apoptosis. This finding was not completely unexpected because it has also been reported that JNK1 is involved in mediating apoptosis in several cancer cells treated with UV radiation or microtubule-interacting agents (40–42), whereas JNK2 has a more specific role in cell survival signaling (21, 43–45).
A recent report has shown that p53-dependent JNK activity might be a general response to chemotherapeutic agents (46). However, it has also been shown that p53 does not appear to be required for JNK-induced apoptosis (47). In our study, JNK activation by DBPT was independent of p53 status in HCT116 cells. Moreover, we found that activation of JNK signaling alone by an MKK7 expression vector was not sufficient to induce cell death in DLD-1 cells. This is consistent with a recent report that expression of a constitutively active JNK kinase-2/JNK1 fusion protein does not induce apoptosis in fibroblasts (48). Our result also showed that excessive activation of JNK by an MKK7 expression vector had little impact on DBPT-induced apoptosis. Together, these data suggest that JNK activation is required but not sufficient to account for DBPT-mediated cytotoxic effect. Other cellular events or apoptosis signaling must be present in addition to JNK activation.
The tumor-selective cytotoxic effect induced by DBPT might be useful in cancer therapy. However, the compound’s mechanism of selectivity and initial targets are not yet known. The selective cell killing observed may have been the result of cellular differences in drug uptake and metabolism or biochemical and physiological differences in activation, transduction, and duration of various signaling pathways. To determine whether DBPT and its analogs can be used to treat human colorectal cancers, we will need to further characterize the compounds’ molecular mechanisms and screen for analogs with better efficacy and selectivity and acceptable in vivo pharmacokinetic properties. Our identification and initial characterization of apoptosis induction by DBPT is only the first step in developing new anticancer agents.
Acknowledgments
We thank Pierrette Lo for editorial review, and Karen M. Ramirez for technical assistance with flow cytometry analysis. This article represents partial fulfillment of the requirements for the Ph.D. degree for J.J.D.
Footnotes
Supported in part by National Cancer Institute grants RO1 CA092487-01A1 and RO1 CA08582-01A1 (to B. F.) and core grant CA16672.
References
- 1.Benson AB, III, Schrag D, Somerfield MR, et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J Clin Oncol. 2004;22:3408–19. doi: 10.1200/JCO.2004.05.063. [DOI] [PubMed] [Google Scholar]
- 2.Chawla AK, Kachnic LA, Clark JW, Willett CG. Combined modality therapy for rectal and colon cancer. Semin Oncol. 2003;30:101–12. doi: 10.1016/s0093-7754(03)00276-8. [DOI] [PubMed] [Google Scholar]
- 3.Adams J. Preclinical and clinical evaluation of proteasome inhibitor PS-341 for the treatment of cancer. Curr Opin Chem Biol. 2002;6:493–500. doi: 10.1016/s1367-5931(02)00343-5. [DOI] [PubMed] [Google Scholar]
- 4.Aklilu M, Kindler HL, Donehower RC, Mani S, Vokes EE. Phase II study of flavopiridol in patients with advanced colorectal cancer. Ann Oncol. 2003;14:1270–3. doi: 10.1093/annonc/mdg343. [DOI] [PubMed] [Google Scholar]
- 5.Iqbal S, Lenz HJ. Integration of novel agents in the treatment of colorectal cancer. Cancer Chemother Pharmacol. 2004;54 (Suppl 1):S32–S39. doi: 10.1007/s00280-004-0884-0. [DOI] [PubMed] [Google Scholar]
