

Abstract
Sarcoidosis is a granulomatous inflammatory disorder of complex etiology with significant linkage to chromosome 5, and marginal linkage was observed to five other chromosomes in African Americans (AAs) in our previously published genome scan. Because genetic factors underlying complex disease are often population specific, genetic analysis of samples with diverse ancestry (i.e., ethnic confounding) can lead to loss of power. Ethnic confounding is often addressed by stratifying on self-reported race, a controversial and less-than-perfect construct. Here, we propose linkage analysis stratified by genetically determined ancestry as an alternative approach for reducing ethnic confounding. Using data from the 380 microsatellite markers genotyped in the aforementioned genome scan, we clustered AA families into subpopulations on the basis of ancestry similarity. Evidence of two genetically distinct groups was found: subpopulation one (S1) comprised 219 of the 229 families, subpopulation two (S2) consisted of six families (the remaining four families were a mixture). Stratified linkage results suggest that only the S1 families contributed to previously identified linkage signals at 1p22, 3p21-14, 11p15, and 17q21 and that only the S2 families contributed to those found at 5p15-13 and 20q13. Signals on 2p25, 5q11, 5q35, and 9q34 remained significant in both subpopulations, and evidence of a new susceptibility locus at 2q37 was found in S2. These results demonstrate the usefulness of stratifying on genetically determined ancestry, to create genetically homogeneous subsets—more reliable and less controversial than race-stratified subsets—in which to identify genetic factors. Our findings support the presence of sarcoidosis-susceptibility genes in regions identified elsewhere but indicate that these genes are likely to be ancestry specific.
Heterogeneity has been cited as a source of many difficulties facing genetic studies of complex disease today.1 The source of heterogeneity may include ambiguous or imprecise definition of the trait of interest (phenotypic heterogeneity),2 more than one locus being involved in the disease expression (locus heterogeneity),3 more than one variant at the same locus contributing to disease (allelic heterogeneity),4 or multiple population-specific loci or alleles predisposing to disease represented in the same sample (sample heterogeneity).5–8 When sample heterogeneity or ethnic confounding is suspected, it is common to stratify a collection of pedigrees by self-reported race before conducting genetic analysis. Although race is a reasonable surrogate for genetic similarity when nothing else is available,9–13 race does not always accurately reflect population of origin or genetic makeup,14–16 particularly in heterogeneous admixed groups. It is known that African Americans (AAs), for example, are admixed with European Americans (EAs) and other populations to varying degrees, as illustrated by Parra et al.,17 who showed that AAs sampled from different geographic regions show different amounts of European admixture, from 6.8% in a Jamaican sample to 22.5% in a sample from New Orleans. Additionally, because race is a social construct, it can be controversial18–23 and, some would argue, should not be used in genetic studies.
In the United States, sarcoidosis (MIM 181000), a multisystem inflammatory disorder that tends to cluster in families, has both a higher incidence and greater severity in AAs than in EAs.24,25 In a U.S. population–based study, the incidence rate was estimated at 12.1 per 100,000 for EA females, 9.6 per 100,000 for EA males, 39.1 per 100,000 for AA females, and 29.8 per 100,000 for AA males.26 A clinically heterogeneous disease, sarcoidosis most frequently affects the lungs but also commonly affects the liver, eye, skin, and lymph nodes and has variable degrees of population-specific severity.27 For example, cardiac involvement in sarcoidosis is more common among Japanese,28 and acute sarcoidosis is more common among Scandinavians,29 whereas chronic sarcoidosis is more common among AAs.27,30 Although the risk factors for sarcoidosis are unknown, studies indicate that genetic components are likely to play an important role,31,32 and disparities in prevalence and severity between populations have not been explained solely by differing environments. Significant linkage evidence from genome scans of a European sample33 and the AA sample34 used here support both the presence and the population specificity of a genetic component to sarcoidosis.
In the present study, we propose stratifying by ancestry similarity estimated from highly polymorphic genetic markers—instead of self-reported race—as a means of reducing sample heterogeneity. We show, in an AA sample ascertained for sarcoidosis, strong evidence of population-specific effects not previously identified in an analysis of the full sample.34
Subjects and Methods
Study Subjects
The study sample from SAGA consists of 519 full- and half-sibling pairs in 229 AA nuclear families, each with at least two affected offspring. The details of the study population—including diagnostic criteria for affected siblings, screening criteria of unaffected siblings to exclude undiagnosed sarcoidosis, and other exclusion criteria—are published elsewhere.32,35 Informed consent was obtained from all subjects, and the institutional review boards at all participating locations approved the research.
