

Abstract
The type II PIP kinases phosphorylate the poorly understood inositol lipid PtdIns5P, producing the multi-functional lipid product PtdIns(4,5)P2. To investigate the regulation of these enzymes by phosphorylation, we partially purified a protein kinase from pig platelets that phosphorylated type IIα PIP kinase on an activation loop threonine residue, T376. Pharmacological studies suggested this protein kinase was protein kinase D (PKD), and in vitro experiments confirmed this identification. A phospho-specific antibody was developed and used to demonstrate phosphorylation of T376 in living cells, and its enhancement under conditions in which PKD was activated. Although we were unable to determine the effects of phosphorylation on PIP kinase activity directly, mutation of T376 to aspartate significantly inhibited enzyme activity. We conclude that the type II PIP kinases are physiological targets for PKD phosphorylation, and that this modification is likely to regulate inositol lipid turnover by inhibition of these lipid kinases.
Keywords: PIP kinase, protein kinase D, inositol lipid, protein phosphorylation
Abbreviations: PKD, protein kinase D; PtdIns, phosphatidylinositol; PY, phosphotyrosine; PS, phosphoserine; PT, phosphothreonine; CalC, calphostin C; OA, okadaic acid; DMEM, Dulbecco’s modified Eagle’s Medium; DMSO, dimethyl sulphoxide; HTLE, Hunter thin-layer electrophoresis system; PIP, phosphatidylinositol phosphate; PtdIns(3,4,5)P3,phosphatidylinositol(3,4,5)trisphosphate; PKC, protein kinase C; PHK, phosphorylase kinase; PBS, phosphate buffered saline; TBS, Tris buffered saline; ECL, enhanced chemiluminescence
1. Introduction
The inositol phospholipids are now recognised as a centrally important family of regulatory molecules found within eukaryotic cells, and the various different family members participate in a large number of diverse cellular processes [1]. The most prevalent species, phosphatidylinositol (PtdIns), has had few specific functions ascribed to it, but its phosphorylated derivatives play a host of important roles within the cell. The best characterised of these lipids, PtdIns(3,4,5)trisphosphate (PtdIns(3,4,5)P3) is a second messenger that acts downstream of numerous tyrosine kinases and G-protein coupled receptors [2]. Similarly, another family member, PtdIns(4,5)P2, is now recognised as a pleiotropic regulatory molecule, with important roles in modulating actin polymerisation, ion movements, protein localisation and gene expression [3, 4]. The other inositol lipids (PtdIns3P, PtdIns4P, PtdIns5P, PtdIns(3,4)P2 and PtdIns(3,5)P2) are less well characterised, but it now seems probable that each distinct species possesses unique and important functions [5].
These different phosphorylated isomers of PtdIns are specifically recognised by binding proteins, such that each lipid interacts with a unique spectrum of protein partners [1, 5]. In many cases, this interaction has an important regulatory function, affecting the activity or subcellular localisation of the target protein. It is through their ability to modulate protein function in this way that the lipids exert their profound effects on cellular processes. Moreover, the existence of specific lipid kinases and phosphatases permits the dynamic regulation of cellular levels of the different inositol lipids. A consistent theme of recent research demonstrates the ability of cells to regulate the localised release, synthesis or destruction of particular inositol lipids via changes in the subcellular localisation or activity of such enzymes [4, 6] or of lipid-sequestering proteins [7]. This can result in considerable flexibility in the regulation of functionally distinct pools of individual lipids, and consequently of the lipid-dependent processes they control.
Not surprisingly, given the existence of multiple lipid species with unique functions, a significant number of different inositol lipid kinases has been identified. Research on the PI 3-kinases, signalling enzymes that uniquely permit phosphorylation of the D3 position of the inositol headgroup, is most advanced, but a distinct group, the phosphatidylinositol phosphate kinases (PIP kinases) is emerging as another important family of enzymes that regulate the synthesis of certain key inositol lipids [3, 4]. The PIP kinases can be subdivided into three groups, based on their substrate preferences: type I (also known as the PI4P 5-kinases), type II (or PI5P 4-kinases) and type III (represented in mammals by the PtdIns3P 5-kinase PIK-fyve). Type I PIP kinases are responsible for the bulk of cellular PtdIns(4,5)P2 production, and acute regulation of their activity or subcellular localisation has been documented in many studies in which levels of this lipid change [4]. In contrast, the type II PIP kinases are less well understood. These enzymes were initially identified on the basis of their ability to synthesise PtdIns(4,5)P2 [8], but it is now clear that they do so by a different route from that catalysed by the type I PIP kinases [9]. Specifically, type II isoforms phosphorylate the low-abundance inositol lipid PtdIns5P at position D4 of the inositol headgroup, whereas type I PIP kinases phosphorylate position D5 of the more abundant PtdIns4P. It is presently not clear to what extent the former pathway does, in fact, contribute to the regulation of cellular PtdIns(4,5)P2 pools.
An alternative possibility is that the type II PIP kinases may actually function to control levels of PtdIns5P, the most incompletely understood member of the inositol lipid family. PtdIns5P levels fluctuate in response to a variety of stimuli [10–13], and it has been proposed to function as a second messenger involved in cytoskeletal rearrangements downstream of the insulin receptor [14], or in regulating transcription [15], but its physiological role has yet to be fully characterised. Alternatively, it is also possible that the type II PIP kinases are involved in producing PtdIns(3,4)P2 from PtdIns3P, a reaction they catalyse in vitro [9], though as the recombinant enzymes exhibit a marked preference for PtdIns5P this possibility is rather less likely.
Recent evidence has suggested that, whatever their preferred substrates inside living cells, the type II PIP kinases are somehow involved in regulating turnover of the lipid second messenger PtdIns(3,4,5)P3. Mice lacking the type IIβ isoform are hypersensitive to insulin [16], and insulin-induced activation of the protein kinase Akt, which occurs in response to insulin-stimulated PtdIns(3,4,5)P3 elevation, is enhanced in these animals. Moreover, overexpression of type II PIP kinases in cultured cells leads to a more rapid cessation of signalling downstream of PtdIns(3,4,5)P3 compared with non-transfected controls [17]. This latter effect has been suggested to result from enhanced PtdIns(3,4,5)P3 5-phosphatase activity, but precise details of the role played by type II PIP kinases in this process are lacking.
We have attempted to elucidate the function of the type II PIP kinases by examining their regulation. Previously, we have demonstrated that these enzymes can be phosphorylated on serine and threonine residues in vivo [18]. Here we demonstrate that protein kinase D (PKD) phosphorylates type II PIP kinases in vitro at a threonine residue in the 'activation loop' [19], and that this modification also occurs in vivo. Mutation of this residue to aspartate profoundly reduced PIP kinase activity, consistent with the possibility that phosphorylation acts to inhibit the activity of the kinase.
