

Abstract
Multilocus analysis of single-nucleotide–polymorphism (SNP) haplotypes may provide evidence of association with disease, even when the individual loci themselves do not. Haplotype-based methods are expected to outperform single-SNP analyses because (i) common genetic variation can be structured into haplotypes within blocks of strong linkage disequilibrium and (ii) the functional properties of a protein are determined by the linear sequence of amino acids corresponding to DNA variation on a haplotype. Here, I propose a flexible Bayesian framework for modeling haplotype association with disease in population-based studies of candidate genes or small candidate regions. I employ a Bayesian partition model to describe the correlation between marker-SNP haplotypes and causal variants at the underlying functional polymorphism(s). Under this model, haplotypes are clustered according to their similarity, in terms of marker-SNP allele matches, which is used as a proxy for recent shared ancestry. Haplotypes within a cluster are then assigned the same probability of carrying a causal variant at the functional polymorphism(s). In this way, I can account for the dominance effect of causal variants, here corresponding to any deviation from a multiplicative contribution to disease risk. The results of a detailed simulation study demonstrate that there is minimal cost associated with modeling these dominance effects, with substantial gains in power over haplotype-based methods that do not incorporate clustering and that assume a multiplicative model of disease risks.
It is widely accepted that population-based disease-marker association studies of samples of unrelated affected cases and unaffected controls have the potential to map genes contributing to complex traits, provided that the causative variants are not extremely rare.1,2 The success of this approach relies on genotyping genetic markers—typically SNPs that are in strong linkage disequilibrium (LD) with the functional polymorphism(s)—generated as a result of the shared ancestry of sampled individuals in the flanking region. Initial association studies focused on genotyping high-density SNPs in candidate genes with a functional basis for disease and/or located in regions highlighted by the results of previous linkage-based analyses. However, with improvements in the efficiency of high-throughput SNP genotyping technology, genomewide scans of hundreds of thousands of markers are now under way with the large sample sizes required to detect the modest genetic effects we expect for complex traits.3
One of the most attractive features of SNPs for complex disease-gene mapping is their abundance throughout the genome. However, single-locus analyses—testing for disease association with each SNP, in turn—do not take into account the background patterns of LD between loci and, hence, may be inefficient even before the issue of multiple testing with many markers is addressed. Data from the International Haplotype Map (HapMap) project suggest that much of the human genome can be arranged in blocks of common SNPs in strong LD with one another.4,5 Haplotype diversity within blocks is very much driven by mutation, rather than by ancestral recombination events. Thus, much of common genetic variation can be structured into haplotypes within blocks that are rarely disturbed by meiosis. Furthermore, Clark6 emphasizes that the functional properties of a protein are determined by the linear sequence of amino acids corresponding to DNA variation on a haplotype. For example, there is evidence that a combination of causal variants in cis in the HPC2/ELAC2 gene increases the risk of prostate cancer.7 This suggests that appropriate multilocus analyses of SNP haplotypes within blocks of strong LD may provide evidence of association for modest genetic effects, even when the individual polymorphisms themselves do not.
The most convenient framework for the development of statistical methodology for multilocus analyses of population-based association studies is the logistic-regression model. It is common to assume a multiplicative model of disease risks, so that paternally and maternally derived alleles contribute independent effects. Under this assumption, the logistic-regression model can be parameterized in terms of the risk (or, more precisely, the odds) of disease for each marker-SNP haplotype. Within this framework, it is straightforward to accommodate covariates that may include environmental and other nongenetic risk factors, polygenic effects, and genotypes at ancestrally informative markers, to allow for underlying population stratification. To allow for unknown phase, we consider all possible pairs of haplotypes consistent with the observed genotype data for each individual.8–11 Each consistent haplotype configuration is weighted in the logistic-regression likelihood by the corresponding phase-assignment probability, easily estimated within blocks of strong LD by statistical algorithms, such as PHASE,12,13 that also take into account additional uncertainty due to missing genotype data.
A major limitation of haplotype-based analyses is lack of parsimony, since we require an odds parameter in the logistic-regression model for each distinct marker-SNP haplotype consistent with the observed genotype data. However, we can take advantage of the expectation that chromosomes carrying the same causal variant tend to share more-recent common ancestry at the underlying functional polymorphism(s) than do a random pair from the population and, thus, are more likely to carry similar haplotypes in the flanking genomic region. Thus, by clustering marker-SNP haplotypes according to their similarity, we can assign the same genetic effect(s) to haplotypes within the same clade, reducing the number of parameters in the logistic-regression model, without substantial loss of information.11,14–21 Morris11 clusters marker-SNP haplotypes according to a Bayesian partition model.22,23 The model is specified by selecting cluster “centers” from the set of distinct haplotypes consistent with the observed genotype data, identified via implementation of the expectation-maximization (EM) algorithm.24 The remaining haplotypes are then assigned to the nearest cluster center, where similarity is defined in terms of marker-allele matches, appropriate for haplotype diversity driven by mutation, such as that expected within blocks of SNPs in strong LD with each other.
In this article, I generalize the approach developed by Morris,11 to allow for more-flexible modeling of marker-SNP haplotype association with disease—in particular, to account for dominance effects, here corresponding to any deviation from multiplicative disease risks. This is achieved by introducing a latent variable to describe the presence or absence of causal variants at the functional polymorphism(s) on each marker-SNP haplotype, in a way similar to that of Clayton et al.25 I assume that each causal variant has the same genetic effects on disease. Thus, the logistic-regression model can be parameterized in terms of (i) the additive effect (or multiplicative risk) of causal variants and (ii) the dominance effect of causal variants over other alleles at the functional polymorphism(s). The Bayesian partition model is then used to describe the correlation between marker-SNP haplotypes and alleles at the functional polymorphism(s). Under this model, each haplotype allocated to the same cluster is assigned the same probability of carrying a causal variant. I develop a reversible-jump Markov chain–Monte Carlo (MCMC) algorithm, GENEBPMv2, to sample over the space of haplotype clusters and the corresponding probabilities that they carry a causal variant at the functional polymorphism(s), in addition to additive and dominance effects of the causal variants and covariate-regression coefficients.
The GENEBPMv2 algorithm can be used to assess the evidence in favor of disease association with polymorphisms in a candidate gene or a small candidate region. However, as the length of the candidate region increases, recombination will play an increasingly important role in driving haplotype diversity, and there will be greater uncertainty and less accuracy in phase assignment from unphased genotype data. I illustrate the method by application to high-density unphased SNP-genotype data across an 890-kb candidate region flanking the CYP2D6 gene, for association with a recessive poor-drug-metabolizer (PDM) phenotype.26 The results of my analysis provide overwhelming evidence of association of the PDM phenotype with polymorphisms in the candidate region and correctly highlight the recessive effect of the underlying causal variants. Further, I identify two clusters of haplotypes, both with high probability of carrying different causal variants in CYP2D6, relative to all other haplotypes. Consequently, I am able to distinguish individuals with PDM carrying two copies of the most common causal variant from those carrying rarer, high-risk mutations at the functional polymorphisms in CYP2D6.
Dominance is often overlooked in haplotype-based association studies because the gain in power over a model of multiplicative disease risks is generally not appreciable, unless there is a strong recessive effect of the causative variant or there is overdominance, and because there is a fear that a less parsimonious model will lose power as a result of the multiple-testing burden. However, I demonstrate, by simulation, that, within the Bayesian framework developed here, there is minimal loss in power from allowance for deviations from a multiplicative model of disease risks with the GENEBPMv2 algorithm. Encouragingly, my results suggest substantial gains in power over existing haplotype-based tests of association that do not allow for dominance and clustering. This clearly demonstrates that, with an appropriate analysis such as that implemented here in the GENEBPMv2 algorithm, dominance effects can and should be included in haplotype-based association studies to increase power without penalty for reduced parsimony.