Cluster Analysis
Percentage of inclusion in clusters with similar genetic ancestry was estimated using the program STRUCTURE.36 STRUCTURE implements an algorithm that defines and places individuals into K clusters on the basis of subpopulation-specific allele frequencies. K is defined in advance but can be varied to find the value of K that provides the most parsimonious fit to the data. The admixture model in STRUCTURE estimates percentage of inclusion in each cluster. For this analysis, STRUCTURE was run using the linkage model,37 which provides an extension to the admixture model to allow for correlations between nearby markers. Tested models included those assuming both two and three underlying subpopulations (K=2 and K=3, respectively), but, since the family cluster configurations did not change with K, we report only the results from the more parsimonious (two-subpopulation) model.
A Marshfield screening set of 380 microsatellite markers from the original genome scan was used for the analysis. This marker set provided reasonable coverage of the genome, yet, with an average spacing of 9 cM, markers were not so closely spaced that they were too highly correlated for an analysis of population structure.37 These markers were not chosen for their ancestral informativity; rather, they were the data available from the genome scan. However, Darvasi and Shifman38 suggest that the Marshfield screening set contains more than enough ancestral informativity to be sufficient for admixture analysis, which is even more sensitive to marker informativity than is the clustering analysis performed here.
Clusters were defined as consisting of families in which at least 94% of each individual’s genome appeared to derive from a common population. Our choice of 94% as the cutoff for defining the subpopulations was based on values calculated from the data, as detailed in the “Results” section. Because we used all members of the family in the analysis, we violated the assumption in STRUCTURE that all individuals are independent. Although violation of this assumption has not been thoroughly investigated, the created bias would affect the estimates of ancestry uniformly across the sample but still allow for accurate clustering of the families, which was our primary goal.
Stratified Linkage Analysis
Multipoint identity-by-descent (IBD) sharing estimates for full- and half-sibling pairs were obtained using GENIBD (S.A.G.E.). Model-free linkage analysis was performed using the Haseman-Elston regression, as implemented in SIBPAL (S.A.G.E.). The original Haseman-Elston method regresses the squared sib-pair trait difference on the estimated IBD sharing between siblings.39 We chose to use the original Haseman-Elston, instead of the revised40 or weighted Haseman-Elston,41 because it is more robust for a small number of concordantly unaffected sibling pairs, as was the case in one of the subpopulations. Therefore, the results shown here for the full sample are slightly different from those in the original report.34 The binary trait of interest was the presence or absence of medically documented sarcoidosis. Since the current implementation of SIBPAL allows only for full siblings—and our sample contained 101 half-sibling pairs—we included a covariate indicative of half-sibling status, as was done in the original genome scan.32 In regions where the nominal P value was <.01, we calculated empirical P values from the full siblings alone, for whom a valid permutation test is available.
Because multiple analyses of the same data can result in increased type I error, we performed a test of interaction between subpopulation membership and allele sharing to confirm the difference in results from the full and stratified samples.42 To do this, we fitted two regression models: (1) a reduced model, in which the sibling trait difference was regressed on an intercept, the estimated allele sharing at the marker of interest, and an indicator variable for subpopulation effect and (2) a full model, which included the reduced model plus an allele sharing by subpopulation-interaction term. The difference in the residual sums of squares for the reduced and full models divided by twice the mean square error for the full model could then be compared with an F distribution with degrees of freedom in the numerator equal to the number of subpopulations (2) and the degrees of freedom in the denominator equal to the difference in the number of siblings and the number of sibships (227).
Analysis of Differences between Subpopulations
We evaluated the difference in the proportion of affected family members in each subpopulation who showed involvement of ocular, bone/marrow, liver, lymph, and skin organ systems. Organ-system involvement was obtained through a standardized review of each affected individual’s medical records. Our test was based on the test for a difference between two sample proportions, for which the test statistic is given as:
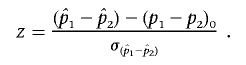 |
In this formulation, (p1-p2)0 is the difference in proportions under the null hypothesis, which, in this case, equals 0, and 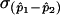 is the SD of the difference in sample proportions. This test statistic (z) can then be compared with a standard normal distribution.43
is the SD of the difference in sample proportions. This test statistic (z) can then be compared with a standard normal distribution.43
To compare the severity of the pulmonary phenotypes in those individuals affected with sarcoidosis, we used three measures—radiographic resolution, percentage of predicted forced vital capacity (FVC) at follow-up, and a pulmonary-severity score with a range from 1 (least severe) to 6 (most severe) based on radiographic staging of disease and pulmonary function. The difference in the percentage of each population with radiographic resolution was evaluated using the above test of difference in proportions. Average values of percentage of predicted FVC and severity score were compared between the two subpopulations with use of a standard t test.