2. Materials and Methods
2.1 Materials
Recombinant mouse active PKD/PKCμ and human PKD2, anti-PKCμ and Visualiser enhanced chemiluminescence (ECL) substrate were from Upstate. Anti-PKD (phosphorylated on S916) was from Cell Signalling Technology. Gö6976, Gö6983 and bis-indolylmaleimide were from Calbiochem. The BCA protein assay kit was from PerBio. Highly species-specific HRP-conjugated anti-rabbit secondary antibody was from Jackson Labs. Protein G sepharose, chromatography columns and ECL reagents were from Amersham. Purified PHK, HRP-anti-rat light chain antibody, phospho-amino acids, TPCK- treated trypsin, PVP-360, BSA and bovine PtdIns(4,5)P2 were from Sigma-Aldrich. MAC 334 and MAC 344 were from the Babraham Institute. Synthetic inositol lipids were from Echelon. Cell culture media, PBS, serum, antibiotics and Lipofectamine 2000 were from Invitrogen. γ-32P-ATP was from Perkin Elmer. ProLong antifade fixative was from Molecular Probes. All other reagents were of laboratory grade.
2.2 Cell culture and lysis
RAW 264 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% FBS, 100 units ml−1 penicillin and 100μg ml−1 streptomycin, in suspension. HeLa S3 cells were maintained in monolayer culture under the same conditions. For most experiments, 2x106 cells were cultured in 3.5cm dishes (to which the RAW 264 cells adhere) overnight before exposure to indicated conditions. In others, cells were grown in 10cm dishes. Cells were lysed by the addition of ice-cold lysis buffer. For immunoprecipitation of type II PIP kinase we obtained clearer results if RIPA buffer (10mM Tris pH 7.2; 150mM NaCl; 1% NP40; 0.5% sodium deoxycholate; 0.1% SDS; 5mM EDTA) was used for lysis, but this compromised immunoprecipitation of PKD: for PKD experiments a different lysis buffer (25mM HEPES pH 7.4; 150mM NaCl; 1% Triton X-100; 2mM EDTA) was used. In both cases, buffers were supplemented with the following phosphatase and protease inhibitors: 50mM NaF, 10mM β-glycerophosphate, 100μM Na3VO4, 100nM OA, 1mM AEBSF, 10μg/ml leupeptin. Lysates were passed ten times through a 25g needle, and frozen at −80°C until required.
For microscopy, HeLa cells were plated on 22mm square glass coverslips in 6-well plates 24 hours prior to transfection (see below).
2.3 Protein purification
Triton X-100 soluble extracts were prepared from isolated pig platelets, fractionated on columns and assayed for protein kinase activity and protein content as previously described [20]. Columns were run on a BioLogic protein purification system (BioRad).
2.4 Protein phosphorylation assay
Recombinant purified protein kinases were mixed with recombinant PIP kinases and kinase buffer (50mM Tris pH7.4; 80mM KCl; 10mM MgCl2; 2mM EGTA; EGTA was omitted in the experiments with PHK) and incubated with the indicated ATP concentrations (containing 10–50μCi γ-32P-ATP per reaction) for 20 min at 30°C in the presence or absence of inhibitors as indicated in the figure legends. In some cases, SDS-PAGE sample buffer (final concentrations: 80mM Tris pH 6.8; 10% glycerol; 2% SDS; 5% β-mercaptoethanol) was added to stop further reaction, and the samples were boiled and subjected to SDS-PAGE. In other experiments, ice cold immunoprecipitation buffer (140mM NaCl; 40mM Tris pH 7.4; 5mM EDTA; 1% triton X-100) was added and the type II PIP kinase immunoprecipitated (see below). Washed immunoprecipitates were prepared for SDS-PAGE by boiling in SDS-PAGE sample buffer. Electrophoresed proteins were transferred to nitrocellulose membranes and the blots exposed to autoradiography film.
2.5 Phosphoprotein analysis
Recombinant type IIα PIP kinase was phosphorylated as described above. Electrophoresed proteins were transferred to PVDF membranes and the phosphorylated type II PIP kinase band excised. The excised membrane containing type II PIP kinase was blocked with 5mg ml−1 PVP-360 in 0.1% acetic acid for 30 min at 37°C, then digested with TPCK-treated trypsin (100μg ml−1 in 40mM ammonium bicarbonate) at 37°C overnight.
For phospho-amino acid analysis, the liberated peptides were dried down and partially digested by incubating at 110°C for 60 min in 5.7M HCl. The acid hydrolysate was dried down again and redissolved in 10μl pH 1.9 buffer (25:78:897 formic acid : acetic acid : water, by volume) containing 62.5mg ml−1 each of phosphotyrosine, phosphoserine and phosphothreonine. Samples were electrophoretically separated in two dimensions on cellulose-coated thin layer chromatography plates using a Hunter Thin Layer Electrophoresis system (HTLE) 7000 (CBS Scientific) according to the manufacturer’s instructions. Dried plates were exposed to autoradiography film. For 2D phosphopeptide mapping, the tryptic peptides were electrophoresed on cellulose-coated plates using the HTLE 7000 in pH 1.9 buffer according to the manufacturer’s instructions, then separated in the second dimension by thin layer chromatography in a solvent system consisting of 1251:96:58:38:558 isobutyric acid : pyridine : acetic acid : n-butanol : water (by volume). Dried plates were exposed to autoradiography film.
2.6 PIP kinase assay
Recombinant type IIα PIP kinase (wild-type or mutant; prepared by expression in E coli from the vector pGEX-4T1 (Amersham Biosciences) as a GST-fusion protein and cleaved with thrombin according to the manufacturer’s instructions) was diluted in kinase buffer and incubated at 30°C in the presence of sonicated lipids (5μM final concentration for PtdIns5P; 50μM for PtdIns3P) and ATP (5μM final, containing 5–10μCi per sample γ-32P-ATP) for the indicated times at 30°C in a total volume of 100μl. Reaction was stopped by the addition of 500μl 1:1 (v:v) chloroform : methanol containing 1μg ml−1 PtdIns(4,5)P2. Samples were acidified with 125μl 2.4M HCl and the lower phase washed twice with 450μl theoretical upper phase (47:48:3 methanol: 1M HCl: chloroform by volume) before extracting to a fresh tube, drying down and running on a thin layer chromatography plate as described previously [21]. The resolved PtdInsP2 spots were visualised by autoradiography. Radiolabel incorporation into individual spots was quantified by scraping and scintillation counting. The protein content of PIP kinase preparations was assessed using the BioRad protein assay to allow comparison of equal amounts of protein.
