

Abstract
The tumor suppressor gene p53 is frequently mutated in cigarette smoke (CS)-related lung cancer. The p53 binding pattern of carcinogenic polycyclic aromatic hydrocarbons (PAHs) found in CS coincides with the p53 mutational pattern found in lung cancer, and PAHs have thus been considered to be major culprits for lung cancer. However, compared with other carcinogenic compounds, such as aldehydes, the amount of PAHs in CS is minute. Acrolein (Acr) is abundant in CS, and it can directly adduct DNA. Acr–DNA adducts, similar to PAH–DNA adducts, induce predominantly G-to-T transversions in human cells. These findings raise the question of whether Acr–DNA adducts are responsible for p53 mutations in CS-related lung cancer. To determine the role of Acr–DNA adducts in p53 mutagenesis in CS-related lung cancer we mapped the distribution of Acr–DNA adducts at the sequence level in the p53 gene of lung cells using the UvrABC incision method in combination with ligation-mediated PCR. We found that the Acr–DNA binding pattern is similar to the p53 mutational pattern in human lung cancer. Acr preferentially binds at CpG sites, and this enhancement of binding is due to cytosine methylation at these sequences. Furthermore, we found that Acr can greatly reduce the DNA repair capacity for damage induced by benzo[a]pyrene diol epoxide. Together these results suggest that Acr is a major etiological agent for CS-related lung cancer and that it contributes to lung carcinogenesis through two detrimental effects: DNA damage and inhibition of DNA repair.
Keywords: DNA damage
The tumor suppressor gene p53 is frequently mutated in human cancers (1, 2), and its mutational patterns often bear the fingerprints of the etiological carcinogens. Most notably it has been found that >50% of alfatoxin B1-associated liver cancers have mutations in codon 249 of the p53 gene and that p53 mutations are concentrated at contiguous pyrimidines in sunlight-associated skin cancers (3, 4). Previously we demonstrated that DNA adducts induced by diol epoxides of polycyclic aromatic hydrocarbons (PAHs), a major category of cigarette smoke (CS) carcinogens, preferentially occur at p53 mutational hotspots in CS-related lung cancers and that adducts formed at these locations are poorly repaired (5–7). The p53 gene is the most frequently mutated tumor suppressor gene in CS-related lung cancers, and its mutational pattern is distinctly different from that found in lung cancers of nonsmokers (Fig. 5, which is published as supporting information on the PNAS web site); PAHs have been shown to be strong carcinogens, and thus PAH-induced DNA damage may shape the p53 mutational pattern in lung cancer and may also represent a strong molecular link between lung cancer and cigarette smoking (1, 2, 5–7). The question of whether PAHs are the major culprits in CS smoke that cause human cancer remains unsettled because CS contains >4,000 compounds, many of which, particularly aldehydes, are not only more cytotoxic than PAHs but can also cause similar kinds of mutations (8, 9).
Acrolein (Acr) is one of the most abundant, reactive, and mutagenic aldehydes in CS; it is found in amounts up to 1,000-fold higher than those of PAHs in CS [10–500 μg per cigarette compared with 0.01–0.05 μg of benzo[a]pyrene (BP) per cigarette] (8). It can be taken up reasonably efficiently by human cells and react directly without metabolic activation with guanine residues in DNA to produce exocyclic DNA adducts, 6-hydroxy-1,N2-propanodeoxyguanosine and 8-hydroxy-1,N2-propanodeoxyguanosine adducts (Acr–dG) (Fig. 1), which are mutagenic and induce predominantly G:C-to-T:A transversion mutations similar to PAHs (9). Although Acr has been shown to be cytotoxic and genotoxic in human cells, as a suspected carcinogen its carcinogenicity in animal models has not been adequately evaluated because of its extremely potent toxic effects, which often result in death (9–11). Nonetheless, it has been found that Acr treatment greatly enhances urinary bladder papilloma occurrence in rats (9). Acr is one of the two major toxic metabolites of the chemotherapeutic agents cyclophosphamide and ifosfamide, and Acr has been long suspected to be an important factor in the induction of secondary human bladder tumors in cyclophosphamide-treated patients (9). Acr–dG DNA adducts have been detected in animal and human tissues (12), and it has been shown that the oral tissues of cigarette smokers have significantly higher Acr–dG levels than those of nonsmokers (13). These findings raise the possibility that Acr may contribute greatly to CS-related mutagenesis and carcinogenesis. To determine this possibility we developed a method using the UvrABC nuclease incision method in combination with the ligation-mediated PCR (LMPCR) technique (UvrABC/LMPCR) to map the Acr–dG adduct distribution at the sequence level in the p53 gene in normal human lung cells and compared it to the p53 binding pattern of BP diol epoxide (BPDE), a major carcinogenic form of BP, in the same cells (5, 8).
Fig. 1.

Chemical structures and detections of Acr–dG. (A) Chemical structure of Acr and Acr–dG adducts. (B) Identification of 32P-labeled Acr–DNA adducts by 2D TLC. Acr-modified DNA isolated from Acr-treated human cells and Acr-treated purified genomic DNA were digested with phosphodiesterase and nuclease P1, labeled with [γ-32P]ATP, and subjected to 2D TLC, as described in Materials and Methods. The standard Acr–dG adducts were obtained by reaction of Acr (0.2 M) with dGMP (2 mM) and then labeled with [γ-32P]ATP, and the major Acr–dG 3 adducts are indicated by circles. (a) DNA isolated from control cells. (b) Acr-modified genomic DNA. (c) DNA from Acr-treated cells. (d) Acr-modified dGMP. (e) Mixture of c and d.