Model and Methods
Consider a case-control sample of unrelated individuals, typed at N marker SNPs in a candidate gene or small candidate region, yielding unphased genotypes G. Alleles at each SNP are coded as “1” for the major allele (i.e., the most frequent in the population) and as “2” for the minor allele. The disease status of individual i is denoted yi=1 if affected and yi=0 if unaffected. I allow for additional covariates, x, each scaled to have zero mean and unit variance. The set of J distinct marker-SNP haplotypes consistent with the observed unphased genotypes is denoted 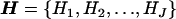 , where Hj denotes the jth most frequent haplotype. Relative haplotype frequencies, h, are estimated by means of maximum likelihood via implementation of the EM algorithm, where hj denotes the frequency of Hj. I denote by
, where Hj denotes the jth most frequent haplotype. Relative haplotype frequencies, h, are estimated by means of maximum likelihood via implementation of the EM algorithm, where hj denotes the frequency of Hj. I denote by 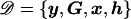 the set of observed data and estimated haplotype frequencies.
the set of observed data and estimated haplotype frequencies.
I assume that alleles at the functional polymorphism(s) in the candidate gene can be classified as high-risk “causative” variants and as low-risk “protective” variants. Each causative variant is assumed to confer the same risk of disease, and likewise for protective variants. I can then model the log-odds of disease within a logistic-regression framework, parameterized in terms of additive and dominance effects of the causative variant, denoted βA and βD, respectively. Under the null model, M0, of no association of disease with polymorphisms in the candidate region, βA=βD=0, whereas the alternative model, M1, corresponds to βA>0 and βD unconstrained. Within the Bayesian paradigm, it is common to evaluate the evidence against the null model by means of the Bayes factor,
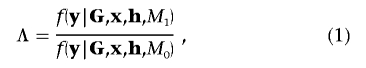 |
where 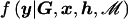 denotes the marginal likelihood of observed phenotype data under model ℳ.
denotes the marginal likelihood of observed phenotype data under model ℳ.
The marginal likelihood of model ℳ can be calculated by integration over model parameters θ, summarized by the directed acyclic graph (DAG) in figure 1. Model parameters include the genetic effects βA and βD but also baseline log-odds of disease, μ, and covariate-regression coefficients, γ. However, since I do not observe genotypes at the functional polymorphism(s) directly, θ must also include parameters to describe the correlation between causal variants and marker-SNP haplotypes in the candidate gene, here taken to be specified by a Bayesian partition model. Then,
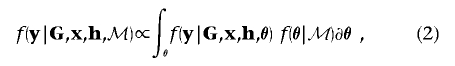 |
where 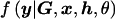 denotes the likelihood of observed phenotype data, given parameters θ, and
denotes the likelihood of observed phenotype data, given parameters θ, and 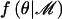 is their joint prior density under model ℳ.
is their joint prior density under model ℳ.
Figure 1. .

DAG representing the model underlying the likelihood, 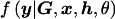 , given by equation (3). The gray nodes represent observed data and estimated relative haplotype frequencies, obtained via implementation of the EM algorithm. The likelihood depends on a number of model parameters, θ—including the baseline risk of disease (μ), covariate-regression coefficients (γ), and genetic effects (β)—of causative variants at the functional polymorphism(s). To evaluate the likelihood, I allow for the correlation between marker-SNP haplotypes (H) and genotypes (Z) at the functional polymorphism(s), by means of a Bayesian partition model. The model is parameterized in terms of the number of clusters of haplotypes (K), the cluster centers (C), and the probability (φ) that haplotypes within each cluster carry a causative variant at the functional polymorphism(s). The parameters, θ, depend on the underlying model of association (ℳ) of disease with marker SNPs. Under the null model, M0, the genetic effects are zero, and there is a single cluster of haplotypes. Under the alternative model of association, M1, I allow for dominance effects of the causative variants at the functional polymorphism(s), and there are at least two clusters in the partition of haplotypes.
, given by equation (3). The gray nodes represent observed data and estimated relative haplotype frequencies, obtained via implementation of the EM algorithm. The likelihood depends on a number of model parameters, θ—including the baseline risk of disease (μ), covariate-regression coefficients (γ), and genetic effects (β)—of causative variants at the functional polymorphism(s). To evaluate the likelihood, I allow for the correlation between marker-SNP haplotypes (H) and genotypes (Z) at the functional polymorphism(s), by means of a Bayesian partition model. The model is parameterized in terms of the number of clusters of haplotypes (K), the cluster centers (C), and the probability (φ) that haplotypes within each cluster carry a causative variant at the functional polymorphism(s). The parameters, θ, depend on the underlying model of association (ℳ) of disease with marker SNPs. Under the null model, M0, the genetic effects are zero, and there is a single cluster of haplotypes. Under the alternative model of association, M1, I allow for dominance effects of the causative variants at the functional polymorphism(s), and there are at least two clusters in the partition of haplotypes.
Bayesian Partition Model
The Bayesian partition model is defined by specifying K cluster centers, ordered and without replacement from the set of haplotypes, H, denoted by 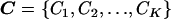 . Haplotype Hj is then assigned to the cluster with the maximum similarity metric, defined as
. Haplotype Hj is then assigned to the cluster with the maximum similarity metric, defined as
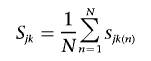 |
for the kth cluster center, Ck. The nth SNP similarity metric, sjk(n), is given by
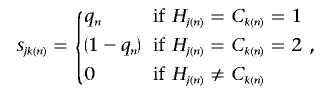 |
where Hj(n) and Ck(n) denote the allele present at SNP n on haplotype Hj and cluster center Ck, respectively, and qn denotes the relative sample frequency of the minor allele in controls. If haplotype Hj is equidistant from more than one cluster center, it is assigned to the center with minimum k. According to the Bayesian partition model, each haplotype assigned to the same clade is then assumed to have the same probability of carrying a causal variant at the functional polymorphism(s), denoted φk for the kth cluster.
The similarity metric, Sjk, treats haplotypes that share rare alleles as less diverse than those that share common alleles, because they are expected to share more-recent common ancestry. Thus, I quantify allele sharing by the complimentary-allele frequency, in the same way as did Durrant et al.20—that is, by 1-qn for minor-allele sharing at SNP n and by qn for major-allele sharing. A number of alternative metrics have been proposed—for example, those that weight SNP-allele matches according to their distance from a putative disease locus in the context of fine mapping18 or that allow for mismatch of alleles due to ancestral mutation or gene-conversion events.21
Likelihood Calculation
To calculate the likelihood term,  , in equation (2), I must consider all possible pairs of marker-SNP haplotypes consistent with the observed unphased genotype data. Then, the likelihood can be expressed as a summation over H, weighted by the corresponding phase-assignment probabilities, given by
, in equation (2), I must consider all possible pairs of marker-SNP haplotypes consistent with the observed unphased genotype data. Then, the likelihood can be expressed as a summation over H, weighted by the corresponding phase-assignment probabilities, given by
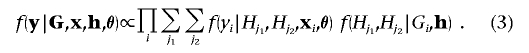 |
Assuming Hardy-Weinberg equilibrium,
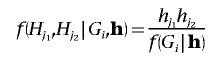 |
and 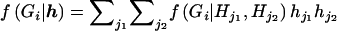 , where
, where 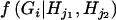 is an indicator variable of the consistency of the genotype Gi with the pair of haplotypes Hj1 and Hj2.
is an indicator variable of the consistency of the genotype Gi with the pair of haplotypes Hj1 and Hj2.