2.7 Preparation of anti-phospho T376 antibodies
The anti-pTVK phospho-specific antibody was produced by Resgen (Invitrogen). Briefly, two rabbits were immunised with the phosphopeptide AAHAAK[phospho-T]VKHGAG, corresponding to amino acid residues 370–382 in the human type IIα PIP kinase sequence (and to residues 380–392 or 384–396 in the IIβ and IIγ isoforms respectively). The antiserum was purified by passage down a column to which the phosphopeptide had been coupled; antibodies eluted from this column were then cross-adsorbed to a second column to which the non-phosphorylated peptide had been coupled, to remove any that could cross-react with the non-phosphorylated protein.
2.8 Western blotting
Western blotting of type II PIP kinase, irrespective of phosphorylation state was carried out using the antibody MAC 344 as described previously [22].
When probing blots with anti-pTVK, PVDF membranes were blocked with 5% BSA/ 0.1% Tween-20 in Tris buffered saline (TBS: 140mM NaCl; 40mM Tris pH 7.4) before incubation with a 1:1000 (by volume) dilution of the antibody at room temperature for 1 hour, or overnight at 4°C. The blots were washed 5 times with 25ml TBS/ 0.1% Tween-20. A highly-species specific anti-rabbit second antibody was applied at 1 in 75,000 (by volume) for 45 min and the blots washed again over 2 hours before detection of antibody binding using the Visualiser reagent.
2.8 Immunoprecipitation
All cell lysates were clarified by microcentrifugation at 10 000g for 10 min at 4°C prior to immunoprecipitation, to remove debris and insoluble material.
PKD was immunoprecipitated using an anti-PKCμ antibody. Antibody was added to clarified lysates and incubated at 4°C for 1 hour. 10μl packed volume of protein G sepharose beads was added and the mixture incubated for a further 2 hours at 4°C with mixing. The beads were pelleted by microcentrifugation for 1 min at 10 000g, washed twice with ice-cold TBS, then boiled in 15μl SDS-PAGE sample buffer.
Type II PIP kinase was immunoprecipitated using the anti-type II PIP kinase antibody MAC 334, cross-linked to protein G sepharose beads, as previously described [22].
2.9 Transfection
RAW 264 cells were transfected with Lipofectamine 2000 according to the manufacturer’s instructions. HeLa S3 cells were transfected by the calcium phosphate precipitation method as previously described [23].
2.10 Fluorescence microscopy
On the day after transfection, HeLa S3 cells were treated as indicated before fixation in 4% paraformaldehyde in 100mM sodium phosphate pH7.4. Fixed cells were washed in Dulbecco’s phosphate buffered saline (PBS), permeabilised with 0.2% Triton X-100 in PBS for 5 minutes, washed, and blocked for 60 minutes with 5% goat serum in PBS. Cells were mounted in ProLong antifade and examined on a Zeiss Axioplan 2 fluorescence microscope.
3. Results
In a previous study we partially purified a protein kinase from pig platelets that phosphorylated recombinant human type IIα PIP kinase at a single serine residue, S304, which we identified as the protein kinase CK2 [20]. During the protein purification experiments that contributed to this study we observed that a significant amount of a protein kinase activity able to phosphorylate recombinant type IIα PIP kinase failed to adhere to a DEAE sephacryl column, unlike the CK2 activity we were pursuing. We subsequently applied this material to a heparin agarose column, to which it stuck, eluting as a single peak of activity in an applied salt gradient of 0-1M NaCl (Fig 1a). The fractions containing the kinase activity were diluted two-fold and subjected to anion exchange chromatography (Fig 1b). The activity was successfully recovered after this column step, apparently eluting as two unequal peaks. Several attempts to purify these activities further resulted in loss of kinase activity.
FIGURE 1.
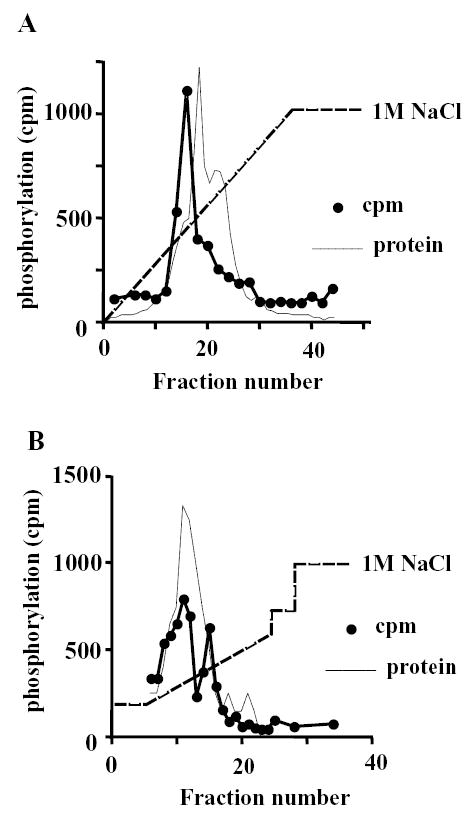
Partial purification of a protein kinase that phosphorylates type IIα PIP kinase.
A) Activity elution from heparin agarose column. The flow-through from a DEAE sephacryl column [20] was loaded onto a 5ml heparin agarose column and the indicated gradient of NaCl applied. 2.5ml fractions were collected and assayed for their ability to phosphorylate recombinant type IIα PIP kinase. The protein elution profile is indicated.
B) Fractions 14–18 from the heparin agarose column were pooled, diluted 2-fold in NaCl-free buffer and loaded onto a Resource Q anion exchange column. The indicated gradient of NaCl was applied. 1ml fractions were collected and assayed for kinase activity using type IIα PIP kinase as a substrate. The protein elution profile is indicated.
Phospho-amino acid analysis demonstrated that the major peak of protein kinase activity from the ion exchange column phosphorylated recombinant type IIα PIP kinase predominantly on threonine residues (Fig 2a). Moreover, two-dimensional tryptic phosphopeptide mapping demonstrated that the majority of radiolabel incorporated into the substrate was located in a single peptide (Fig 2b; arrow), which migrated with an Rf value of 0.47 in the thin-layer chromatography step of the mapping process. Although other peptides are also reproducibly labelled, we found that the intensity of labelling of these sites occurs to a far lesser degree than that of the major phosphopeptide, and was somewhat variable between experiments. The most prominent of these additional phosphopeptides is indicated by an asterisk in Fig 2b. This peptide is considerably less mobile in the solvent system used than is the major phosphopeptide, with an Rf in this experiment of 0.34.
FIGURE 2.