Acr is very reactive toward nucleophiles, including thiol-containing proteins; intracellularly the majority of Acr is covalently bonded with proteins (9). Two major endogenously produced aldehydes, trans-4-hydroxy-2-nonenal (4-HNE) and malondialdehyde, can bond with proteins and have been found to cause an inhibitory effect on nucleotide excision repair (NER) (14, 15). We therefore also examined the effect of Acr on DNA repair using host cell reactivation and in vitro DNA damage-specific repair synthesis assays (14, 15).
Results
Acr Can Directly Modify DNA to Form Propanodeoxyguanine Adducts in Vitro and in Vivo.
It is well known that Acr can react directly with guanine residues in purified DNA to produce four isomeric exocyclic DNA adducts, two minor stereoisomeric Acr–dG adducts (Acr–dG 1 and 2), and two major stereoisomeric 8-hydroxy-1,N2-propanodexoyguaosine adducts (Acr–dG 3 and 4) (16) (F. L. Chung and C. Rizzo, personal communication) (Fig. 1). To ensure that the Acr–DNA adducts formed in vivo resulted from the direct interaction of Acr with genomic DNA, we identified the type of DNA adducts formed in cultured lung cells treated with Acr and in purified genomic DNA treated with Acr using the 32P postlabeling and 2D TLC method. The results in Fig. 1 show that Acr–dG 3 is the major Acr–dG adduct found both in Acr-treated purified genomic DNA and in DNA from cells treated with Acr. Three isomeric Acr–dG adducts are found in Acr-modified dGMP, with the major adduct (Acr–dG 3) being the same as that found in Acr-treated cells and genomic DNA. No Acr–dG 1 or Acr–dG 2 adducts were found in Acr-treated cells or Acr-modified genomic DNA. Although a small quantity of Acr–dG 4 adducts was found in Acr-treated cells, this type of adduct was not found in Acr-modified genomic DNA. Using relatively high concentrations of Acr (1.1 M) for modifications of calf thymus DNA, Chung et al. (16) detected Acr–dG 1 and 2 adducts. These two types of adducts were not evident in Acr-cultured Chinese hamster ovary cells, liver samples from mice and rats, and human liver and oral tissue samples; in contrast, Acr–dG 3 adducts were detected in these samples (12, 13, 17). Structural constraints in genomic DNA and chromatin structure may hinder formation of Acr–dG 1 and 2 adduct isomers. Our results are consistent with well established findings that Acr can react directly without metabolic activation with guanine residues in DNA to produce exocyclic propanodeoxyguanosine DNA adducts (12, 13, 16–18).
UvrABC Is Able to Incise Acr–dG Adducts Quantitatively and Specifically.
To assess the contribution of Acr-induced DNA damage to the p53 mutation pattern in lung cancer it is necessary to map the Acr–DNA adduct distribution at the sequence level in the p53 gene of lung cells treated with Acr. Previously, using the UvrABC/LMPCR method, we successfully mapped various types of bulky carcinogen-induced DNA damage at the sequence level in the p53 and ras genes (5, 19–21). The rationale of this approach is based on the finding that, under proper conditions, UvrABC can incise bulky DNA adducts specifically and quantitatively, and the extent of UvrABC incision therefore represents the extent of adduct formation rather than UvrABC sequence preferences (22). Because radioactively labeled Acr is not available, we assessed the quantitative relationship between UvrABC incision and Acr–DNA adduct formation in supercoiled DNA. We found that the number of UvrABC incisions was proportional to the concentration of Acr used for DNA modification, indicating that UvrABC is able to cut Acr–dG adducts quantitatively (Fig. 6A, which is published as supporting information on the PNAS web site). To determine the specificity of UvrABC cutting, 5′ or 3′ single-end, 32P-labeled p53 DNA fragments were reacted with UvrABC nuclease, and the resultant DNAs were separated by electrophoresis in a denatured DNA sequencing gel. The results show that UvrABC makes the typical dual incisions 7 nt 5′ and 4 nt 3′ to an Acr–dG adduct, similar to what we have found for most bulky carcinogen-induced DNA damage (22) (Fig. 6 B and C). The results also show that the kinetics of UvrABC cutting at different sequences are similar, if not identical (Fig. 7 A and C, which is published as supporting information on the PNAS web site). It should be noted that we have found that at the end of incubation UvrABC remains active. Based on these results we concluded that UvrABC is able to cut Acr–dG adducts quantitatively and specifically and that the extent of UvrABC cutting at different sequences represents the extent of Acr–DNA adduct formation at the sequence.
Acr–DNA Adducts Are Preferentially Formed at the Lung Cancer p53 Mutational Hotspots.