I must then consider the three possible genotype categories at the functional polymorphism(s), denoted by 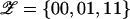 , corresponding, respectively, to two protective variants (not necessarily copies of the same allele), to one protective variant and one causative variant, and to two causative variants (not necessarily copies of the same allele). Thus, denoting the genotype of the ith individual at the functional polymorphism(s) by Zi, it follows that
, corresponding, respectively, to two protective variants (not necessarily copies of the same allele), to one protective variant and one causative variant, and to two causative variants (not necessarily copies of the same allele). Thus, denoting the genotype of the ith individual at the functional polymorphism(s) by Zi, it follows that
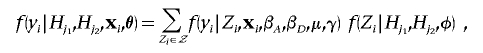 |
where
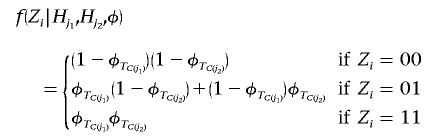 |
and 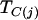 denotes the cluster assignment of haplotype Hj in partition C. Finally, within a logistic-regression framework,
denotes the cluster assignment of haplotype Hj in partition C. Finally, within a logistic-regression framework,
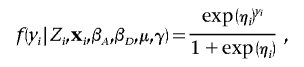 |
where the linear component, ηi, is given by
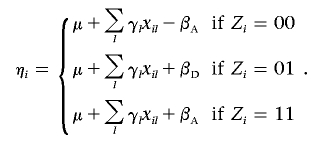 |
Prior-Density Function
The Bayes factor (Λ) in favor of disease association with polymorphisms in the candidate gene depends crucially on the prior-density function of parameters, 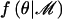 , under each model ℳ. Logistic-regression model parameters are assumed a priori to be independent of the partition of haplotypes. The baseline log-odds of disease is assumed to have a uniform prior distribution, whereas covariate-regression coefficients are assumed to have independent standard normal prior distributions, irrespective of the model of association. Under the null model, M0, the genetic effects βA=βD=0 and the haplotype clustering is irrelevant to disease risk, so that
, under each model ℳ. Logistic-regression model parameters are assumed a priori to be independent of the partition of haplotypes. The baseline log-odds of disease is assumed to have a uniform prior distribution, whereas covariate-regression coefficients are assumed to have independent standard normal prior distributions, irrespective of the model of association. Under the null model, M0, the genetic effects βA=βD=0 and the haplotype clustering is irrelevant to disease risk, so that
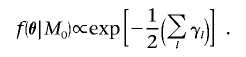 |
Conversely, under the alternative model, M1, the genetic effects are assumed a priori to have standard normal distributions, subject to the constraint βA>0. In defining the Bayesian partition model, each haplotype in H has equal prior probability of selection as one of the K cluster centers, where each cluster has independent uniform prior probability of carrying a causal variant at the functional polymorphism(s). Furthermore, the unconditional prior density of the number of clusters, K>1, has a geometric distribution, such that 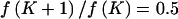 . Thus,
. Thus,
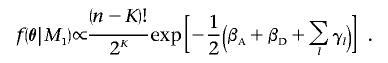 |
I investigate the properties of the Bayes factor, Λ, for these prior-density functions by simulation, explained below.
MCMC Algorithm
I have developed a Metropolis-Hastings MCMC algorithm27,28 to approximate the posterior density of parameters under model ℳ, given by
in the integrand of equation (2). The dimensionality of θ depends on the number of clusters, K, of haplotypes. This can be addressed by incorporating a birth-death process for the number of clusters via implementation of a reversible-jump step in the MCMC algorithm.29 At each stage of the algorithm, a new candidate set of parameter values, θ′, is proposed by making a small change to the current set, θ, as detailed in appendix B. The proposed set of parameter values is then accepted in place of θ, with probability proportional to 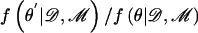 ; otherwise, the current set is retained.
; otherwise, the current set is retained.
The MCMC algorithm is run for an initial burn-in period to allow convergence from a randomly selected set of starting values of θ, assessed using standard diagnostics.30 After convergence, each set of parameter values accepted, or retained, by the algorithm represents a random draw from the posterior density 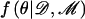 . Autocorrelation between consecutive draws is reduced by recording the sampled set of parameter values only at every tth iteration of the algorithm, for some suitably large t.
. Autocorrelation between consecutive draws is reduced by recording the sampled set of parameter values only at every tth iteration of the algorithm, for some suitably large t.
To approximate the Bayes factor in equation (1), I perform two independent runs of the MCMC algorithm—one under the null model, M0, and one under the alternative model, M1. Over R recorded MCMC outputs for each independent run,
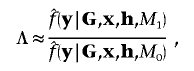 |
where
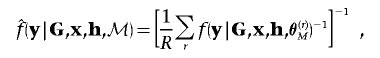 |
and 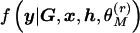 denotes the likelihood in equation (3), recorded in the rth output under model ℳ.
denotes the likelihood in equation (3), recorded in the rth output under model ℳ.
Interpretation
The Bayes factor, Λ, reflects the strength of evidence in favor of disease association with polymorphisms in the candidate gene. By convention, 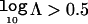 corresponds to positive evidence of association, whereas
corresponds to positive evidence of association, whereas 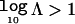 and
and 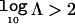 correspond to strong and decisive evidence, respectively.31 The posterior probability of association can then be approximated by
correspond to strong and decisive evidence, respectively.31 The posterior probability of association can then be approximated by
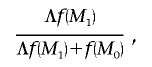 |
where 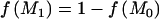 is the prior probability against the null model, reflecting beliefs about association between disease and polymorphisms in the candidate gene before the data is looked at. This probability might take into account the functional relevance of the gene or the results of previous linkage and association studies of the same disease. The issue of multiple testing with many candidate genes or regions can also be addressed by increasing the prior probability in favor of the null model for each.32
is the prior probability against the null model, reflecting beliefs about association between disease and polymorphisms in the candidate gene before the data is looked at. This probability might take into account the functional relevance of the gene or the results of previous linkage and association studies of the same disease. The issue of multiple testing with many candidate genes or regions can also be addressed by increasing the prior probability in favor of the null model for each.32
Software Availability
The GENEBPMv2 software has been developed to (i) obtain maximum-likelihood estimates of the relative frequencies of haplotypes consistent with a sample of observed SNP genotypes, via application of the EM algorithm; (ii) implement the MCMC algorithm to sample over the space of covariate-regression coefficients under the null model of no association; (iii) implement the reversible-jump MCMC algorithm to sample over the space of haplotype clusters and the corresponding probabilities that they carry the causal variant at the functional polymorphism(s), in addition to the additive and dominance effects of the causal variant and covariate-regression coefficients under the alternative model of association; and (iv) estimate Λ and summarize the output of the MCMC algorithm. The GENEBPMv2 software is available, as a suite of Linux executables, on request from the author.
Results
In this section, I demonstrate the utility of the proposed method by applying the GENEBPMv2 algorithm to the detection of association of the PDM phenotype with polymorphisms in the CYP2D6 gene. I also present the results of a simulation study to investigate the performance of the GENEBPMv2 algorithm to detect disease association with polymorphisms in candidate regions of up to 100 kb in length.
Example Application: CYP2D6
The gene CYP2D6 on human chromosome 22q13 has an established role in drug response and is known to be involved in the metabolism of ∼20% of commonly prescribed compounds.33 Four functional polymorphisms have been identified in the gene; the most common mutation, G1846A, occurs with relative frequency of 20.7% in the general population.34 The PDM phenotype acts in a recessive fashion and is manifested in individuals homozygous or compound heterozygous for causal variants at these functional polymorphisms.
Hosking et al.26 genotyped 1,108 individuals at 32 SNP markers across an 890-kb region flanking CYP2D6, to evaluate the efficacy of mapping methods to identify the gene. By the typing of the sample at the four known functional polymorphisms, 41 individuals were predicted to have the PDM phenotype. Single-locus analysis of the markers identified 10 SNPs displaying strong association with the PDM phenotype and residing in a block of strong LD that includes CYP2D6. Here, I apply the GENEBPMv2 software to the 32 marker SNPs (excluding the functional polymorphisms) to demonstrate (i) evidence of the recessive effect of causal variants in CYP2D6, by allowing for deviations from a multiplicative model of PDM phenotype risks, and (ii) clustering of haplotypes carrying the same causal variant in CYP2D6.
Implementation of the EM algorithm identified 906 haplotypes consistent with the observed marker-SNP genotype data. There were 17 common haplotypes with estimated relative frequency of at least 1%. I performed two independent runs of the MCMC algorithm: once under the null model (βA=βD=0) and once under the alternative model (βA>0 and βD unconstrained). Each run of the MCMC algorithm consisted of an initial 100,000 iteration burn-in period, to allow convergence from a random starting parameter set. In the subsequent 1,000,000-iteration sampling period, output of the algorithm was recorded every 1,000th iteration.