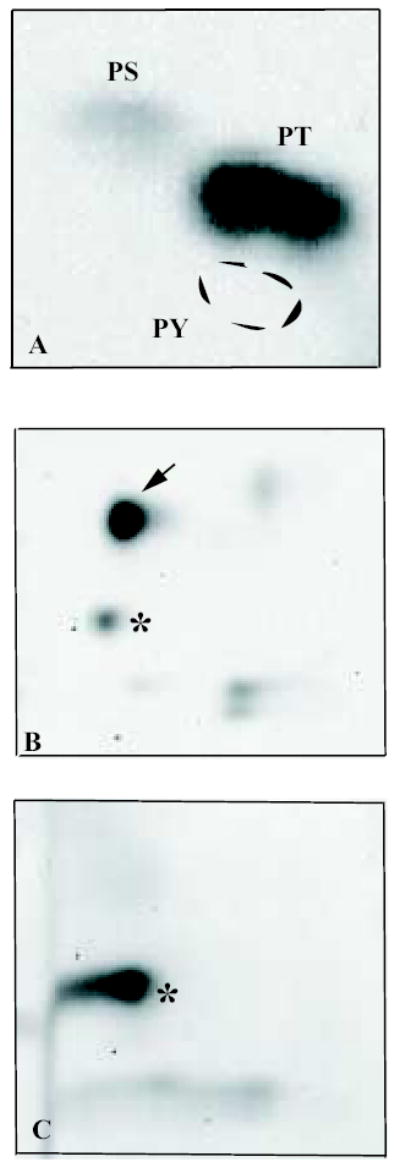
Threonine phosphorylation of type IIα PIP kinase.
A) Phospho-amino acid analysis of type IIα PIP kinase phosphorylated by the partially purified protein kinase activity. 32P labelled spots corresponding to phospho-serine (PS) and phosphothreonine (PT), and the position of migration of the phospho-tyrosine standard (PY), are indicated.
B) 2D phospho-peptide map of type IIα PIP kinase phosphorylated by the partially purified protein kinase. The arrow indicates the phosphopeptide corresponding to the major phosphorylation site. The spot marked by the asterisk corresponds to that indicated in the same way in C: we consistently observe enhanced phosphorylation of this residue in T376 mutants.
C) 2D phospho-peptide map of the T376A mutant of type IIα PIP kinase demonstrating loss of the major phospho-peptide. The spot marked by the asterisk corresponds to the peptide indicated in B (Rf value in this experiment 0.37).
Based on the mobility of the major phosphopeptide in the two-dimensional separation system used, in combination with the amino acid sequence of the type IIα PIP kinase [24], we were able to identify certain threonine residues as potential candidates for the major phosphorylation site. Site-directed mutagenesis was used to convert these residues to alanine, and the effects on type IIα PIP kinase phosphorylation investigated by 2D mapping. Mutation of one such residue, threonine 376 (T376) caused the disappearance of the major phosphorylation site (Fig 2c). This cannot simply be due to misfolding of the protein, leading to a failure of the protein kinase to interact with it, as the T376A mutant can be expressed in bacteria as a GST fusion protein, and retains some PIP kinase activity (not shown). The tryptic peptide corresponding to this site is only three amino acids in length, and contains a single threonine residue and we therefore conclude that the predominant target of the partially purified protein kinase activity is T376. Interestingly, this residue and the amino acid sequence around it are completely conserved in all three mammalian type II PIP kinase isoforms, α, β and γ [24–26].
Disappointingly, no strong protein kinase candidates were identified as being likely to phosphorylate T376 when the sequence was analysed by the program Scansite [27]. However, as the sequence was a reasonable match for a protein kinase Cδ target we investigated the sensitivity of the partially purified activity to inhibitors of protein kinase C (PKC). Although sensitive to the broad-spectrum inhibitor K252a (Fig 3a), the kinase activity was insensitive to the PKC inhibitor calphostin C (CalC). Interestingly, however, it was inhibited by another PKC inhibitor, Gö6976. Sensitivity to the latter in combination with resistance to other PKC inhibitors is a feature of several protein kinases, including protein kinase D 1 (PKD1, also known as PKCμ) [28], CHK1 and phosphorylase kinase (PHK) [29]. CHK1, a tumour suppressor protein involved in cell-cycle regulation, is unlikely to be present in significant amounts in platelets, but this cell type is known to contain PKD [30]. We therefore tested whether PKD and PHK could phosphorylate recombinant type IIα PIP kinase in vitro. 17μg PHK in the presence or absence of 1μM Ca2+ (an activator) was unable to phosphorylate type IIα PIP kinase, though a modest amount of autophosphorylation of a protein band of around 200 Kd was observed, demonstrating that the kinase preparation used was active (Fig 3b). In contrast, 0.15μg recombinant PKD catalysed a robust phosphorylation of type IIα PIP kinase (Fig 3b). Importantly, this phosphorylation occurred predominantly on T376 (Fig 3c).
FIGURE 3.
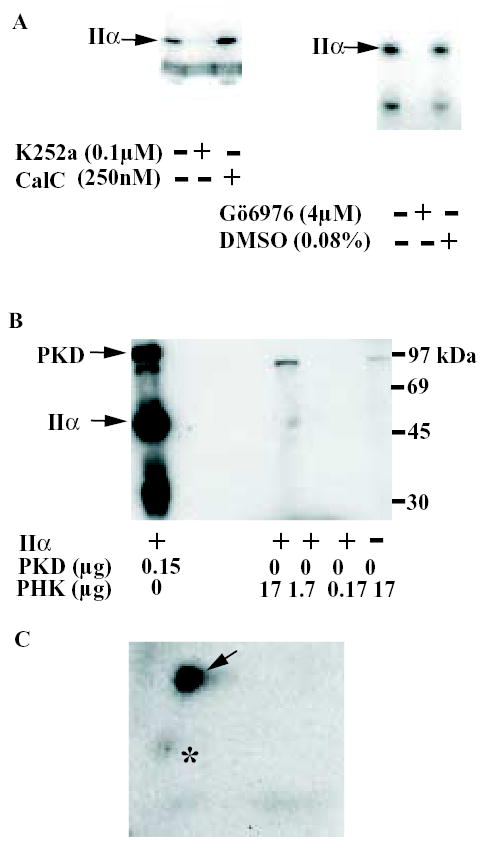
Identification of the partially purified protein kinase as PKD.
A) Effect of protein kinase inhibitors on the protein kinase activity.
The ability of the partially purified protein kinase activity to phosphorylate recombinant type IIα PIP kinase in the presence of the kinase inhibitors K252a, calphostin C (CalC) and Gö6976 at the indicated concentrations was determined. DMSO was used as a vehicle control. The ATP concentration used in the experiment shown in the left-hand panel was 5μM; that in the right-hand panel was 50μM.