Having established that the UvrABC nuclease is able to incise Acr–dG adducts specifically and quantitatively, we then used the UvrABC/LMPCR method to map the Acr–DNA binding spectrum in the coding strand of exons 5, 7, and 8 of the p53 gene in normal human lung cells. Normal human bronchial epithelial (NHBE) cells and normal human lung fibroblasts (NHLF) were treated with different concentrations of Acr for 6 h, and genomic DNA was isolated to map the Acr–DNA binding pattern. For comparison purposes we also mapped the distribution of BPDE–DNA adducts in the p53 gene of NHBE cells treated with BPDE using the same UvrABC/LMPCR method. The results in Fig. 2A, lanes 3–5, show that Acr–dG preferentially formed at codons 152, 154, 156, 157, and 158 in exon 5; codons 248 and 249 in exon 7; and codons 273 and 282 in exon 8 of the p53 gene. The Acr–DNA adduct distributions in exons 5, 7, and 8 of the p53 gene in the NHBE cells are very similar, but not quite identical, to the BPDE–DNA adduct distribution, as shown in Fig. 2B. BPDE preferentially forms DNA adducts at only CpG sites in codons 156, 157, and 158 of exon 5; codon 248 of exon 7; and codon 273 of exon 8 in the p53 gene in NHBE cells, which is consistent with previously published reports (5, 6). Codons 157, 158, 248, 249, 273, and 282 of the p53 gene are the mutational hotspots in CS-related lung cancer, and codons 249 and 273 of the p53 gene are mutational hotspots in lung cancers of both cigarette smokers and nonsmokers (Fig. 5). Whereas Acr–dG adducts preferentially form at codon 249, BPDE–dG adducts do not (Fig. 2). It is worth noting that we have previously found that six of the activated cigarette PAHs either do not bind or weakly bind to this codon (5, 7).
Fig. 2.

Acr–dG and BPDE–dG distributions in the p53 gene. (A and B) Acr–dG and BPDE–dG adduct distribution in exons 5, 7, and 8 of the p53 gene of normal human lung cells treated with Acr (A) and BPDE (B). In A, NHBE cells and NHLF were treated with 20 μM Acr for 6 h, and in B, NHBE cells were treated with 1 μM BPDE for 30 min. Genomic DNA was then isolated, the DNA adduct distribution was mapped by the UvrABC/LMPCR method, and the DNA was separated by electrophoresis. A/G and T/C are Maxam and Gilbert reaction products (26). (C) Comparisons of the frequency of Acr–dG adduct distribution along the p53 gene in NHBE cells with the frequency of the p53 mutations in CS-related lung cancer (International Agency for Research on Cancer p53 Mutation database, http://www-p53.iarc.fr).
We also mapped the distribution of Acr–DNA adducts along the p53 gene in NHLF treated with Acr. The results show that the Acr–DNA adduct distribution pattern in the p53 gene in NHLF was almost identical to that found in NHBE cells (Fig. 2A). These results clearly demonstrate that the specific binding spectrum of Acr in NHBE cells is due to the intrinsic binding specificity of Acr rather than the specificity of cell types.
The levels of Acr–dG formation in exons 5, 7, and 8 of the p53 gene of NHBE cells were compared with the mutation distribution in this gene in CS-related lung cancer obtained from the p53 database (Fig. 2C). The histograms of both show remarkable resemblance, which indicates that Acr–dG adducts may contribute to CS-related lung cancer and that Acr, instead of PAHs, from CS may be the etiological agent that causes mutations at codon 249 of the p53 in lung cancers of both cigarette smokers and nonsmokers. Acr is abundant in secondhand smoke and is also rich in cooking fumes (8, 9), which may be the major sources of Acr exposure for nonsmokers.
C5 Cytosine Methylation Enhances Acr–dG Binding at CpG Sites in the p53 Gene.
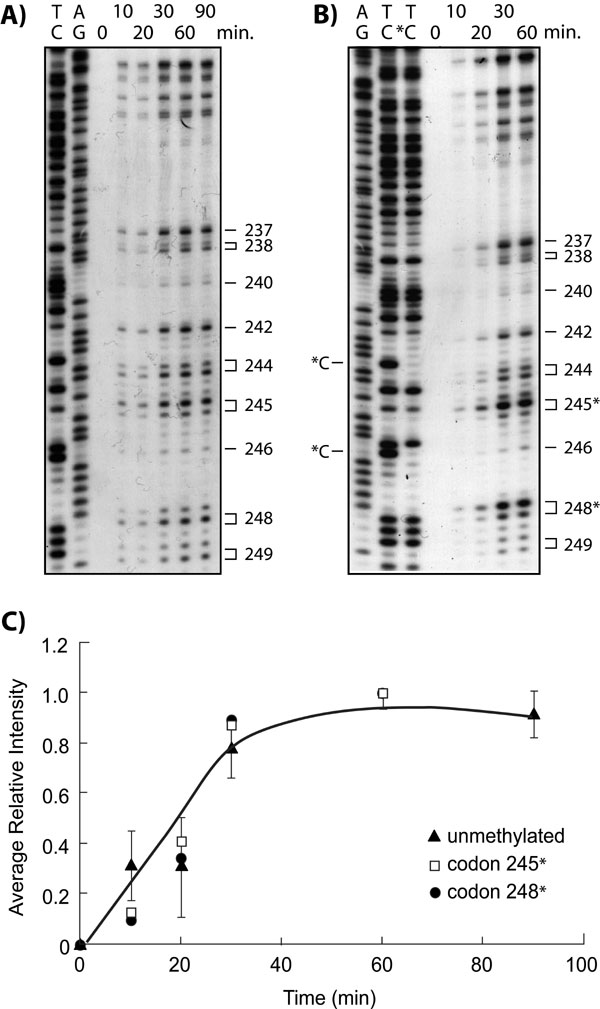
Results in Fig. 2 show that, except for codon 249, all of the preferential sites of Acr–dG binding are guanines within CpG sites. Why does Acr preferentially bind at codons containing CpG sequences? In human genomic DNA cytosines at CpG sequences are frequently methylated at the C5 position, and this methylation may affect the stereostructure of DNA and/or nucleosomal structure in a manner that consequently affects bulky carcinogen binding (23–25). Indeed, we have found that C5 cytosine methylation at CpG sites enhances the binding of bulky chemicals at the adjacent guanines (25). Two approaches were undertaken to ascertain the reason behind the selectivity of Acr binding at CpG-containing codons in the p53 gene. First, we determined the Acr–dG binding pattern in the p53 gene by directly modifying NHBE genomic DNA and compared it to the pattern that resulted from treating intact NHBE cells; we found that both patterns of Acr–dG formation in the p53 gene are very similar, if not identical (Fig. 8, which is published as supporting information on the PNAS web site). These results thus rule out nucleosomal structure playing a major role in determining the Acr–dG binding pattern in the p53 gene. Second, we determined the Acr binding pattern in p53 DNA fragments with or without C5 cytosine methylation at CpG sequences. 