Results
Cluster Analysis
Two genetically distinct subpopulations of families were found, via STRUCTURE, in our AA collection. A majority of the sample, 547 individuals in 219 families (454 sibling pairs), clustered together, and each derived >94% (average 99.4% [range 94.7%–99.9%]) of its genome from the same ancestry. We will refer to this group as “subpopulation one” (S1). Twenty-eight individuals in six families (54 sibling pairs) clustered with >99% (average 99.9% [range 99.7%–99.9%]) of their ancestry deriving from a unique subpopulation, henceforth called “subpopulation two” (S2). Individuals in the four remaining families were estimated to share an average of 80.1% (range 72.7%–86.8%) of their ancestry from S1 and 19.9% (range 13.2%–27.3%) of their ancestry from S2. Because of the high level of genetic heterogeneity within these families, they were excluded from further analysis. The pedigrees in S2 tended to be larger and also to have a higher proportion of discordant sibling pairs compared with the pedigrees in S1 (table 1). To verify that this difference in mean family size was not the source of the observed substructure, we reran STRUCTURE after randomly removing siblings from the larger sibships, such that no sibship size was >3. This resulted in only a slight change, with two of the original six S2 families no longer clustering in that group. This suggests that, although family size may have influenced the clustering, the sibship size was not sufficient to explain the subdivisions—even though 24 of the S1 families also had a sibship size >3—since all of the S1 families remained clustered together.
Table 1. .
Pedigree Information by Subpopulation
| Subpopulation |
||
| Sample Characteristic | S1 | S2 |
| No. of sibships | 219 | 6 |
| Mean sibship size | 2.44 | 4.67 |
| No. of all sib pairs | 454 | 54 |
| No. (%) of half-sib pairs | 111 (24.4) | 5 (9.3) |
| No. (%) of concordant unaffected sib pairs | 15 (3.3) | 2 (3.7) |
| No. (%) of concordant affected sib pairs | 298 (65.6) | 27 (50.0) |
| No. (%) of discordant sib pairs | 141 (31.1) | 25 (46.3) |
| No. (%) of sibships with size ⩾4 | 24 (11.0) | 5 (83.3) |
We also reran STRUCTURE after removing those markers shown, a priori, to be linked to sarcoidosis (P<.01 in the original scan), so that our family clustering was not driven by similarities in linked regions. Only one family no longer obviously belonged to S1 and one no longer obviously belonged to S2, indicating that, although linkage to disease may play a role in clustering, it, like family size, is unlikely to bias our results.
Stratified Linkage Analysis
The results of the stratified linkage analyses of S1 and S2, together with the results from the original genome scan including all families, are shown in figure 1. The stratified analysis suggests that the marginal significance (P=.05) of several of the regions identified in the original scan is due only to S1 (1p22, 11p15, and 17q21) or S2 (5p15-13 and 20q13) families, whereas other regions remain significant (P⩽.05) in both subpopulations (2p25, 3p24, 5q11, 5q35, and 9q34). Evidence of two new sarcoidosis susceptibility loci with linkage only in the S2 families (2q37 and 3p21-14) was also found.
Figure 1. .

Results of stratified linkage analysis. In each plot, the solid line represents the results from the original (full sample) linkage analysis, plotted as the −log(P value) versus the distance from the first marker. The dashed line represents the results from S1. The dotted line represents the results from S2. The horizontal lines represent P values of 1.0×10-3 (lower) and 2.2×10-5 (upper). The values to the right of the plots represent the corresponding LOD scores.
Several of the signals originally identified in the genome scan at a significance level of P=.05 are now significant at P=.01 in one or more subpopulation, despite smaller sample sizes (table 2). In some cases (for 3p21-14, 5p15-13, 17q21, and 20q13), this increase in significance was several orders of magnitude. The peak on 17q21 was not even highlighted in the original scan,34 because the significance was only marginal. In 4 of 12 cases, the difference in the effects between the combined and stratified samples was significantly different at α=.05 (table 2), further demonstrating the gain of reducing sample heterogeneity in this way. These pronounced linkage signals are likely unique to the stratum being analyzed, since the reduced sample size of the stratum would lower the power to detect the effect size that was observed in the full sample.