- 6.Jemal A, Tiwari RC, Murray T, et al. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 7.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–80. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 8.Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–44. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kyriakis JM, Banerjee P, Nikolakaki E, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–60. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 10.Butterfield L, Zentrich E, Beekman A, Heasley LE. Stress- and cell type-dependent regulation of transfected c-Jun N-terminal kinase and mitogen-activated protein kinase kinase isoforms. Biochem J. 1999;338 (Pt 3):681–6. [PMC free article] [PubMed] [Google Scholar]
- 11.Yang DD, Kuan CY, Whitmarsh AJ, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–70. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 12.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–6. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 13.Stadheim TA, Kucera GL. c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for mitoxantrone- and anisomycin-induced apoptosis in HL-60 cells. Leuk Res. 2002;26:55–65. doi: 10.1016/s0145-2126(01)00099-6. [DOI] [PubMed] [Google Scholar]
- 14.Verheij M, Bose R, Lin XH, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–9. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 15.Osborn MT, Chambers TC. Role of the stress-activated/c-Jun NH2-terminal protein kinase pathway in the cellular response to adriamycin and other chemotherapeutic drugs. J Biol Chem. 1996;271:30950–5. doi: 10.1074/jbc.271.48.30950. [DOI] [PubMed] [Google Scholar]
- 16.Chauhan D, Li G, Hideshima T, et al. JNK-dependent release of mitochondrial protein, Smac, during apoptosis in multiple myeloma (MM) cells. J Biol Chem. 2003;278:17593–6. doi: 10.1074/jbc.C300076200. [DOI] [PubMed] [Google Scholar]
- 17.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 18.Hatai T, Matsuzawa A, Inoshita S, et al. Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J Biol Chem. 2000;275:26576–81. doi: 10.1074/jbc.M003412200. [DOI] [PubMed] [Google Scholar]
- 19.Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–4. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 20.Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 21.Wojtaszek PA, Heasley LE, Siriwardana G, Berl T. Dominant-negative c-Jun NH2-terminal kinase 2 sensitizes renal inner medullary collecting duct cells to hypertonicity-induced lethality independent of organic osmolyte transport. J Biol Chem. 1998;273:800–4. doi: 10.1074/jbc.273.2.800. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Su B, Sah VP, et al. Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J Biol Chem. 1998;273:5423–6. doi: 10.1074/jbc.273.10.5423. [DOI] [PubMed] [Google Scholar]
- 23.Gu J, Kagawa S, Takakura M, et al. Tumor-specific transgene expression from the human telomerase reverse transcriptase promoter enables targeting of the therapeutic effects of the Bax gene to cancers. Cancer Res. 2000;60:5359–64. [PubMed] [Google Scholar]
- 24.Teraishi F, Kadowaki Y, Tango Y, et al. Ectopic p21(sdi1) gene transfer induces retinoic acid receptor beta expression and sensitizes human cancer cells to retinoid treatment. International Journal of Cancer. 2003;103:833–9. doi: 10.1002/ijc.10892. [DOI] [PubMed] [Google Scholar]
- 25.Ambudkar SV, Dey S, Hrycyna CA, et al. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–98. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 26.Shtil AA, Mandlekar S, Yu R, et al. Differential regulation of mitogen-activated protein kinases by microtubule-binding agents in human breast cancer cells. Oncogene. 1999;18:377–84. doi: 10.1038/sj.onc.1202305. [DOI] [PubMed] [Google Scholar]
- 27.Stone AA, Chambers TC. Microtubule inhibitors elicit differential effects on MAP kinase (JNK, ERK, and p38) signaling pathways in human KB-3 carcinoma cells. Exp Cell Res. 2000;254:110–9. doi: 10.1006/excr.1999.4731. [DOI] [PubMed] [Google Scholar]
- 28.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 29.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meek DW. Multisite phosphorylation and the integration of stress signals at p53. Cell Signal. 1998;10:159–66. doi: 10.1016/s0898-6568(97)00119-8. [DOI] [PubMed] [Google Scholar]