3′ 32P-end-labeled DNA fragments of exon 7 and 5′ 32P-end-labeled DNA fragments of exon 5 of the p53 gene obtained by PCR amplification were subjected to SssI methylase treatment in the presence of S-adenosylmethionine to methylate all cytosines at CpG sites. DNA fragments with and without methylation treatment were then modified with Acr, and the adduct distributions were mapped by the UvrABC incision method. The extent of cytosine methylation was determined by Maxam and Gilbert chemical cleavage reactions (26). Because hydrazine is unable to modify C5-methylated cytosines, both the 5′- and 3′-phosphodiester bonds of each methylated cytosine are refractory to piperidine hydrolysis and no cytosine ladders are observed at methylated cytosines (26). As shown in Fig. 3A and B, under the methylation conditions we used all of the CpG sites in the DNA fragments are methylated, and the intensities of the UvrABC incision bands of Acr–DNA adducts are enhanced 2- to 4-fold at almost all CpG-containing codons, such as codons 152, 154, 156, 157, 158, and 159 (Fig. 3A) and codons 245 and 248 (Fig. 3B) in methylated versus unmethylated p53DNA fragments. In contrast, the intensities of UvrABC incision bands of Acr–DNA adducts did not change significantly at the non-CpG-containing codons in methylated DNA fragments. To rule out the possibility that this enhancement of UvrABC cutting at methylated CpG sites is due to UvrABC having a higher cutting efficiency toward Acr adducts at methylated CpG sites versus other non-CpG sequences, the kinetics of UvrABC cutting for methylated CpG sequences and other sequences was determined in methylated p53 exon 7 DNA fragments to determine whether the kinetics of UvrABC incision was in fact different for methylated CpG sites versus other sequences. We found UvrABC incision at all Acr-binding sites, in both methylated CpG sequences (codons 245 and 248) and other unmethylated sequences, to be a function of incubation time and to plateau after 30 min of incubation, which leads us to conclude that the enhancement of UvrABC cutting at methylated CpG sites is due to preferential Acr binding as opposed to preferential UvrABC cutting for Acr adducts at methylated CpG sites (Fig. 7). Taken together, these results suggest that the strong binding of Acr at CpG-containing codons in the p53 gene in human lung cells is due to 5-C cytosine methylation at these sequences. The results also suggest that the weaker Acr–dG formation at codons such as 158, 175, and 245 that contain CpG sequences is likely due to cytosines at these sequences not being methylated.
Fig. 3.

The effect of 5C cytosine methylation at CpG sites on Acr–dG adduct formation. Cytosines at CpG sites of 5′ 32P-labeled exon 5 (A) and 3′ 32P-labeled exon 7 (B) of p53 DNA fragments were methylated by SssI CpG methylase, and the DNA fragments with and without methylation treatment were modified with Acr (30 μM, 10-h incubation), treated with UvrABC nucleases, and separated by electrophoresis as previously described (25). A/G and T/C are Maxam and Gilbert reaction products. T/*C represents Maxam and Gilbert reaction products from methylated DNA fragments. *C represents the methylated cytosine, and the codon number of the bands corresponding to CpG sites is indicated by an asterisk.
Acr Treatment Reduces the Ability of Cells to Repair BPDE–DNA Adducts.
The carbonyl group and olefinic bond of Acr make this molecule reactive toward not only nucleic acids but also thiol-containing proteins (9). It is conceivable that Acr binding may or may not affect the functions of the proteins. Previously we have found that aldehydes, such as 4-HNE and malondialdehyde, can greatly reduce cellular NER capacity (14, 15). We therefore determined the effect of Acr treatment on NER capacity using the well established host cell reactivation assay and in vitro DNA damage-specific repair synthesis assay (14, 15). We found that, similar to 4-HNE and malondialdehyde, Acr can greatly inhibit NER for BPDE-induced DNA damage (Fig. 4). This inhibitory effect is much more pronounced in cell lysates directly treated with Acr than in whole cells treated with Acr (Fig. 4), suggesting that the Acr may interact with components in growth medium, that the cellular membrane may serve as a barrier to the uptake of Acr, and that the inhibitory effect is due to the interactions of Acr with repair proteins.
Fig. 4.

Inhibition of the repair of BPDE–DNA adducts in human cells by Acr. (A) Repair inhibition determined by host cell reactivation assay. BPDE-modified luciferase reporter and unmodified β-galactosidase plasmids were cotransfected into NHLF treated with different concentrations of Acr for 1 h, and luciferase and β-galactosidase activities were measured 20 h after transfection. The relative repair capacity was calculated as the percentage of the relative luciferase activity of the plasmids in Acr-treated cells compared with untreated cells after normalization of the transfection frequency with β-galactosidase activity. (B–D) Repair inhibition determined by in vitro DNA repair synthesis assay. BPDE-modified pUC18 and unmodified pBR322 plasmids were used as DNA substrates for in vitro DNA repair synthesis assay. (B) NHLF were treated with different concentrations of Acr for 1 h, and the cell extracts were used for repair assay. (C) Different concentrations of Acr were added directly into cell extracts prepared from untreated NHLF immediately before the start of repair assay. (Upper) Photograph of an ethidium bromide-stained gel. (Lower) Autoradiograph of the same gel. (D) The relative repair capacity was calculated as the percentage of the repair activity in Acr-treated samples to untreated samples. The data represent three independent experiments, and the error bars represent the standard deviation.