Table 2. .
Peak P Values by Subpopulation
|
P for |
|||||
| Region | Marker | Full Sample | S1 | S2 | Differencea |
| 1p22 | D1S551 | .056 | .036 | .20 | .49 |
| 2p25 | TPO | 4.64×10-3 | .026 | 3.10×10-3 | .06 |
| 2q37 | D2S1363 | .134 | .272 | .034 | .31 |
| 3p24 | D3S3038 | .043 | .043 | .057 | .22 |
| 3p21-14 | D3S1285 | .178 | .639 | 4.58×10-4 | 1.44×10-4 |
| 5p15-13 | D5S817 | .019 | .225 | 4.18×10-5 | 4.52×10-4 |
| 5q11 | D5S2500 | 5.00×10-4 | .010 | 4.68×10-3 | .10 |
| 5q35 | D5S1456 | .052 | .023 | .112 | .45 |
| 9q34 | D9S1825 | 6.26×10-3 | .044 | .024 | 3.19×10-3 |
| 11p15 | D11S1984 | 8.86×10-3 | .038 | .329 | .96 |
| 17q21 | D17S2180 | .014 | 9.22×10-3 | .856 | .42 |
| 20q13 | D20S480 | .010 | .054 | 4.71×10-5 | 1.96×10-3 |
For an F test of significant interaction between allele-sharing and subpopulation membership.
Empirical P values were calculated for those regions showing a nominal P value <.01. These values were quite similar to the asymptotic P values; therefore, only the asymptotic P values are reported.
To assess these findings further, we looked at the mean allele sharing (π) between concordant and discordant sibling pairs at the peaks showing a P value of <.01 (TPO, D3S1285, D5S817, D5S2500, and D20S480 in S2; D5S2500 and D17S2180 in S1). Since the mean allele sharing for the discordant sibling pairs at all these peaks remains significantly <0.5 (P<.02 in all cases) and is thus less than is expected under the null hypothesis, these are not likely to be false-positive results. The peaks at TPO, D3S1285, and D5S817 in S2 appear to be driven primarily by sharing between concordantly affected sibling pairs, since the mean allele sharing in the concordantly affected sibling pairs is much >0.5 (>0.6 in all cases). At D5S2500 in S2, the signal appears to be driven by the decreased sharing among discordant pairs; the sharing for the concordant pairs was only slightly >0.5, but the sharing between discordant pairs was estimated to be only 0.2994. The peaks at D5S2500 and D17S2180 in S1 show mean allele sharing much >0.5 in concordantly unaffected sibling pairs (⩾0.6). The peak in S2 on chromosome 20 (D20S480) shows mean allele sharing in both concordantly affected sibling pairs and concordantly unaffected sibling pairs >0.5 (>0.62 in both cases).
Phenotypic Differences between Subpopulations
Evaluation of phenotypic differences showed that the affected individuals in the families of S2 had more ocular involvement and that the affected members of the S1 families had more liver and lymph involvement; patients in the S2 families also had a lower percentage of predicted FVC at diagnosis but a higher percentage of predicted FVC at follow-up (table 3). However, only the difference in liver involvement and FVC at follow-up were statistically significant at the .05 level, indicating that the linkage results seen here are due to subpopulation, not phenotypic differences. To further assess this, we compared the linkage peaks from this study with those found for liver and FVC at follow-up in a subphenotype-specific analysis (authors' unpublished data). We found only slight overlap, supporting our previous assertion of subpopulation—not phenotypic—differences.
Table 3. .
Phenotypic Differences among Affected Persons by Subpopulation
| Phenotype | S1 | S2 | Pa |
| No. affected:unaffected (% unaffected) | 485:62 (11) | 20:8 (29) | |
| Pulmonary related: | |||
| Percentage of radiographic resolution | 25.0 | 15.8 | .82 |
| Percentage of predicted FVC at follow-up | 86.4 | 95.7 | .04 |
| Severity scoreb (average) | 2.81 | 2.65 | .34 |
| Extrathoracic organ involvement (%): | |||
| Ocular | 34.7 | 52.9 | .06 |
| Bone/marrow | 7.0 | 5.9 | .43 |
| Liver | 22.2 | 0 | <.001 |
| Lymph | 27.2 | 11.8 | .08 |
| Skin | 42.6 | 31.6 | .17 |
P value of test of differences between subpopulations.