- 32.Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13:332–7. doi: 10.1016/s0955-0674(00)00216-7. [DOI] [PubMed] [Google Scholar]
- 33.Loganzo F, Discafani CM, Annable T, et al. HTI-286, a synthetic analogue of the tripeptide hemiasterlin, is a potent antimicrotubule agent that circumvents P-glycoprotein-mediated resistance in vitro and in vivo. Cancer Res. 2003;63:1838–45. [PubMed] [Google Scholar]
- 34.Oyaizu H, Adachi Y, Taketani S, et al. A crucial role of caspase 3 and caspase 8 in paclitaxel-induced apoptosis. Mol Cell Biol Res Commun. 1999;2:36–41. doi: 10.1006/mcbr.1999.0146. [DOI] [PubMed] [Google Scholar]
- 35.Panvichian R, Orth K, Day ML, et al. Paclitaxel-associated multimininucleation is permitted by the inhibition of caspase activation: a potential early step in drug resistance. Cancer Res. 1998;58:4667–72. [PubMed] [Google Scholar]
- 36.Sasaki J, Ramesh R, Chada S, et al. The anthelmintic drug mebendazole induces mitotic arrest and apoptosis by depolymerizing tubulin in non-small cell lung cancer cells. Molecular Cancer Therapeutics. 2002;1:1201–9. [PubMed] [Google Scholar]
- 37.Suzuki A, Kawabata T, Kato M. Necessity of interleukin-1beta converting enzyme cascade in taxotere-initiated death signaling. Eur J Pharmacol. 1998;343:87–92. doi: 10.1016/s0014-2999(97)01520-3. [DOI] [PubMed] [Google Scholar]
- 38.Xiao D, Pinto JT, Soh JW, et al. Induction of apoptosis by the garlic-derived compound S-allylmercaptocysteine (SAMC) is associated with microtubule depolymerization and c-Jun NH(2)-terminal kinase 1 activation. Cancer Res. 2003;63:6825–37. [PubMed] [Google Scholar]
- 39.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–9. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 40.Ham YM, Choi JS, Chun KH, Joo SH, Lee SK. The c-Jun N-terminal kinase 1 activity is differentially regulated by specific mechanisms during apoptosis. J Biol Chem. 2003;278:50330–7. doi: 10.1074/jbc.M302997200. [DOI] [PubMed] [Google Scholar]
- 41.Butterfield L, Storey B, Maas L, Heasley LE. c-Jun NH2-terminal kinase regulation of the apoptotic response of small cell lung cancer cells to ultraviolet radiation. J Biol Chem. 1997;272:10110–6. doi: 10.1074/jbc.272.15.10110. [DOI] [PubMed] [Google Scholar]
- 42.Wang TH, Wang HS, Ichijo H, et al. Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signal-regulating kinase pathways. J Biol Chem. 1998;273:4928–36. doi: 10.1074/jbc.273.9.4928. [DOI] [PubMed] [Google Scholar]
- 43.Dai Y, Rahmani M, Grant S. Proteasome inhibitors potentiate leukemic cell apoptosis induced by the cyclin-dependent kinase inhibitor flavopiridol through a SAPK/JNK- and NF-kappaB-dependent process. Oncogene. 2003;22:7108–22. doi: 10.1038/sj.onc.1206863. [DOI] [PubMed] [Google Scholar]
- 44.Hess P, Pihan G, Sawyers CL, Flavell RA, Davis RJ. Survival signaling mediated by c-Jun NH(2)-terminal kinase in transformed B lymphoblasts. Nat Genet. 2002;32:201–5. doi: 10.1038/ng946. [DOI] [PubMed] [Google Scholar]
- 45.Potapova O, Anisimov SV, Gorospe M, et al. Targets of c-Jun NH(2)-terminal kinase 2-mediated tumor growth regulation revealed by serial analysis of gene expression. Cancer Res. 2002;62:3257–63. [PubMed] [Google Scholar]
- 46.Zhang H, Shi X, Zhang QJ, et al. Nocodazole-induced p53-dependent c-Jun N-terminal kinase activation reduces apoptosis in human colon carcinoma HCT116 cells. J Biol Chem. 2002;277:43648–58. doi: 10.1074/jbc.M203214200. [DOI] [PubMed] [Google Scholar]
- 47.Chen YR, Tan TH. The c-Jun N-terminal kinase pathway and apoptotic signaling (review) Int J Oncol. 2000;16:651–62. doi: 10.3892/ijo.16.4.651. [DOI] [PubMed] [Google Scholar]
- 48.Tang G, Minemoto Y, Dibling B, et al. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414:313–7. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]