Acr–dG Adducts Induce G-to-T Transversion Mutations in Human Cells.
It has long been recognized that CS-related lung cancers are rich in G-to-T transversions in the p53 gene (>30% versus 10% in other human cancers) (2). This phenomenon has been attributed to mutations induced by bulky carcinogens in CS, including PAHs (5–7, 27). To determine the types of mutations induced by Acr–dG, we modified shuttle vector pSP189 DNA containing the supF gene with Acr (100 μM) and transfected the plasmid into NHLF for replication. Replicated plasmid was then transformed into indicator Escherichia coli cells, mutant white colonies were collected, and Acr-induced mutations in the supF gene were sequenced. We found that Acr modification resulted in greatly enhancing mutation frequency (from 4 × 10−4 to 120 × 10−4) and that >50% of the base substitutions induced by Acr–dG adducts are G-to-T transversions (28 of 55 total base substitution mutants sequenced). These results further support the hypothesis that Acr in CS contributes significantly to p53 mutagenesis in lung cancer.
Discussion
CS is the major cause of lung cancer deaths, and 90% of all lung cancers in the United States are CS-related (28, 29). CS contains >4,000 compounds, many of which, including PAHs, N-nitrosamines, aromatic amines, and metals, are not only mutagenic but also well established carcinogens in animal models (8, 29, 30). Therefore, it is reasonable to assume that these compounds contribute to CS-related lung carcinogenesis in humans. Previously, using the UvrABC/LMPCR method, we found that diol epoxides of potent PAH carcinogens found in CS preferentially form DNA adducts at p53 mutational hotspots in CS-related lung cancer, such as codons 156, 157, 158, 245, 273, and 282 (5, 7). In addition, BPDE–DNA adducts formed at these sites are poorly repaired (6). Because the p53 gene is the most frequently mutated gene in CS-related lung cancer and the p53 mutational spectra in lung cancer of smokers and nonsmokers are distinctly different (1, 2) (Fig. 5), our findings strongly suggest that targeted DNA damage determines the p53 mutational spectrum in CS-related lung cancer. These results, however, do not exclude the possibility that many other CS compounds also contribute to lung carcinogenesis. In fact, these results raise the possibility that any DNA-damaging compounds present in CS that preferentially bind to the CS-related lung cancer p53 mutational hotspots are lung cancer etiological agents.
Acr is one of the most abundant compounds generated in CS; the amount of Acr in a single cigarette, depending on the manufacturer, ranges from 10 to 500 μg (8, 31). The total amount of PAHs present in CS, in contrast, is in the range of just a few micrograms (8). Acr has been shown to interact with nucleophiles, including DNA and proteins in cells (32). Unlike PAHs, where only metabolically activated forms can form adducts with DNA, Acr can directly interact with DNA and form DNA adducts (18, 23). Similar to PAH–DNA adducts, Acr–DNA adducts induce mainly G:C-to-T:A transversion mutations (10, 33–35), the major type of mutations found in the p53 gene in CS-related lung cancer. Although the carcinogenicity of Acr in the lung has not been studied because of the severe toxicity associated with Acr treatment in animals, i.p. injection of Acr has been shown to cause bladder cancer in rats (36). These results indicate that Acr is indeed a carcinogenic substance. Although the effects of CS on Acr–dG formation in the lung tissue of cigarette smokers has not been determined, it has been reported that the level of Acr–dG DNA adducts in the oral tissue of smokers is in the range of a few micromoles per mole of guanine, which is far above the level of PAH–DNA adducts found in the oral or lung tissue of cigarette smokers (12, 13). It is very likely that the level of Acr–dG in lung tissue is similar to that found in the oral tissues of cigarette smokers. If this is the case, then, based on Acr abundance in CS, its reactivity toward DNA, and mutagenicity of Acr–DNA adducts, Acr is potentially one of the major etiological agents of CS-induced lung cancer. Our current results demonstrate that, similar to PAHs, Acr preferentially binds to p53 mutational hotspots of CS-related lung cancer in human cells, including NHBE cells. These results strongly suggest that Acr–dG adducts, as well as PAH–DNA adducts, contribute greatly to these mutations in lung cancer.
We have found that all of the Acr preferential binding guanines, except for codon 249, in the p53 gene are located at CpG sequences and that C5 cytosine methylation at CpG sites can greatly enhance Acr–dG adduct formation. This C5 cytosine methylation has been previously shown to also enhance guanine adduction at CpG sites by BPDE, aflatoxin B1 8,9-epoxide, and N-acetoxy-acetylaminofluorene, even though the binding positions by these agents on guanines are different (25). The precise mechanism of how C5 methylation enhances adduction to guanines at these sites has yet to be elucidated. It is worth noting that these CpG sites are in the coding region of the p53 gene and are also very distant from promoter region. The function and the extent of methylation at these CpG sites in the p53 gene in NHBE and lung fibroblasts are also unknown and may vary among different individuals. If this is the case perhaps these variations may contribute to the different susceptibilities of individuals to CS-induced lung cancer.