B) PKD, but not PHK, phosphorylate recombinant type IIα PIP kinase.
Recombinant type IIα PIP kinase was incubated with the indicated amounts of purified PHK or recombinant PKD at 30°C in the presence of γ-32P-ATP. The samples were then mixed with SDS-PAGE sample buffer, subjected to SDS-PAGE and transferred to a nitrocellulose membrane before being exposed to autoradiography film.
C) 2D phospho-peptide map of type IIα PIP kinase phosphorylated by recombinant PKD. The arrow indicates the T376 phosphopeptide and the asterisk the minor phospho-peptide indicated in the same way in Fig 2b.
To investigate whether this phosphorylation site is of physiological relevance, we developed a phospho-specific antibody that recognised type IIα PIP kinase only when phosphorylated at T376. The characterisation of this antibody, anti-pTVK, is shown in Fig 4a. In Western blots anti-pTVK recognises recombinant type IIα PIP kinase that has previously been incubated with recombinant active PKD in the presence of ATP, but fails to recognise the non-phosphorylated enzyme (Fig 4a). Moreover, pre-incubation with the phosphopeptide used in development of the antiserum, but not a non-phosphorylated peptide of identical sequence, abolishes antibody binding in Western blots. Importantly, the antibody also recognises a band of the correct molecular weight in type II PIP kinase immunoprecipitates from unstimulated RAW 264 cells (Fig 4b, left-hand panel), which is exactly co-incident with the migration of type II PIP kinase immunoreactivity (right-hand panel), demonstrating that T376 is phosphorylated in vivo. As shown in Fig 4b (left-hand panel), several other bands of higher molecular weight are present in anti-pTVK probed blots of type II PIP kinase immunoprecipitates. Those larger than 97kDa are due to non-specific interaction of antibodies with material in the preparation of anti-PIP kinase immunoprecipitation beads. The lower molecular weight bands (around 69kDa) appear to be due to interaction of the antibodies with proteins from the cell lysates that adhere to the beads. The intensity of these bands is somewhat variable between experiments, and unlike that of the type II PIP kinase band (see below), does not vary with exposure of the cells to different conditions. Their clear separation on blots from the type II PIP kinase band means that they do not interfere with the results of experiments that use the anti-pTVK antibody to investigate T376 phosphorylation, and they have been omitted from subsequent figures for clarity.
FIGURE 4.

PKD phosphorylation of type II PIP kinase in vivo.
A) Type IIα PIP kinase was incubated in the presence of 500μM ATP and the presence or absence of PKD at 30°C for 60 min, then subjected to SDS-PAGE. Proteins were transferred to PVDF membranes, blocked with 5% BSA / 0.1% Tween-20 in TBS and probed with 7μg anti-pTVK antibody applied in a total volume of 5.1ml, either without any pre-incubation (left-hand panel), or following incubation overnight at 4°C in the presence of 70μg of either the phospho-peptide AAHAAK[phospho-T]VKHGAG (central panel) or its non-phosphorylated equivalent (right-hand panel).
B) Type II PIP kinase was immunoprecipitated from RAW 264 cells using cross-linked MAC 334 resin. The washed immunoprecipitate was subjected to SDS-PAGE and Western blotting and the blot probed with anti-pTVK antibody (left hand panel). The same blot was subsequently stripped and re-probed using MAC 344 to demonstrate the presence of type II PIP kinase immunoreactivity. The arrow indicates the type II PIP kinase band.
Consistent with the idea that PKD is responsible for the type II PIP kinase phosphorylation, we observed that PKD was constitutively active in these cells, as assessed by phosphorylation of its autophosphorylation site, Serine 916 [31] (Fig 5a). However, we also found that treatment of the cells with hydrogen peroxide, which activates PKD [31], further increased autophosphorylation. Significantly, H2O2 treatment also promoted increased phosphorylation of both native type II PIP kinase (Fig 5b), and of FLAG-tagged type IIα PIP kinase transfected into the cells (Fig 5c), though it had no impact on the intensity of the higher molecular weight bands in the anti-pTVK-probed Western blots (not shown). Furthermore, Gö6976, but not Gö6983 (another PKC inhibitor to which PKD is insensitive) inhibits type II PIP kinase phosphorylation in vivo (Fig 5d). All these pieces of evidence support the idea that type II PIP kinase is a physiological substrate for PKD.
FIGURE 5.
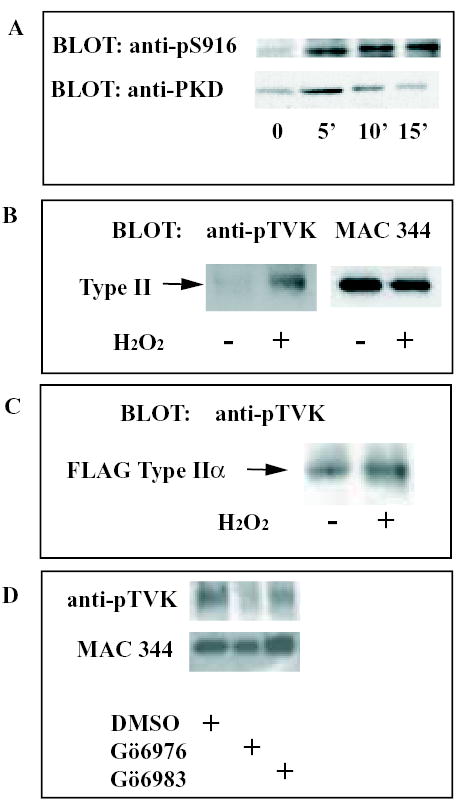
PKD phosphorylation of type II PIP kinase in vivo.
A) RAW 264 cells were incubated in the presence or absence of 1mM H2O2 for the indicated times before lysis. Total PKD was immunoprecipitated from the lysates and the immunoprecipitates analysed by Western blotting. The upper autoradiograph demonstrates the degree of autophosphorylation of PKD on S916, an indication of the degree of activation of the kinase. The lower autoradiograph is the same blot re-probed with an antibody specific to PKD irrespective of its phosphorylation state. We have repeatedly found that PKD is constitutively active at a low level in RAW 264 cells, and that serum deprivation does not abolish this activity (not shown).
B) Type II PIP kinase was immunoprecipitated from lysates of RAW 264 cells incubated in the presence or absence of 1mM H2O2 for 10 min as indicated. The blot was probed with anti-pTVK (left), followed by MAC 344 (right).