Figure 2 presents a summary of output from the sampling period of the MCMC algorithm (1,000 recorded outputs) under the alternative model of association. Figure 2A and 2B presents the prior and posterior distributions of the additive and dominance effects of causative variants at functional polymorphisms in CYP2D6, with figure 2C demonstrating the posterior correlation between them. Finally, figure 2D presents the posterior distribution of the number of clusters, here ranging from 3 to 9, with a mode of 4. The 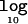 Λ is 48.436, which provides overwhelming evidence in favor of association of the PDM phenotype with polymorphisms in the candidate region. The posterior mean (±SD) additive and dominance effects of causal variants in CYP2D6 are estimated as 4.17 (±0.52) and −2.26 (±0.67), respectively. The dominance effect is negative, which correctly highlights the recessive nature of deviations from the multiplicative model of disease risks.
Λ is 48.436, which provides overwhelming evidence in favor of association of the PDM phenotype with polymorphisms in the candidate region. The posterior mean (±SD) additive and dominance effects of causal variants in CYP2D6 are estimated as 4.17 (±0.52) and −2.26 (±0.67), respectively. The dominance effect is negative, which correctly highlights the recessive nature of deviations from the multiplicative model of disease risks.
Figure 2. .

Summary of the output of a single run of the MCMC algorithm under the general alternative model of association with the PDM phenotype for 32 marker SNPs across an 890-kb region flanking the CYP2D6 gene. A, Prior and posterior distribution of additive effects of causal variants. B, Prior and posterior distribution of dominance effects of causal variants. C, Correlation between the posterior distribution of additive and dominance effects of the causal variants. D, Posterior distribution of the number of clusters of haplotypes.
Figure 3 presents a summary of the posterior partition of common marker-SNP haplotypes across the 890-kb region flanking the CYP2D6 gene under the alternative model, identifying four clear clusters, labeled “A–D.” The dendogram was constructed using standard average-linkage hierarchical-clustering techniques35 based on a posterior measure of pairwise similarity, given by the proportion of MCMC outputs in which each pair of haplotypes appears in the same cluster of the Bayesian partition model. To interpret the clustering, I estimate the posterior probability, ψj, that haplotype Hj carries a causal variant at the functional polymorphisms in CYP2D6 (table 1). Over R outputs of the MCMC algorithm,
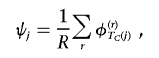 |
where φ(r)TC(j) denotes the probability that haplotype Hj carries a causal variant at the functional polymorphisms recorded in the rth output. Marker-SNP haplotypes in cluster A have a 98% probability of carrying a causal variant, whereas those in cluster B have a 15%–16% probability, both considerably higher than the baseline risk of 1%–2% in clusters C and D. Note that, for a case-control study, ψj does not represent the population risk of carrying a causal variant on haplotype Hj, since the sample is enriched for affected individuals and, thus, for high-risk alleles.
Figure 3. .

Dendogram of the 17 common haplotypes from a single run of the MCMC algorithm under the general alternative model of association with the PDM phenotype for 32 marker SNPs across an 890-kb region flanking the CYP2D6 gene. Haplotypes are coded according to their relative frequency, where 1 denotes the most common. Haplotypes within the four clusters, labeled “A–D,” are similar in terms of allele matching and their risk of carrying causal variants for the PDM phenotype.
Table 1. .
Approximate Posterior Probability That Hj Carries a Causal Variant at the Functional Polymorphisms in CYP2D6
| Marker-SNP Haplotype Hj | Clustera | j | hj (%) | 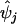 |
| 11111111121121111121111121222111 | A | 3 | 3.42 | .979 |
| 11111111121121111121111121221111 | A | 5 | 2.42 | .979 |
| 11111111121121111121111121111111 | A | 6 | 2.35 | .979 |
| 22122111111212211112111111111111 | B | 11 | 1.66 | .143 |
| 22122111111212211112111111111221 | B | 17 | 1.10 | .143 |
| 11111111111111122211111112221111 | C | 2 | 4.66 | .009 |
| 22122111212121121211111111112111 | C | 7 | 2.34 | .015 |
| 11111111121111122211111112221111 | C | 9 | 1.71 | .009 |
| 22122111212121121211111111111121 | C | 10 | 1.71 | .012 |
| 11111222112111122211111112221111 | C | 16 | 1.20 | .009 |
| 11111111111212211112111111221111 | D | 1 | 6.90 | .017 |
| 11111111111212211112111111111121 | D | 4 | 3.02 | .019 |
| 11111111111212211112111111112111 | D | 8 | 1.90 | .020 |
| 11111111111212211112111111221112 | D | 12 | 1.65 | .018 |
| 11111111111212211112111111111111 | D | 13 | 1.52 | .017 |
| 11111111111212211112111111111221 | D | 14 | 1.40 | .020 |
| 11111111111212211122111111221111 | D | 15 | 1.33 | .022 |
Clusters correspond to four clades identified in the dendogram of haplotypes (fig. 3).
As a final stage in the analysis, I investigated the relatedness of the 41 individuals with the PDM phenotype, illustrated by the dendogram presented in figure 4, constructed using hierarchical-clustering techniques based on the output of the MCMC algorithm for the alternative model. Similarity between a pair of individuals is given by the posterior mean number of haplotypes they share from the same cluster of the Bayesian partition model over all MCMC outputs. For the rth output, the mean sharing is calculated over all possible haplotype configurations consistent with the observed genotype data of the pair of individuals, weighted by the corresponding phase-assignment probabilities. For each combination of phase assignments, sharing is scored as “2” if the individuals share both pairs of haplotypes from the same cluster(s), as “1” if they share one pair of haplotypes from the same cluster, and as “0” otherwise.
Figure 4. .

Denodogram of the 41 cases from a single run of the MCMC algorithm under the general alternative model of association with the PDM phenotype for 32 marker SNPs across an 890-kb region flanking the CYP2D6 gene. The dendogram is constructed to illustrate the relatedness of individuals in terms of the posterior mean number of haplotypes they share from the same cluster (fig. 3). Individuals with PDM are coded according to their genotype at functional polymorphisms in the CYP2D6 gene, where 1 is the G1846A mutation, 2 is the delA2548 mutation, and 3 is the delT1707 mutation.
Figure 4 indicates the genotype of each individual at functional polymorphism(s) in CYP2D6: “1” corresponds to the common G1846A mutation, whereas “2” and “3” correspond to rarer mutations delA2548 and delT1707, respectively. The dendogram distinguishes, with remarkable accuracy, individuals with different CYP2D6 genotypes. The 32 individuals carrying the 1/1 genotype form a tight cluster, with posterior mean sharing of haplotypes from the same cluster close to 2, as would be expected. All 32 of these individuals carry two haplotypes from cluster A, which suggests that this clade is highly associated with the G1846A mutation in CYP2D6. The seven individuals carrying the 1/2 genotype also form a tight cluster. In addition to carrying one haplotype from cluster A, all seven of these individuals also carry one haplotype from cluster B, which suggests that this rarer clade is highly associated with the delA2548 mutation in CYP2D6. Morris et al.36 analyzed the same data in an attempt to fine map functional polymorphisms in the CYP2D6 gene, using the COLDMAP software. Their analysis was also able to distinguish cases carrying zero, one, or two copies of the most common mutation in the gene, although the clustering was not quite so clear cut.
Simulation Study
For simulation purposes, I consider a range of complex disease models for different causative variant frequencies at a single functional polymorphism. For each simulation model, I generate 1,000 replicates of unphased marker-SNP genotype data for unrelated cases and controls. Each replicate is obtained as follows:
-
1.
Generate an ancestral recombination graph37 for a population of 20,000 SNP haplotypes from a realization of the coalescent process with recombination, simulated using the MS software.38 I assume scaled mutation and recombination rates of 4 per 10 kb of the region.39 For an effective population size of 10,000 individuals, this corresponds to a mutation rate of 10-8 per site, per chromosome, and per generation, and a uniform recombination rate of 1 cM per Mb.
-
2.
Select the functional polymorphism at random from all SNPs, so that the causative variant will occur at a rate close to the predefined frequency.