We found that codon 249 in the p53 gene is a preferential binding site for Acr even though it is not a CpG-containing site and the Acr binding at this position is not affected by CpG methylation at the surrounding sequences (codon 248). Codon 249 is a mutational hotspot in lung and liver cancers (2) (Fig. 5). Intriguingly, we found that all of the PAHs we tested and AFB1–8,9-epoxides, the etiological agents for liver cancer, do not preferentially bind to codon 249 (3, 5, 7, 25). Recently, however, we found that 4-HNE, another α,β-unsaturated aldehyde and a major lipid peroxidation product that is also able to interact with DNA to form exocyclic propanodeoxyguanosine adducts, preferentially forms DNA adducts at this codon (37). 4-HNE–dG adducts have been found to induce G:C-to-T:A mutations in human cells (38). Increasing evidence suggests that lipid peroxidation and chronic oxidative stress play important roles in human carcinogenesis; although the mechanisms involved are unclear, it is possible that aldehydes resulting from endogenous lipid peroxidation and other sources, such as Acr, 4-HNE, malondialdehyde, and crotonaldehyde, may contribute greatly to mutations at codon 249 in human cancer.
We have also found that Acr can greatly reduce the NER capacity. NER is the major repair pathway for bulky DNA damage, including PAH–DNA adducts and exocyclic propanodeoxyguanine adducts (22, 39). It has been found that the NER capacity in individuals who have a genetic defect in NER genes, such as xeroderma pigmentosum patients, is reduced to 10–20% that of normal individuals; these patients have 2,000-fold and 20- to 30-fold higher cancer incidence in skin and internal organs, respectively (40). NER gene knockout animals have a predisposition for spontaneous and chemically induced carcinogenesis (41). It has been strongly suggested that PAHs present in CS and in the environment are the agents responsible for the lung carcinogenesis, and the DNA damage induced by activated metabolites of PAHs initiate carcinogenesis (5, 7, 42). Our findings that Acr greatly inhibit cellular repair capacity to remove BPDE–DNA adducts in human lung cells strongly suggest that Acr also contributes significantly to lung carcinogenesis by its inhibitory effects on DNA repair in addition to damaging DNA directly. Our findings that the Acr–dG adduct distribution is similar to the mutational spectrum in CS-related lung cancer and that Acr can cause a significant inhibitory effect on DNA repair raise the possibility that Acr is an equally potent, if not more potent, lung cancer etiological agent as PAHs, considering the abundant amount of Acr in CS and in the environment. We propose that the carcinogenicity of Acr is derived from two detrimental effects: damaging DNA and reducing DNA repair capacity. These two effects may in turn lead to more mutations, which can be induced by both PAHs and Acr, to trigger carcinogenesis.
Worldwide, >2 million people die of CS-related cancer annually (43). Although smoking cessation is the most effective way to reduce these cigarette-induced deaths, this approach is unrealistic in the short term. Identifying the etiological agents for CS-induced cancer and designing methods to eliminate them from CS provide us with the simplest and most realistic solution. Although Acr carcinogenicity requires further epidemiological confirmation and studies in proper animal models, immediate measures to reduce this contaminant both in CS and in the environment seem warranted.
Materials and Methods
Cell Culture, Carcinogen Treatment, and Genomic DNA Isolation.
NHBE cells were cultured in medium provided by Clonetics (San Diego, CA). NHLF (CCL-202) and lung adenocarcinoma cells (A549) (American Type Culture Collection, Manassas, VA) were grown in MEM supplemented with 10% FBS. Stock solutions of Acr (Sigma-Aldrich, St. Louis, MO) and BPDE (Chemsyn Science Laboratories, Lenexa, KS) were prepared immediately before use. Cells at 70% confluency were washed with phosphate buffer (PBS/70 mM NaCl/2 mM KCl/1 mM KH2PO4, pH 7.4) and treated with different concentrations of Acr (0–100 μM) in serum-free culture medium for 6 h or different concentrations of BPDE for 30 min at 37°C in the dark. After treatment, the genomic DNA was isolated as previously described (5–7). For in vitro modifications, genomic DNA was isolated from untreated cells and dissolved in H2O, mixed with different concentrations of Acr, and incubated at 37°C for 12 h. After repeated phenol and diethyl ether extractions, the DNA was then precipitated with ethanol and dissolved in TE buffer (10 mM Tris, pH 7.5/1 mM EDTA).
Acr–DNA Adduct Analysis.
Acr–DNA adducts formed in cells treated with Acr (0–100 μM) and in purified genomic DNA modified with Acr (0–100 μM) were analyzed by the 32P postlabeling and the 2D TLC method on polyethyleneimine cellulose sheets (Anatech, Newark, DE), as described by Eder and Budiawan (44). The solvents used were as follows: D1, 0.7 M ammonium formate (pH 3.5); D2, 0.3 M ammonium sulfate (pH 7.5). The chromatograms were visualized by autoradiography, the Acr–DNA adducts (although all 32P-labeled Acr–dG adducts were in 3′,5′-bisphosphate forms, for the sake of simplicity they remain labeled as Acr–dG adducts) were excised, and the radioactivity was measured. Acr–DNA adduct levels were calculated by determining the relative adduct labeling, which is the ratio of labeled adduct nucleotides to labeled total nucleotides. The specific radioactivity of [γ-32P]ATP, determined by labeling a known amount of dGMP (deoxyadenosine 3′-phosphate) (Sigma), was used for the relative labeling calculations. The well characterized Acr–dG 2 and Acr–dG 3 adducts and oligomer containing Acr–dG 3 [kindly provided by Fung-Lung Chung (Georgetown University, Washington, DC) and Carmelo Rizzo and Larry Marnett (Vanderbilt University, Nashville, TN), respectively] were used as standards (16).