C) RAW 264 cells were transfected with FLAG-tagged type IIα PIP kinase. Cells were treated with or without 1mM H2O2 for 10 min before lysis and immunoprecipitation of type II PIP kinase. The blot was probed with anti-pTVK.
D) Following a 1h pre-treatment with the relevant compound, RAW 264 cells were exposed to 1mM H2O2 for 10 min in the presence of DMSO (0.2%), Gö6976 (20μM) or Gö6983 (20μM). Western blots of type II PIP kinase immunoprecipitates were probed with anti-pTVK and, subsequently, MAC 344 antibodies as indicated.
As stated above, the PKD phosphorylation site in type IIα PIP kinase is completely conserved in all three type II isoforms. To confirm that PKD also phosphorylates the type IIβ isoform, we incubated this protein with ATP in the presence or absence of PKD, and used anti-pTVK to confirm that it, too, is a substrate for PKD (not shown). Interestingly, the crystal structure of type IIβ PIP kinase has been solved, and the threonine residue corresponding to T376 in the IIα sequence, T386, is situated in a region topologically analogous to the activation loop of typical protein kinases [19]. The activation loop is an extremely important part of the protein kinase structure, controlling access of substrate to the active site, and many protein kinases are regulated by phosphorylation of this region [32]. However, it should be noted that the corresponding domain of type II PIP kinases is significantly shorter than that of protein kinases [32], and the original study that elucidated the type IIβ PIP kinase structure emphasised that this region was not necessarily functionally analogous to the activation loop of protein kinases [19]. Despite this caveat, site-directed mutagenesis studies have shown that this region is indeed involved in determining substrate specificity and activity in PIP kinases [33, 34]. Firstly, mutagenesis of individual amino acids within the activation loop can have profound effects on enzyme activity [34]. Moreover, the N terminal portion of the activation loop, which does not include the PKD-targetted threonine residue of the type II enzymes, determines whether a PIP kinase will phosphorylate PtdIns4P or PtdIns5P [34]. In fact, the presence of glutamate (which occupies this position in type I PIP kinases) or alanine (from the type IIs) at position 381 in type IIβ PIP kinase is sufficient to dictate whether the enzyme is a PtdIns4P or PtdIns5P kinase. Furthermore, the ability of a PIP kinase to phosphorylate PtdIns4P correlates closely with its localisation to the plasma membrane when transfected into cells [33, 34].
These studies support the idea that the functional role of the activation loop has been conserved between protein kinases and the distantly related type II PIP kinases, and suggested to us that PKD phosphorylation may alter type II PIP kinase activity. To investigate this, we used site-directed mutagenesis to convert T376 of type IIα to aspartate to mimic the effects of phosphorylation by introducing a negative charge.
Expression of T376D-IIα as a GFP-fusion protein in HeLa cells revealed it to display identical subcellular localisation to that of the wild-type enzyme (Fig 6a). This is consistent with the fact that T376 lies outside the region of the activation loop that allows PIP kinases to interact with PtdIns4P. The mutant protein is still capable of phosphorylating PtdIns5P, and the mutation does not enhance the ability of type IIα PIP kinase to utilise PtdIns3P (Fig 6b). However, mutation markedly reduced type IIα PIP kinase activity (Fig 6b and c) to 23.3 ± 2.1% of control (mean ± s.e.m; n=4) using PtdIns5P as a substrate (Fig 6d).
FIGURE 6.
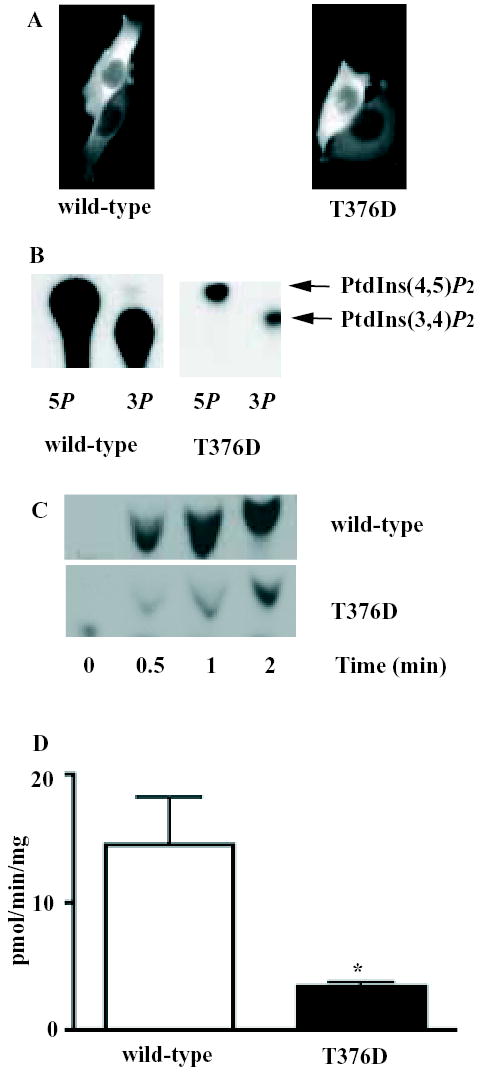
The T376D mutation inhibits type IIα PIP kinase activity but does not affect subcellular localisation or substrate preference.
A) HeLa S3 cells were transfected with GFP-tagged wild-type type IIα PIP kinase, or with the GFP-tagged T376D mutant, fixed and examined by fluorescence microscopy. In both cases, the GFP-tagged protein displays a diffuse cytoplasmic localisation.
B) Bacterially expressed wild-type and T376D IIα PIP kinase were assayed for their ability to phosphorylate PtdIns5P or PtdIns3P (indicated as 5P or 3P respectively). Reaction was stopped after 5min, and the lipid products resolved by thin-layer chromatography. The position of migration of PtdIns(4,5)P2 (produced from PtdIns5P) and of PtdIns(3,4)P2 (produced from PtdIns3P) are shown. Note that the assays using PtdIns3P contained more lipid substrate (50μM cf 5μM for PtdIns5P).
C) PtdIns5P was phosphorylated using 32.5ng per sample of type IIα PIP kinase (wild-type or T376D) for the indicated times, and the lipid product resolved by thin-layer chromatography.
D) Histogram of means ± s.e.m. of wild-type (n=3) and T376D (n=4) type IIα PIP kinase activity, expressed as pmol PtdIns(4,5)P2 produced per minute per μg protein. The asterisk represents statistical significance at the 95% level (Student's unpaired t-test).