-
3.
Select markers at random from the remaining SNPs, with probability
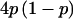 , where p is the minor-allele frequency (MAF). This distribution reflects the bias in MAF towards common SNPs in public databases, such as the International HapMap project.4,5
, where p is the minor-allele frequency (MAF). This distribution reflects the bias in MAF towards common SNPs in public databases, such as the International HapMap project.4,5 -
4.
Generate a diploid individual by sampling a pair of haplotypes, at random and with replacement, from the population of 20,000 chromosomes. Generate the disease phenotype of the individual according to their genotype at the functional polymorphism and the predefined simulation disease model. Repeat this step until the required number of cases and controls have been simulated.
-
5.
Retain the unphased genotype of each individual only at the marker SNPs.
For each replicate of data, I obtain maximum-likelihood estimates of marker-SNP haplotype frequencies via implementation of the EM algorithm. I then perform two independent runs of the MCMC algorithm: once under the null model of no association (βA=βD=0) and once under a general alternative model allowing for nonmultiplicative disease risks (βA>0 and βD unconstrained). Each run of the MCMC algorithm consists of an initial 100,000 iteration burn-in period, with output recorded every 1,000th iteration in the subsequent 1,000,000-iteration sampling period. For each replicate of data, output from each run of the MCMC algorithm is used to approximate Λ.
Table 2 presents summary statistics to assess the properties of the GENEBPMv2 algorithm under the null simulation model, in which all individuals have the same risk of disease, regardless of their genotype at the functional polymorphism. Results are presented for a range of different candidate regions and sample sizes. As expected, the LD between marker SNPs decreases as the length of the candidate region—and, hence, the distance between them—increases. As a consequence, there is greater haplotype diversity in larger candidate regions. For larger sample sizes, the mean number of haplotypes increases, since there is greater opportunity to observe rare haplotypes. The mean number of common haplotypes, however, is unaffected by sample size. The mean number of clusters in the partition of haplotypes increases with the length of the candidate region but decreases with sample size. This reflects the increased haplotype diversity in large candidate regions but the reduced variability in cluster membership for larger sample sizes. The mean 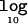 Λ is close to zero, irrespective of the length of the candidate region and sample size. The proportions of replicates with positive and strong evidence of association are ∼7%–8% and ∼1%–2%, respectively.
Λ is close to zero, irrespective of the length of the candidate region and sample size. The proportions of replicates with positive and strong evidence of association are ∼7%–8% and ∼1%–2%, respectively.
Table 2. .
Summary Statistics over 1,000 Replicates of Data Generated According to M0
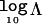 |
|||||||
| No. of Markers, Candidate-Region Size, and No. of Cases/Controls |
Mean MAF | Mean r2a | Mean No. of Haplotypes (Common)b | Mean No. of Clusters | Mean | >.5c | >1c |
| 5 SNPs in 30 kb: | |||||||
| 200/200 | .239 | .189 | 9.31 (4.89) | 2.85 | −.033 | .073 | .020 |
| 500/500 | .237 | .185 | 9.88 (4.86) | 2.78 | −.061 | .073 | .010 |
| 1,000/1,000 | .237 | .181 | 10.2 (4.86) | 2.72 | −.064 | .072 | .021 |
| 10 SNPs in 50 kb: | |||||||
| 200/200 | .239 | .145 | 25.0 (6.36) | 2.91 | −.015 | .070 | .018 |
| 500/500 | .244 | .150 | 27.7 (6.45) | 2.84 | −.043 | .076 | .015 |
| 1,000/1,000 | .238 | .142 | 29.3 (6.43) | 2.75 | −.060 | .083 | .023 |
| 20 SNPs in 100 kb: | |||||||
| 200/200 | .241 | .102 | 72.4 (4.58) | 2.93 | .001 | .076 | .016 |
| 500/500 | .242 | .102 | 85.7 (4.56) | 2.85 | −.039 | .068 | .011 |
| 1,000/1,000 | .240 | .101 | 95.9 (4.48) | 2.77 | −.016 | .080 | .011 |
Between pairs of marker SNPs.
Mean number of haplotypes consistent with observed genotype data; in parentheses, mean number of common haplotypes (>5% relative sample frequency).
Proportion of replicates with positive (>0.5) and strong (>1) evidence of association.
I next consider a simulation model of disease-marker association, parameterized in terms of (i) the population frequency of the causal variant and (ii) the genotype relative risks (GRRs) of individuals homozygous and heterozygous for the causal variant, with the homozygous protective variant genotype as baseline. Figure 5 presents the mean number of clusters in the partition of haplotypes, as a function of the disease model for candidate regions of 30 and 100 kb in length, typed at 5 and 20 SNPs, respectively, each for samples of 1,000 cases and 1,000 controls. As expected, the mean number of clusters increases with the strength of association between disease and the functional polymorphism. The number of clusters is greatest in candidate regions 100 kb in length, presumably because of the increased haplotype diversity (table 2). Nevertheless, this still reflects improved parsimony in comparison with the number of distinct haplotypes consistent with the observed genotype data.
Figure 5. .

Mean number of clusters in the partition of haplotypes as a function of the disease model for samples of 1,000 cases and 1,000 controls for causative-allele frequencies of 0.05, 0.2, and 0.5 in a candidate region of 30 kb, typed at 5 SNPs (A), and for a causative-allele frequency of 0.2 in candidate regions of 30 and 100 kb, typed at 5 and 20 SNPs, respectively (B).
Figure 6 presents the mean 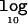 Λ in favor of disease association as a function of the disease model for candidate regions of 30 and 100 kb, typed at 5 and 20 marker SNPs, respectively, each for samples of 1,000 cases and 1,000 controls. The results are entirely as expected, where the magnitude of Λ increases with causal-variant frequency and with the strength of association between disease and the functional polymorphism. Furthermore, for a fixed causal-variant frequency, the mean
Λ in favor of disease association as a function of the disease model for candidate regions of 30 and 100 kb, typed at 5 and 20 marker SNPs, respectively, each for samples of 1,000 cases and 1,000 controls. The results are entirely as expected, where the magnitude of Λ increases with causal-variant frequency and with the strength of association between disease and the functional polymorphism. Furthermore, for a fixed causal-variant frequency, the mean 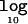 Λ is generally higher in large candidate regions with greater haplotype diversity. This presumably reflects increased precision in clustering of haplotypes carrying the causal variant, despite the effects of recombination on the similarity metric and phase-reconstruction process.
Λ is generally higher in large candidate regions with greater haplotype diversity. This presumably reflects increased precision in clustering of haplotypes carrying the causal variant, despite the effects of recombination on the similarity metric and phase-reconstruction process.
Figure 6. .

Mean 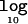 Λ as a function of the disease model for samples of 1,000 cases and 1,000 controls for causative-allele frequencies of 0.05, 0.2, and 0.5 in a candidate region of 30 kb, typed at 5 SNPs (A), and for a causative-allele frequency of 0.2 in candidate regions of 30 and 100 kb, typed at 5 and 20 SNPs, respectively (B). Also shown is the increase in the mean
Λ as a function of the disease model for samples of 1,000 cases and 1,000 controls for causative-allele frequencies of 0.05, 0.2, and 0.5 in a candidate region of 30 kb, typed at 5 SNPs (A), and for a causative-allele frequency of 0.2 in candidate regions of 30 and 100 kb, typed at 5 and 20 SNPs, respectively (B). Also shown is the increase in the mean 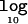 Λ by allowing for deviations from a multiplicative model of disease risks for samples of 1,000 cases and 1,000 controls for causative-allele frequencies of 0.05, 0.2, and 0.5 in a candidate region of 30 kb, typed at 5 SNPs (C), and for a causative-allele frequency of 0.2 in candidate regions of 30 and 100 kb, typed at 5 and 20 SNPs, respectively (D).
Λ by allowing for deviations from a multiplicative model of disease risks for samples of 1,000 cases and 1,000 controls for causative-allele frequencies of 0.05, 0.2, and 0.5 in a candidate region of 30 kb, typed at 5 SNPs (C), and for a causative-allele frequency of 0.2 in candidate regions of 30 and 100 kb, typed at 5 and 20 SNPs, respectively (D).