Preparation of 32P End-Labeled p53 DNA Fragments and C5 Cytosine Methylation at CpG Sites.
DNA fragments of 247-bp 5′ 32P-end-labeled p53 exon 5 and 141-bp 3′ 32P-end-labeled p53 exon 7 were prepared according to the previously described method (19). These 32P-end-labeled p53 DNA fragments were subjected to SssI methylase (New England Biolabs, Beverly, MA) treatment in the presence of S-adenosylmethionine according to the manufacturer's instructions to methylate all cytosines at CpG sites.
Acr Modification of Supercoiled Plasmids and DNA Fragments and UvrABC Incision Assay of Acr–dG Adducts.
Supercoiled pGEM plasmids, purified as described previously (14), and 32P-labeled DNA fragments were modified with different concentrations of Acr solution and purified as described above; after ethanol precipitation DNA was dissolved in TE buffer. Methods for UvrA, UvrB, and UvrC protein purifications, UvrABC nuclease incision assays, separations of the resultant DNA, and quantification of band intensity were the same as previously described (5–7, 22).
Mapping DNA Adduct Distribution in the p53 Gene in Human Genomic DNA by Using the UvrABC/LMPCR Method.
The UvrABC/LMPCR method was the same as described previously (5, 20). Each experiment was repeated three times with very similar results. The calculation of the relative intensities of DNA adduct formation at different codons in the p53 gene was performed as previously described (5, 20). We used various concentrations of Acr for treating cells in this study and found that concentrations did not qualitatively affect the Acr–DNA binding patterns. Thus, for the sake of clarity, results from one concentration were used for the quantitative determinations.
Determination of the Effect of Acr Treatment on DNA Repair.
Host cell reactivation and in vitro DNA repair synthesis assays were performed as previously described to determine the effects of Acr treatment on DNA repair in human cells (14, 15).
Determination of Mutations Induced by Acr–DNA Adducts.
The methods used for mutation detection and for mutational spectrum determination were the same as previously described (38). Briefly, shuttle vector pSP189 plasmid DNA was modified with different concentrations of Acr and transfected into NHLF for replication. Plasmids were recovered 72 h after transfection, and replicated plasmids were then transformed into MB7070 E. coli indicator cells. The supF gene in plasmids isolated from mutant white colonies were then sequenced.
Supplementary Material
Acknowledgments
We thank Drs. Yen-Yee T. Nydam and Cathy Klein for critical review and Joyce Clemente and Helen Duss for manuscript preparation. This study was supported by National Institutes of Health Grants ES03124, ES10344, ES00260, CA99007, and CA114541.
Abbreviations
- CS
cigarette smoke
- PAH
polycyclic aromatic hydrocarbon
- Acr
acrolein
- Acr–dG
6- or 8-hydroxy-1,N2-propanodeoxyguanosine
- BP
benzo[a]pyrene
- BPDE
BP diol epoxide
- 4-HNE
trans-4-hydroxy-2-nonenal
- MDA
malondialdehyde
- NHBE
normal human bronchial epithelial
- NHLF
normal human lung fibroblast
- LMPCR
ligation-mediated PCR
- NER
nucleotide excision repair.
Footnotes
The authors declare no conflict of interest.
References
- 1.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 2.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 3.Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Nature. 1991;350:427–428. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 4.Brash DE. Trends Genet. 1997;13:410–414. doi: 10.1016/s0168-9525(97)01246-8. [DOI] [PubMed] [Google Scholar]
- 5.Denissenko MF, Pao A, Tang M-s, Pfeifer GP. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 6.Denissenko M, Pao A, Pfeifer G, Tang M-s. Oncogene. 1998;16:1241–1249. doi: 10.1038/sj.onc.1201647. [DOI] [PubMed] [Google Scholar]
- 7.Smith LE, Denissenko MF, Bennett WP, Amin S, Tang M-s, Pfeifer GP. J Natl Cancer Inst. 2000;92:803–811. doi: 10.1093/jnci/92.10.803. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman D, Hecht SS. In: Handbook of Experimental Pharmacology. Cooper CS, Grover PL, editors. Heidelberg: Springer; 1990. pp. 70–74. [Google Scholar]
- 9.Gomes R, Meek ME, Eggleton M. Concise International Chemical Assessment Document No 43. Geneva: World Health Organization; 2002. [Google Scholar]
- 10.Curren RD, Yang LL, Conklin PM, Grafstrom RC, Harris CC. Mutat Res. 1988;209:17–22. doi: 10.1016/0165-7992(88)90104-2. [DOI] [PubMed] [Google Scholar]