Unfortunately, we were unable to determine directly whether phosphorylation produces the same effects on type II PIP kinase activity as mutation, as we were unable to obtain stoichiometric phosphorylation of the type II PIP kinase under conditions that preserved its activity: we experienced considerable attrition of the type II PIP kinase activity after prolonged incubation in both the presence and absence of PKD (not shown). Under conditions that did permit an assessment of PIP kinase activity following incubation with or without protein kinase, the degree of phosphorylation by PKD was less than 10%, and we were unable to detect a significant effect on either lipid kinase activity or substrate preference. This is probably due to the fact that PKD requires phosphorylation by activating kinases for full activity [28].
4. Discussion
In the work presented here, we demonstrate the phosphorylation in vivo of type II PIP kinases at a conserved threonine residue located within the activation loop, and identify the protein kinase involved as PKD. PKD is a signalling kinase that is activated, and that alters its subcellular location, in response to a variety of stimuli (for recent reviews see [28, 35]). However, the physiological functions of PKD remain unclear, and few of its substrates have been identified to date. Although it has been implicated in a number of cellular processes, including regulation of cell division, apoptosis and Golgi function, its specific role in these has not yet been fully elucidated. In our hands, PKD is constitutively active within RAW 264 cells, maintained in 10% FBS. Serum removal for 16 hours does not abolish PKD activity (not shown). However, H2O2 causes further activation of the enzyme. Interestingly, activation of PKD by H2O2 may be physiologically relevant: RAW 264 cells are a mouse monocyte/macrophage cell line, and H2O2, which is released by activated neutrophils, stimulates macrophages ([36] and references therein).
The target site for PKD-mediated phosphorylation has previously been identified as an amino acid residue that is important for type II PIP kinase activity. Mutation of type IIβ T386 to alanine causes an 80% inhibition of the enzyme [34], and we have confirmed that the conversion of T376 of IIα to alanine replicates this effect (not shown). This study demonstrates that mutation of T376 to aspartate cause inhibition of a similar magnitude. These observations emphasise the significance of this amino acid residue for PIP kinase activity: clearly the activity of the enzyme will not tolerate modifications at this site, consistent with the idea that it plays an important role in enzyme regulation. We suggest that, physiologically, such regulation is achieved by phosphorylation of this key residue. In accord with the idea that the type II PIP kinases are negatively regulated by phosphorylation in vivo, we have previously demonstrated that hyperphosphorylation of platelet type II PIP kinase, provoked by OA, correlates with marked reduction of PIP kinase activity in anti-type II immunoprecipitates [18]. Although some of the immunoprecipitated PIP kinase activity was due to co-precipitation of a type I PIP kinase [21], this cannot account for the extent of inhibition seen (>60%; [18]). In the absence of phosphatidic acid (an activator of type I PIP kinases [37], which was not included in these assays [18]) only a modest amount of PtdIns4P kinase activity is present in type II PIP kinase immunoprecipitates (see supplementary figure 1): its loss would not account for the observed degree of inhibition.
SUPPLEMENTARY FIGURE 1.
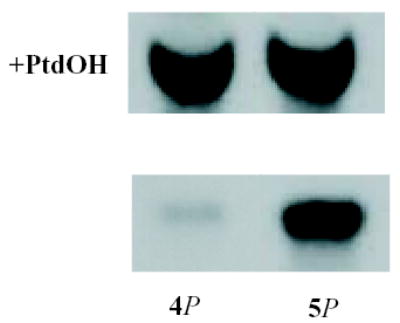
Phosphatidic acid stimulation of type I PIP kinase activity in anti-type II immunoprecipitates.
Type II PIP kinase was immunoprecipitated from unstimulated human platelet lysates. The washed immunoprecipitates were assayed for PIP kinase in the presence of PtdIns4P (4P; 10μM) or PtdIns5P (5P; 10μM) as indicated. Assays using PtdIns4P were carried out in the presence or absence of 10μM phosphatidic acid (PtdOH) as indicated.
In addition to identifying the kinase, its target site and the probable impact of phosphorylation, we have also demonstrated that type II PIP kinase is phosphorylated at this site in living cells, and that stimulation of PKD increases this phosphorylation. This study therefore suggests that the type II PIP kinases are downstream targets of PKD. It is interesting to speculate that PKD, by its phosphorylation of these enzymes, may regulate inositol lipid turnover. Interestingly, a previous report showed that PKD associates and co-immunoprecipitates with a PIP kinase from Cos-7 cells [38], though in this case the PIP kinase activity phosphorylated PtdIns4P but not PtdIns5P, implying it to be a type I isoform. As type I and type II PIP kinases associate in vivo [21] it is possible that such an interaction may be involved in recruiting the type II PIP kinase for phosphorylation by PKD, but this possibility is beyond the scope of the present study.
The physiological consequences of type II PIP kinase phosphorylation by PKD remain to be established. However, the marked reduction in kinase activity seen in the T376D mutant suggests that phosphorylation of this residue has an inhibitory effect on the enzyme. The effect of PKD activation within the cell would thus be to inhibit type II PIP kinases, probably resulting in decreased conversion of PtdIns5P to PtdIns(4,5)P2. Whether the effect of this is to deplete a functionally important PtdIns(4,5)P2 pool, or alternatively to enhance PtdIns5P levels, are key questions to be addressed by future work.
Acknowledgments
This work was supported by an MRC Career Development Award to KAH, and by the Royal Society and the Wellcome Trust (RFI). We thank Professor Peter Parker for supplying a preliminary test sample of recombinant PKD, and Professor Doreen Cantrell for providing reagents used in preliminary experiments. We also wish to thank Drs Jon Clarke, David Brough, Mike Schell and Liz Fitzgerald for helpful discussions, and Sarah Gumusgoz and Andy Letcher for technical support.