Score of 1–6; results are based on follow-up chest x-ray and percentage of predicted FVC.
Discussion
Heterogeneity has been blamed for lack of replication, difficulty in fine mapping, and other challenges present in genetic studies of complex human diseases. Although strategies to address heterogeneity in linkage analysis have been developed, most were not feasible for this data set. Falk,44 for example, showed that including a heterogeneity parameter into model-based linkage methods allowed a more accurate estimate of the recombination fraction. However, model-free linkage methods are considered more appropriate for studies of complex diseases in which the underlying genetic model is unknown, as was the case here. The model-free two-level Haseman-Elston45 test can be used to model heterogeneity but requires the estimation of additional parameters—which requires increased sample size—and does not ultimately identify for follow-up of those families most likely to be linked to that region. Stratification by race, a simple alternative, was not feasible in our sample, because all participants reported that they belong to the same race. We therefore applied genetic ancestry estimated from the genome-scan data, to reduce sample heterogeneity.
Our approach was not without certain assumptions and restrictions. Ancestrally informative markers were not available for this analysis. In theory, such markers would have improved our ability to cluster families into appropriate subpopulations. However, the Marshfield screening set has been shown to be sufficiently informative for admixture analysis,38 a method much more contingent on ancestry informativity than the methods used here, and, in our case, it was a readily available data set. We also chose to use all members of the families, not just a single individual, violating the assumption of independence in STRUCTURE. For the reasons mentioned above, however, the bias created by this violation did not greatly affect the clustering of the families, which was our primary goal. We also recognize that S2 was a small sample of only six families (54 sib pairs), but empirical and asymptotic P values for all regions of interest were in close agreement. Finally, we recognize that use of the same families to generate the clusters as those used in assessing linkage might cause bias, since similarities in clusters could be driven by similarities in the linked regions of the genomes of the affected individuals. However, because we included unaffected individuals and conducted a whole-genome scan (not an examination of only selected candidate genes), we think this potential bias is minimal. Additionally, the results of the clustering after removal of the linked markers shows that the linked markers only minimally affected the clustering. Despite these limitations, by stratifying our self-reported racially homogeneous sample on the basis of genetically determined common ancestry, our ability to detect linkage was strengthened. We identified a novel linkage signal and an increase in the significance of several previously identified signals. Certainly, one can argue that stratification of a sample, even random stratification, could result in increased significance for linkage in some locations. However, one would not expect, by chance alone, that 10 of the 11 signals found in the stratum would be in the same location as those previously identified.
Finally, the results of this study highlight the limitation of using race as a classification tool for genetic studies. Some view race as a social construct, with little genetic relevance, that can potentially lead to discrimination and should be avoided (reviewed by the Race, Ethnicity, and Genetics Working Group46). Others contend that self-identified race is highly correlated with ethnicity,12 so that when no other data are available, it is a reasonable surrogate, but it may not always be accurate or practical. For example, 39.6% of AAs did not know all of their biological grandparents and therefore could not classify them by ancestry.14 Similarly, individuals within a family of mixed race often classified themselves as belonging to only one.47 Lastly, as demonstrated here and reported by others,48 a study sample drawn from a single self-reported race may contain several subpopulations that are genetically unique (for example, South African vs. West African in an AA sample or Scandinavian vs. Mediterranean in a European American sample) or that vary in degree of admixture with other populations. Although we are not suggesting that this type of stratified analysis can provide greater inference about linkage in all samples, particularly those that are not admixed, when the ancestral populations are known to be extremely diverse, as is the case for Africans,49 we have shown that addressing genetic substructure is both a feasible and a useful exercise. Although we do not assert that this type of analysis will eliminate all discrepancies in linkage results—nor are we able, at this stage, to definitively attribute our differing results to population substructure—this type of analysis shows great promise for the reduction of random variability in samples in which genetic heterogeneity due to population substructure is suspected.
In conclusion, this study suggests that genetic clustering via methods such as that implemented in STRUCTURE can effectively create genetically homogeneous subpopulations for a linkage analysis. Our results support the presence of population-specific sarcoidosis genes on chromosomes 1p22, 3p21-14, 5p15-13, 11p15, 17q21, and 20q13 and suggest a previously unknown sarcoidosis-susceptibility locus at 2q37. Studies to better classify S1 and S2 families by population of ancestry (such as African, EA, American Indian, Latino, etc.), as well as admixture-mapping analyses, are currently under way to further elucidate genes for sarcoidosis in AAs.