- 11.Grafstrom RC, Dypbukt JM, Willey JC, Sundqvist K, Edman C, Atzori L, Harris CC. Cancer Res. 1988;48:1717–1721. [PubMed] [Google Scholar]
- 12.Nath RG, Chung F-L. Proc Natl Acad Sci USA. 1994;91:7491–7495. doi: 10.1073/pnas.91.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nath RG, Ocando JE, Guttenplan JB, Chung F-L. Cancer Res. 1998;58:581–584. [PubMed] [Google Scholar]
- 14.Feng Z, Hu W, Tang M-s. Proc Natl Acad Sci USA. 2004;101:8598–8602. doi: 10.1073/pnas.0402794101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Z, Hu W, Marnett L, Tang M-s. Mutat Res. 2006 doi: 10.1016/j.mrfmmm.2006.06.003. in press. [DOI] [PubMed] [Google Scholar]
- 16.Chung F-L, Young R, Hecht SS. Cancer Res. 1984;44:990–995. [PubMed] [Google Scholar]
- 17.Foiles PG, Akerkar SA, Miglietta LM, Chung F-L. Carcinogenesis. 1990;11:2059–2061. doi: 10.1093/carcin/11.11.2059. [DOI] [PubMed] [Google Scholar]
- 18.Nath RG, Chen HJC, Nishikawa A, Young-Sciame R, Chung F-L. Carcinogenesis. 1994;15:979–984. doi: 10.1093/carcin/15.5.979. [DOI] [PubMed] [Google Scholar]
- 19.Feng Z, Hu W, Rom WN, Beland FA, Tang M-s. Biochemistry. 2002;41:6414–6421. doi: 10.1021/bi020093s. [DOI] [PubMed] [Google Scholar]
- 20.Feng Z, Hu W, Chen JX, Pao A, Li H, Rom W, Hung MC, Tang M-S. J Natl Cancer Inst. 2002;94:1527–1536. doi: 10.1093/jnci/94.20.1527. [DOI] [PubMed] [Google Scholar]
- 21.Feng Z, Hu W, Rom WN, Beland FA, Tang M-s. Carcinogenesis. 2002;23:1721–1727. doi: 10.1093/carcin/23.10.1721. [DOI] [PubMed] [Google Scholar]
- 22.Tang M-s. In: Technologies for Detection of DNA Damage and Mutation. Pfeifer G, editor. New York: Plenum; 1996. pp. 139–152. [Google Scholar]
- 23.Tornaletti S, Pfeifer GP. Oncogene. 1995;10:1493–1499. [PubMed] [Google Scholar]
- 24.Johnson WS, He QY, Tomasz M. Bioorg Med Chem. 1995;3:851–860. doi: 10.1016/0968-0896(95)00067-q. [DOI] [PubMed] [Google Scholar]
- 25.Chen JX, Zheng Y, West M, Tang M-s. Cancer Res. 1998;58:2070–2075. [PubMed] [Google Scholar]
- 26.Maxam AM, Gilbert W. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 27.Pfeifer GP, Hainaut P. Mutat Res. 2003;526:39–43. doi: 10.1016/s0027-5107(03)00013-7. [DOI] [PubMed] [Google Scholar]
- 28.World Health Organization. Tobacco or Health: A Global Status Report. Geneva: World Health Organization; 1997. [Google Scholar]
- 29.Hoffmann D, Hoffmann I, El-Bayoumy K. Chem Res Toxicol. 2001;14:767–790. doi: 10.1021/tx000260u. [DOI] [PubMed] [Google Scholar]
- 30.Hecht SS, Carmella SG, Murphy SE, Foiles PG, Chung F-L. J Cell Biochem Suppl. 1993;17F:27–35. doi: 10.1002/jcb.240531005. [DOI] [PubMed] [Google Scholar]
- 31.Fujioka K, Shibamoto T. Environ Toxicol. 2006;21:47–54. doi: 10.1002/tox.20153. [DOI] [PubMed] [Google Scholar]
- 32.Esterbauer H, Schaur RJ, Zollner H. Free Radical Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 33.Kawanishi M, Matsuda T, Nakayama A, Takebe H, Matsui S, Yagi T. Mutat Res. 1998;417:65–73. doi: 10.1016/s1383-5718(98)00093-x. [DOI] [PubMed] [Google Scholar]
- 34.Kanuri M, Minko IG, Nechev LV, Harris TM, Harris CM, Lloyd RS. J Biol Chem. 2002;21:18257–18265. doi: 10.1074/jbc.M112419200. [DOI] [PubMed] [Google Scholar]
- 35.Yang IY, Chan G, Miller H, Huang Y, Torres MC, Johnson F, Moriya M. Biochemistry. 2002;41:13826–13832. doi: 10.1021/bi0264723. [DOI] [PubMed] [Google Scholar]
- 36.Cohen SM, Garland EM, St John M, Okamura T, Smith RA. Cancer Res. 1992;52:3577–3581. [PubMed] [Google Scholar]
- 37.Hu W, Feng Z, Eveleigh J, Iyer G, Pan J, Amin S, Chung F-L, Tang M-s. Carcinogenesis. 2002;23:1781–1789. doi: 10.1093/carcin/23.11.1781. [DOI] [PubMed] [Google Scholar]
- 38.Feng Z, Hu W, Amin S, Tang M-s. Biochemistry. 2003;42:7848–7854. doi: 10.1021/bi034431g. [DOI] [PubMed] [Google Scholar]
- 39.Sancar A, Tang M-S. Photochem Photobiol. 1993;57:905–921. doi: 10.1111/j.1751-1097.1993.tb09233.x. [DOI] [PubMed] [Google Scholar]
- 40.Cleaver JE. Nat Rev Cancer. 2005;5:564–573. doi: 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- 41.van Steeg H, Mullenders LH, Vijg J. Mutat Res. 2000;450:167–180. doi: 10.1016/s0027-5107(00)00023-3. [DOI] [PubMed] [Google Scholar]
- 42.Harvey RG. Chemistry and Carcinogenicity. Oxford: Oxford Univ Press; 1991. p. 396. [Google Scholar]
- 43.Stewart BW, Kleihues P, editors. World Cancer Report. Geneva: IARC Press, World Health Organization; 2003. [Google Scholar]
- 44.Eder E, Budiawan Cancer Epidemiol. 2001;10:883–888. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}