Loss of information by ignoring dominance
For each replicate of data, I perform a third independent run of the MCMC algorithm, this time under the alternative model of association assuming multiplicative disease risks (βA>0 and βD=0). Figure 6 presents the increase in the mean 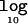 Λ, by allowing for deviations from this multiplicative model, as a function of the disease model for candidate regions of 30 and 100 kb, typed at 5 and 20 marker SNPs, respectively, each for samples of 1,000 cases and 1,000 controls. For the lower causal-variant frequency (i.e., 0.05), affected individuals tend to be heterozygous, rather than homozygous, for the causal variant. As a result, it becomes difficult to disentangle the additive and dominance effects of the causal variant, resulting in minimal gains by allowing for deviations from a multiplicative model of disease risks. However, for higher causal-variant frequencies (e.g., 0.2 and 0.5), affected individuals homozygous for the causal variant are more common, and there is, consequently, a greater loss of information by ignoring dominance, except in situations where the disease risks are approximately multiplicative—for example, a heterozygous GRR of 2 and a homozygous GRR of 5.
Λ, by allowing for deviations from this multiplicative model, as a function of the disease model for candidate regions of 30 and 100 kb, typed at 5 and 20 marker SNPs, respectively, each for samples of 1,000 cases and 1,000 controls. For the lower causal-variant frequency (i.e., 0.05), affected individuals tend to be heterozygous, rather than homozygous, for the causal variant. As a result, it becomes difficult to disentangle the additive and dominance effects of the causal variant, resulting in minimal gains by allowing for deviations from a multiplicative model of disease risks. However, for higher causal-variant frequencies (e.g., 0.2 and 0.5), affected individuals homozygous for the causal variant are more common, and there is, consequently, a greater loss of information by ignoring dominance, except in situations where the disease risks are approximately multiplicative—for example, a heterozygous GRR of 2 and a homozygous GRR of 5.
Comparison with existing methods
For each replicate of data, I also perform a standard likelihood-ratio test of association of disease with marker SNPs in the candidate region, using the haplotype-based methodology developed by Zaykin et al.9 that does not allow for clustering or dominance. Disease status is modeled in a logistic-regression framework, parameterized in terms of the multiplicative risk of disease of each haplotype. To allow for unknown phase, all possible pairs of haplotypes consistent with the observed genotype data are considered, weighted in the logistic-regression model by the corresponding phase-assignment probability calculated from the maximum-likelihood estimates of the haplotype frequencies already obtained via implementation of the EM algorithm. Rare haplotypes, occurring with estimated relative sample frequency of <5%, are pooled to improve parsimony.
Figure 7 presents the power of the GENEBPMv2 algorithm to detect association with the use of a 5% significance threshold, as a function of the disease model, for candidate regions of 30 and 100 kb in length, typed at 5 and 20 marker SNPs, respectively, each for samples of 1,000 cases and 1,000 controls. The significance threshold was determined from the null distributions of Λ for samples of 1,000 cases and 1,000 controls and was 0.639 and 0.678 for candidate regions of 30 and 100 kb, respectively. As expected, power increases with the frequency of the causative variant and with the strength of association between the disease and the functional polymorphism. There is also greater power to detect association with 20 SNPs in a candidate region of 100 kb, compared with 5 SNPs in 30 kb, despite the increase in haplotype diversity, the increased uncertainty in the phase-assignment process, and the effects of recombination on the haplotype-similarity metric.
Figure 7. .

Power of the GENEBPMv2 algorithm to detect association, at a 5% significance threshold, as a function of the disease model for samples of 1,000 cases and 1,000 controls for causative-allele frequencies of 0.05, 0.2, and 0.5 in a candidate region of 30 kb, typed at 5 SNPs (A), and for a causative-allele frequency of 0.2 in candidate regions of 30 and 100 kb, typed at 5 and 20 SNPs, respectively (B). Also shown is the increase in power to detect association, at a 5% significance threshold, for the GENEBPMv2 algorithm over a standard haplotype-based likelihood-ratio test for samples of 1,000 cases and 1,000 controls for causative-allele frequencies of 0.05, 0.2, and 0.5 in a candidate region of 30 kb, typed at 5 SNPs (C), and for a causative-allele frequency of 0.2 in candidate regions of 30 and 100 kb, typed at 5 and 20 SNPs, respectively (D).
Figure 7 also presents the gain in power of the GENEBPMv2 algorithm over the standard likelihood-ratio test of association of disease with marker SNPs in the candidate region, again evaluated using a 5% significance threshold. The GENEBPMv2 algorithm is generally as powerful, with noticeable increases in power for causative variants of frequency 0.2. The difference in power is most noteworthy for candidate regions of 100 kb. With increased marker-SNP haplotype diversity, it is most likely that the causative variant is carried by several closely related rare haplotypes that can be identified through clustering in the Bayesian partition model but that will be lost through pooling by frequency.
Discussion
In the context of association with a binary trait, “dominance” refers to any deviation from a multiplicative model of disease risks. Allowing for dominance in haplotype-based studies requires one parameter in the logistic-regression model for each observed diplotype (i.e., pair of haplotypes). Methods developed under this diplotype model will lack power to detect association in a standard frequentist-analysis framework, unless the deviation from multiplicative disease risks is extreme. Lin et al.40 describe a general-likelihood approach to test for association of a single target haplotype with disease, allowing for dominance. Their codominant model, which allows for deviations from multiplicative risks of the target haplotype, is less powerful than a multiplicative model for detecting association of three SNPs in the XRCC1 gene with breast cancer. Furthermore, they cannot simultaneously consider the joint effects of all haplotypes in the gene without correcting for multiple testing of each target one by one, which is clearly suboptimal.
To overcome the problem of lack of parsimony, I use a Bayesian partition model to cluster SNP haplotypes according to their similarity, which is used as a proxy for recent shared ancestry. Each haplotype allocated to the same clade is assigned the same probability of carrying a causal variant at the functional polymorphism(s). By assuming that each causal variant has the same genetic effect on disease, the logistic-regression model can be parameterized in terms of an additive component (the multiplicative contribution to risk) and a dominance component (any nonmultiplicative contribution to risk). A similar approach has been used by Waldron et al.,21 in the context of fine mapping, by clustering phased SNP haplotypes into just two clades, high- and low-risk, according to the Bayesian partition model. In this way, inclusion of dominance effects of causal variants at the functional polymorphism(s) requires only a single additional parameter over a model of multiplicative disease risks. One of the main advantages of this framework is flexibility, since the logistic-regression model could easily be extended to incorporate interaction with nongenetic risk factors and epistasis between functional polymorphisms in two candidate regions, without introducing the large numbers of additional parameters that would be required in existing haplotype-based methods.
I have developed a Bayesian reversible-jump MCMC algorithm, GENEBPMv2, to sample from the posterior distribution of haplotype clusters and the corresponding probabilities that they carry a causal variant at the functional polymorphism(s), in addition to the additive and dominance effects of the causal variant and any additional covariate-regression parameters, given observed phenotype and genotype data. I allow for unphased genotype data by considering all possible haplotype configurations, weighted in the logistic-regression model by the corresponding phase-assignment probabilities. These probabilities are estimated by maximum likelihood via implementation of an EM algorithm, although other, more sophisticated haplotype-reconstruction techniques, such as PHASE,12,13 could also be used. Output from the MCMC algorithm can be used to estimate the Bayes factor in favor of association, together with the posterior distribution of additive and dominance effects of the underlying causal variants. The current implementation of the algorithm allows for up to 100 SNPs, or 2,000 distinct haplotypes, consistent with the observed genotype data. Typically, analysis of 20 SNPs, typed in 1,000 cases and 1,000 controls across a 100-kb candidate region, requires <15 min of computation time with a dedicated Pentium IV work station.