References
- 1.Parker PJ. Biochem Soc Trans. 2004;32(Pt 6):893–898. doi: 10.1042/BST0320893. [DOI] [PubMed] [Google Scholar]
- 2.Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Trends Biochem Sci. 1997;22(7):267–272. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 3.Hinchliffe KA, Ciruela A, Irvine RF. Biochim Biophys Acta. 1998;1436:87 – 104. doi: 10.1016/s0005-2760(98)00140-4. [DOI] [PubMed] [Google Scholar]
- 4.Doughman RL, Firestone AJ, Anderson RA. J Membr Biol. 2003;194(2):77–89. doi: 10.1007/s00232-003-2027-7. [DOI] [PubMed] [Google Scholar]
- 5.Payrastre B, Missy K, Giuriato S, Bodin S, Plantavid M, Gratacap M. Cell Signal. 2001;13(6):377–387. doi: 10.1016/s0898-6568(01)00158-9. [DOI] [PubMed] [Google Scholar]
- 6.Comer FI, Parent CA. Cell. 2002;109(5):541–544. doi: 10.1016/s0092-8674(02)00765-1. [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin S, Murray D. Nature. 2005;438(7068):605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 8.Bazenet CE, Ruano AR, Brockman JL, Anderson RA. J Biol Chem. 1990;265(29):18012–18022. [PubMed] [Google Scholar]
- 9.Rameh LE, Tolias KF, Duckworth BC, Cantley LC. Nature. 1997;390(6656):192–196. doi: 10.1038/36621. [DOI] [PubMed] [Google Scholar]
- 10.Morris JB, Hinchliffe KA, Ciruela A, Letcher AJ, Irvine RF. FEBS Lett. 2000;475:57–60. doi: 10.1016/s0014-5793(00)01625-2. [DOI] [PubMed] [Google Scholar]
- 11.Clarke J, Letcher AJ, D'santos CS, Halstead JR, Irvine RF, Divecha N. Biochem J. 2001;357 doi: 10.1042/0264-6021:3570905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sbrissa D, Ikonomov OC, Deeb R, Shisheva A. J Biol Chem. 2002;277(49):47276–47284. doi: 10.1074/jbc.M207576200. [DOI] [PubMed] [Google Scholar]
- 13.Roberts HF, Clarke JH, Letcher AJ, Irvine RF, Hinchliffe KA. FEBS Lett. 2005;579(13):2868–2872. doi: 10.1016/j.febslet.2005.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Sbrissa D, Ikonomov OC, Strakova J, Shisheva A. Endocrinology. 2004;145(11):4853–4865. doi: 10.1210/en.2004-0489. [DOI] [PubMed] [Google Scholar]
- 15.Gozani O, Karuman P, Jones DR, Ivanov D, Cha J, Lugovskoy AA, Baird CL, Zhu H, Field SJ, Lessnick SL, Villasenor J, Mehrotra B, Chen J, Rao VR, Brugge JS, Ferguson CG, Payrastre B, Myszka DG, Cantley LC, Wagner G, Divecha N, Prestwich GD, Yuan J. Cell. 2003;114(1):99–111. doi: 10.1016/s0092-8674(03)00480-x. [DOI] [PubMed] [Google Scholar]
- 16.Lamia KA, Peroni OD, Kim YB, Rameh LE, Kahn BB, Cantley LC. Mol Cell Biol. 2004;24(11):5080–5087. doi: 10.1128/MCB.24.11.5080-5087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carricaburu V, Lamia KA, Lo E, Favereaux L, Payrastre B, Cantley LC, Rameh LE. Proc Natl Acad Sci USA. 2003;100:9867 – 9872. doi: 10.1073/pnas.1734038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinchliffe KA, Irvine RF, Divecha N. Biochem J. 1998;329(Pt 1):115–119. doi: 10.1042/bj3290115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao VD, Misra S, Boronenkov IV, Anderson RA, Hurley JH. Cell. 1998;94:829 – 839. doi: 10.1016/s0092-8674(00)81741-9. [DOI] [PubMed] [Google Scholar]
- 20.Hinchliffe KA, Ciruela A, Letcher A, Divecha N, Irvine RF. Curr Biol. 1999;9:983 – 986. doi: 10.1016/s0960-9822(99)80429-1. [DOI] [PubMed] [Google Scholar]
- 21.Hinchliffe KA, Giudici ML, Letcher AJ, Irvine RF. Biochem J. 2002;363(Pt 3):563–570. doi: 10.1042/0264-6021:3630563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinchliffe KA, Irvine RF, Divecha N. Embo J. 1996;15(23):6516–6524. [PMC free article] [PubMed] [Google Scholar]
- 23.Ciruela A, Hinchliffe KA, Divecha N, Irvine RF. Biochem J. 2000;346:587–591. [PMC free article] [PubMed] [Google Scholar]
- 24.Divecha N, Truong O, Hsuan JJ, Hinchliffe KA, Irvine RF. Biochem J. 1995;309(Pt 3):715–719. doi: 10.1042/bj3090715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castellino AM, Parker GJ, Boronenkov IV, Anderson RA, Chao MV. J Biol Chem. 1997;272(9):5861–5870. doi: 10.1074/jbc.272.9.5861. [DOI] [PubMed] [Google Scholar]
- 26.Itoh T, Ijuin T, Takenawa TJ. Biol Chem. 1998;273(32):20292 – 20299. doi: 10.1074/jbc.273.32.20292. [DOI] [PubMed] [Google Scholar]
- 27.Obenauer JC, Cantley LC, Yaffe MB. Nucleic Acids Res. 2003;31(13):3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rykx A, De Kimpe L, Mikhalap S, Vantus T, Seufferlein T, Vandenheede JR, Van Lint J. FEBS Lett. 2003;546(1):81–86. doi: 10.1016/s0014-5793(03)00487-3. [DOI] [PubMed] [Google Scholar]
- 29.Davies SP, Reddy H, Caivano M, Cohen P. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stafford MJ, Watson SP, Pears CJ. Blood. 2003;101(4):1392–1399. doi: 10.1182/blood-2002-08-2384. [DOI] [PubMed] [Google Scholar]
- 31.Waldron RT, Rozengurt E. J Biol Chem. 2000;275(22):17114–17121. doi: 10.1074/jbc.M908959199. [DOI] [PubMed] [Google Scholar]
- 32.Scheeff ED, Bourne PE. PLoS Comput Biol. 2005;1(5):e49. doi: 10.1371/journal.pcbi.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunz J, Wilson MP, Kisseleva M, Hurley JH, Majerus PW, Anderson RA. Mol Cell. 2000;5(1):1–11. doi: 10.1016/s1097-2765(00)80398-6. [DOI] [PubMed] [Google Scholar]
- 34.Kunz J, Fuelling A, Kolbe L, Anderson RA. J Biol Chem. 2002;277(7):5611–5619. doi: 10.1074/jbc.M110775200. [DOI] [PubMed] [Google Scholar]
- 35.Rozengurt E, Rey O, Waldron RT. J Biol Chem. 2005;280(14):13205–13208. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 36.Cho M, Hunt TK, Hussain MZ. Am J Physiol Heart Circ Physiol. 2001;280(5):H2357–2363. doi: 10.1152/ajpheart.2001.280.5.H2357. [DOI] [PubMed] [Google Scholar]
- 37.Jenkins GH, Fisette PL, Anderson RA. J Biol Chem. 1994;269(15):11547–11554. [PubMed] [Google Scholar]
- 38.Nishikawa K, Toker A, Wong K, Marignani PA, Johannes F-J, Cantley LC. J Biol Chem. 1998;273(36):23126–23133. doi: 10.1074/jbc.273.36.23126. [DOI] [PubMed] [Google Scholar]