The results of this simulation study suggest that there is minimal cost associated with modeling the dominance effects of causative variants at the functional polymorphism(s) within the Bayesian MCMC framework presented here. In fact, there is an increase in the Bayes factor in candidate regions of up to 100 kb when the causative variants are common. Furthermore, I demonstrate increased power of the GENEBPMv2 algorithm over a standard likelihood-ratio test of association of disease with marker SNPs in the candidate region, using the haplotype-based methodology developed by Zaykin et al.9 that does not allow for clustering or dominance. These results clearly demonstrate that, with an appropriate analysis, such as GENEBPMv2, dominance effects can and should be included in haplotype-based association studies to increase power without substantial penalty for reduced parsimony.
Analysis of marker-SNP haplotypes is appropriate within candidate genes or small candidate regions subject to limited ancestral recombination. It is possible that even these small regions will be spanned by a number of blocks of SNPs in strong LD, interrupted by hotspots of recombination. Nevertheless, the results of this simulation study suggest that the GENEBPMv2 algorithm performs well in candidate regions of up to 100 kb, despite the fact that a block model of LD was not used to generate the data. In fact, the GENEBPMv2 algorithm performed best in candidate regions with increased haplotype diversity, particularly in comparison with existing haplotype-based methods that do not allow for dominance or clustering. Further investigation is required to assess the detrimental effects of recombination in genetic regions of >100 kb. Of course, haplotype-based analysis across several megabases or a complete chromosome in a genome scan would be inappropriate because of the effects of recombination on the clustering process and the expected inaccuracies in phase assignment. Exceptions to this rule might include (i) tagging SNPs selected within LD blocks, with each block analyzed independently, and (ii) aggressively selected tagging SNPs, which are often chosen to be tested in specific combinations, as haplotypes, in the subsequent association study.41,42 Furthermore, haplotype-based analyses may provide additional information with high-density genotyping in a follow-up study of associated regions from an initial genome scan. The pattern of haplotype clustering may help to refine the likely location of the underlying functional polymorphism(s) and may identify cases with high probability of carrying causal variants to be sequenced for novel mutations.
Acknowledgments
A.P.M. acknowledges financial support from the Leverhulme Trust and the Wellcome Trust. A.P.M. thanks Louise Hosking and Chun-Fang Xu, from GlaxoSmithKline, for providing the CYP2D6 data. A.P.M. also thanks Prof. David Balding, from Imperial College, and an anonymous reviewer, for their helpful comments in preparing the revised version of this article.
Appendix A: Glossary of Notation
- yi
Phenotype of individual i, where 0 indicates unaffected status and 1 indicates affected status
- Gin
Genotype of individual i at marker SNP n
- xil
Response of individual i for the lth covariate
- Hj
The jth most frequent marker-SNP haplotype consistent with genotype data
- hj
Estimated relative frequency of haplotype Hj
- βA
Additive effect of causal variants at the functional polymorphism(s)
- βD
Dominance effect of causal variant at the functional polymorphism(s)
- Zi
Genotype of individual i at the functional polymorphism(s)
- K
Number of clusters of marker-SNP haplotypes
- Ck
Marker-SNP haplotype center of cluster k
- φk
Probability that the haplotype in the kth cluster carries causal variant at the functional polymorphism(s) in partition C
- TC(j)
Cluster assignment of haplotype Hj in partition C
- γl
Logistic-regression coefficient for the lth covariate
- μ
Baseline log-odds of disease
- θ
Model parameters
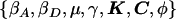
- 𝒟
Observed data and estimated haplotype frequencies
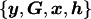
- ℳ
Model of association between disease and polymorphisms in the candidate gene, where M0 indicates the model with no association and M1 indicates the model with association
- Λ
Bayes factor in favor of disease association with polymorphisms in the candidate gene
Appendix B: Details of the MCMC Algorithm
I have developed a reversible-jump Metropolis-Hastings MCMC algorithm to approximate the posterior-density function, 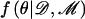 , for model ℳ (eq. [4]), where
, for model ℳ (eq. [4]), where 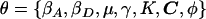 and, for observed data,
and, for observed data, 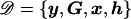 . For each iteration of the algorithm, a new set of parameter values, θ′, is proposed according to predetermined weights, w, chosen to optimize mixing and convergence (table A1). The proposed parameter values are substituted for the current set, provided that
. For each iteration of the algorithm, a new set of parameter values, θ′, is proposed according to predetermined weights, w, chosen to optimize mixing and convergence (table A1). The proposed parameter values are substituted for the current set, provided that
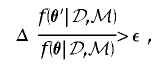 |
where ε is a standard uniform random variable and Δ denotes the Hastings ratio of proposal probabilities,
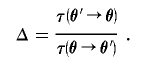 |
Otherwise, the current set of parameter values is retained. The possible changes to the parameter set are summarized below, where ε is a standard uniform random variable.
Table A1. .
Summary of Possible Changes to the Current Parameter Set in the Reversible-Jump MCMC Algorithm[Note]
Relative Weights  |
||||||
| Change (j) | Proposal | Parameters | K=1 | K=2 | 2<K<n | K=n |
| 1 | Cluster birth | K, C, φ | 0 | .25 | .25 | 0 |
| 2 | Cluster death | K, C, φ | 0 | 0 | .25 | .25 |
| 3 | Cluster-center swap | C | 0 | .10 | .10 | 0 |
| 4 | Cluster center | C | 0 | .10 | .10 | 0 |
| 5 | Causal-variant probability | φ | 0 | .10 | .10 | .10 |
| 6 | Baseline log-odds of disease | μ | .05 | .05 | .05 | .05 |
| 7 | Causal-variant additive effect | βA | 0 | .05 | .05 | .05 |
| 8 | Causal-variant dominance effect | βD | 0 | .05 | .05 | .05 |
| 9 | Covariate-regression coefficient | γ | .05 | .05 | .05 | .05 |
Total weight 
|
.10 | .75 | 1.00 | .55 | ||
Note.— Relative weights for changes under the null model, M0, are given by K=1. Relative weights for changes under the alternative model, M1, are given by K>1.
Change 1: Propose a Cluster Birth
The proposed number of clusters is given by K=K+1. Select a position, k*, at random for the new cluster in the list of ordered cluster centers. Select at random from H a haplotype, Hj, that is not already a cluster center, so that C′k*=Hj. Generate a new probability that haplotypes in the new cluster carry the causal variant at the functional polymorphism(s), φ′k*, from a uniform distribution. Then,
and
To ensure reversibility, 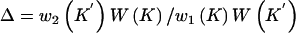 .
.
Change 2: Propose a Cluster Death
The proposed number of clusters is given by K=K-1. Select a cluster, k*, at random for death. The proposed cluster centers and probabilities of carrying the causal variant at the functional polymorphism(s) are then given by
and
To ensure reversibility, 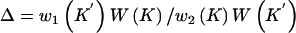 .
.
Change 3: Propose a Cluster-Center Swap
The following proposal procedure is performed K times. Select a pair of clusters, k1 and k2, at random. The proposed cluster-center swap is given by
and
Δ=1.
Change 4: Propose a Cluster-Center Change
The following proposal procedure is performed K times. Select a cluster, k, at random. Select at random from H a haplotype, Hj, that is not already a cluster center, so that C′k=Hj. Δ=1.
Change 5: Propose a New Cluster Causal-Variant Probability
The following proposal procedure is performed K times. Select a cluster, k, at random. The proposed probability that haplotypes in cluster k carry the causal variant at the functional polymorphism(s) is given by 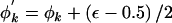 . Δ=1 but, to ensure reversibility,
. Δ=1 but, to ensure reversibility,
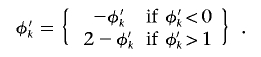 |
Change 6: Propose a New Baseline Log-Odds of Disease
The proposed parameter is given by μ′=μ+νM(ε-0.5), where νM denotes the maximum change in the parameter value. Δ=1.
Change 7: Propose a New Additive Effect of the Causal Variant
The proposed parameter is given by 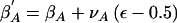 , where νA denotes the maximum change in the parameter value. Δ=1.
, where νA denotes the maximum change in the parameter value. Δ=1.
Change 8: Propose a New Dominance Effect of the Causal Variant
The proposed parameter is given by 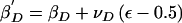 , where νD denotes the maximum change in the parameter value. Δ=1.
, where νD denotes the maximum change in the parameter value. Δ=1.
Change 9: Propose a New Covariate-Regression Coefficient
The following proposal procedure is performed L times. Select a covariate, l, at random. The proposed regression coefficient for the selected covariate is given by 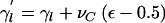 , where νC denotes the maximum change in the parameter value. Δ=1.
, where νC denotes the maximum change in the parameter value. Δ=1.
References
- 1.Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- 2.Zondervan KT, Cardon LR (2004) The complex interplay among factors that influence allelic association. Nat Rev Genet 5:89–100 10.1038/nrg1270 [DOI] [PubMed] [Google Scholar]
- 3.Thomas DC, Haile RW, Duggan D (2005) Recent developments in genomewide association scans: a workshop summary and review. Am J Hum Genet 77:337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- 5.The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320 10.1038/nature04226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark AG (2004) The role of haplotypes in candidate gene studies. Genet Epidemiol 27:321–333 10.1002/gepi.20025 [DOI] [PubMed] [Google Scholar]
- 7.Tavtigian S, Simard J, Teng D, Abtin V, Baumgard M, Beck A, Camp N, et al (2001) A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet 27:172–180 10.1038/84808 [DOI] [PubMed] [Google Scholar]
- 8.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG (2002) Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered 53:79–91 10.1159/000057986 [DOI] [PubMed] [Google Scholar]
- 10.Stram DO, Pearce CL, Bretsky P, Freedman M, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, Thomas DC (2003) Modelling and EM estimation of haplotype-specific relative risks from genotype data for a case-control study of unrelated individuals. Hum Hered 55:179–190 10.1159/000073202 [DOI] [PubMed] [Google Scholar]
- 11.Morris AP (2005) Direct analysis of unphased SNP genotype data in population-based association studies via Bayesian partition modelling of haplotypes. Genet Epidemiol 29:91–107 10.1002/gepi.20080 [DOI] [PubMed] [Google Scholar]
- 12.Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephens M, Donnelly P (2003) A comparison of Bayesian methods for haplotype reconstruction from population genetic data. Am J Hum Genet 73:1162–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Templeton AR, Boerwinkle E, Sing CF (1987) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. I. Basic theory and an analysis of alcohol dehydrogenase activity in Drosophila. Genetics 117:343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Templeton AR, Sing CF, Kessling A, Humphries S (1988) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. II. The analysis of natural populations. Genetics 120:1145–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132:619–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Templeton AR, Sing CF (1993) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. Nested analyses with cladogram uncertainty and recombination. Genetics 134:659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molitor J, Marjoram P, Thomas D (2003) Application of Bayesian spatial statistical methods to the analysis of haplotype effects and gene mapping. Genet Epidemiol 25:95–105 10.1002/gepi.10251 [DOI] [PubMed] [Google Scholar]
- 19.Molitor J, Marjoram P, Thomas D (2003) Fine-scale mapping of disease genes with multiple mutations via spatial clustering techniques. Am J Hum Genet 73:1368–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durrant C, Zondervan KT, Cardon LR, Hunt S, Deloukas P, Morris AP (2004) Linkage disequilibrium mapping via cladistic analysis of SNP haplotypes. Am J Hum Genet 75:35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waldron ERB, Whittaker JC, Balding DJ (2006) Fine mapping of disease genes by haplotype clustering. Genet Epidemiol 30:170–179 10.1002/gepi.20134 [DOI] [PubMed] [Google Scholar]
- 22.Knorr-Held L, Rasser G (2000) Bayesian detection of clusters and discontinuities in disease maps. Biometrics 56:13–21 10.1111/j.0006-341X.2000.00013.x [DOI] [PubMed] [Google Scholar]
- 23.Denison DGT, Holmes CC (2001) Bayesian partitioning for estimating disease risk. Biometrics 57:143–149 10.1111/j.0006-341X.2001.00143.x [DOI] [PubMed] [Google Scholar]
- 24.Excoffier L, Slatkin M (1995) Maximum likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12:921–927 [DOI] [PubMed] [Google Scholar]
- 25.Clayton D, Chapman J, Cooper J (2004) The use of unphased multilocus genotype data in indirect association studies. Genet Epidemiol 27:415–428 10.1002/gepi.20032 [DOI] [PubMed] [Google Scholar]
- 26.Hosking LK, Boyd PR, Xu CF, Nissum M, Cantone K, Purvis IJ, Khakkar R, Barnes MR, Liberwith U, Hagen-Mann K, Ehm MG, Riley JH (2002) Linkage disequilibrium mapping identifies a 390kb region association with CYP2D6 poor drug metabolising activity. Pharmacogenomics J 2:165–175 10.1038/sj.tpj.6500096 [DOI] [PubMed] [Google Scholar]
- 27.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E (1953) Equation of state calculations by fast computing machines. J Chem Phys 21:1087–1092 10.1063/1.1699114 [DOI] [Google Scholar]
- 28.Hastings WK (1970) Monte-Carlo sampling methods using Markov chains and their applications. Biometrika 57:97–109 10.2307/2334940 [DOI] [Google Scholar]
- 29.Green PJ (1995) Reversible jump Markov chain Monte Carlo computation and Bayesian model determination. Biometrika 82:711–732 10.2307/2337340 [DOI] [Google Scholar]
- 30.Gammerman D (1997) Markov chain Monte Carlo: stochastic simulation for Bayesian inference. Chapman and Hall, London [Google Scholar]
- 31.Kass RE, Raftery AE (1995) Bayes factors and model uncertainty. J Am Stat Assoc 90:773–795 10.2307/2291091 [DOI] [Google Scholar]
- 32.Farrall M, Morris AP (2005) Gearing up for genome-wide gene-association studies. Hum Mol Genet 14:R157–R162 10.1093/hmg/ddi273 [DOI] [PubMed] [Google Scholar]
- 33.Evans WE, Relling MV (1999) Pharmacogenomics: translating functional genomics into rational therapeutics. Science 286:487–491 10.1126/science.286.5439.487 [DOI] [PubMed] [Google Scholar]
- 34.Sachse C, Brockmoller J, Bauer S, Roots I (1997) Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotype consequences. Am J Hum Genet 60:284–295 [PMC free article] [PubMed] [Google Scholar]
- 35.Hartigan JA (1975) Clustering algorithms. Wiley, New York [Google Scholar]
- 36.Morris AP, Whittaker JC, Xu C-F, Hosking L, Balding DJ (2003) Multipoint linkage-disequilibrium mapping narrows location interval and identifies mutation heterogeneity. Proc Natl Acad Sci USA 100:13442–13446 10.1073/pnas.2235031100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griffiths RD, Marjoram P (1997) An ancestral recombination graph. In: Donnelly P, Tavare S (eds) Progress in population genetics and human evolution. Springer-Verlag, New York, pp 257–270 [Google Scholar]
- 38.Hudson RR (2002) Generating samples under a Wright-Fisher neutral model. Bioinformatics 18:337–338 10.1093/bioinformatics/18.2.337 [DOI] [PubMed] [Google Scholar]
- 39.Nordborg M (2001) Coalescent theory. In: Balding DJ, Bishop M, Cannings C (eds) Handbook of statistical genetics. Wiley, Chichester, pp 179–212 [Google Scholar]
- 40.Lin DY, Zeng D, Millikan R (2005) Maximum likelihood estimation of haplotype effects and haplotype-environment interactions in association studies. Genet Epidemiol 29:299–312 10.1002/gepi.20098 [DOI] [PubMed] [Google Scholar]
- 41.Weale ME, Depondt C, Macdonald SJ, Smith A, Lai PS, Shorvon SD, Wood NW, Goldstein DB (2003) Selection and evaluation of tagging SNPs in the neuronal-sodium-channel gene SCN1A: implications for linkage-disequilibrium gene mapping. Am J Hum Genet 73:551–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Bakker PIW, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D (2005) Efficiency and power in genetic association studies. Nat Genet 37:1217–1223 10.1038/ng1669 [DOI] [PubMed] [Google Scholar]