Acknowledgments
We thank all family members who participated in the SAGA study, and we thank the reviewers for their insightful comments. This study was supported by National Heart, Lung, and Blood Institute training grant HL07567 and National Cancer Institute training grant CA094186, as well as National Institutes of Health grants UO1 HL060263, RO1 GM28356, and P41 RR003655 and National Center for Research Resources grant RR03655.
Members of SAGA are as follows: Clinical Centers Core—Cleveland Clinic: Mani S. Kavuru, Chenett Johnson, and Nancy Ivansek; Emory University: Samuel M. Aguayo, Gloria Westney, Kathleen A. S. Cannella, and Francesca Cordi; Georgetown University Medical Center: David L. Rabin, Henry Yeager, Jr., Lisa Stewart, Kemi Odegbile, and Tameka Weerasingna; Henry Ford Health System: Michael C. Iannuzzi, Benjamin A. Rybicki, Wanda Bibbs, and Marcie Major; Johns Hopkins University School of Medicine: David R. Moller and Rebecca Robinson; Medical University of South Carolina: Marc A. Judson and Angela Anderson; Mount Sinai Medical Center: Alvin S. Teirstein, Louis DePalo, and Romena Rouf; National Jewish Medical and Research Center: Lee S. Newman, Cecile Rose, Juliana Barnard, Lori Silveira, Rosalind Dudden, Ben Lee, and Lynn Morehead; University of Cincinnati Medical Center: Robert P. Baughman and Donna B. Winget; University of North Carolina: James F. Donohue, Julie Montenegro, and Jeanie Mascarella; University of Pennsylvania and Medical College of Pennsylvania–Hahnemann University Medical Centers: Milton D. Rossman and Jackie Regovich. Data Coordinating Center—George Washington University: Kathryn Hirst, Sarah Fowler, Robin Laney, and Laure Elghormli. Genetics Core—Henry Ford Health System (DNA Core Laboratory): Mary Maliarik; Case Western Reserve University (Genotyping Laboratory and Genetic Analysis): Robert C. Elston, Sudha Iyengar, Courtney Gray-McGuire, and Christopher Millard. National Heart, Lung, and Blood Institute—Sandra Hatch. Data Safety and Monitoring Board—David Center (Chair), Rodney Go, Christine Grady, Talmadge King, Jr., and William Nichols. Study Chairman: Michael C. Iannuzzi.
Web Resources
The URLs for data presented herein are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for sarcoidosis)
- S.A.G.E.–Statistical Analysis for Genetic Epidemiology, http://darwin.cwru.edu/sage/ (for version 5.0)
References
- 1.Vieland VJ, Wang K, Huang J (2001) Power to detect linkage based on multiple sets of data in the presence of locus heterogeneity: comparative evaluation of model-based linkage methods for affected sib pair data. Hum Hered 51:199–208 10.1159/000053343 [DOI] [PubMed] [Google Scholar]
- 2.Rao S, Olson JM, Moser KL, Gray-McGuire C, Bruner GR, Kelly J, Harley JB (2001) Linkage analysis of human systemic lupus erythematosus-related traits: a principal component approach. Arthritis Rheum 44:2807–2818 [DOI] [PubMed] [Google Scholar]
- 3.Cox NJ, Frigge M, Nicolae DL, Concannon P, Hanis CL, Bell GI, Kong A (1999) Loci on chromosome 2 (NIDDM1) and 15 interact to increase susceptibility to diabetes in Mexican Americans. Nat Genet 21: 213–215 10.1038/6002 [DOI] [PubMed] [Google Scholar]
- 4.Rybicki BA, Walewski JL, Maliarik MJ, Kian H, Iannuzzi MC, ACCESS Research Group (2005) The BTNL2 gene and sarcoidosis susceptibility in African Americans and whites. Am J Hum Genet 77:491–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogdie MN, Bakker SC, Fisher SE, Francks C, Yang MH, Canto RM, Loo SK, van der Meulen E, Pearson P, Buitelaar J, Monaco A, Nelson SF, Sinke RJ, Smalley SL (2006) Pooled genome-wide linkage data on 424 ADHD ASPs suggests genetic heterogeneity and a common risk locus at 5p13. Mol Psychiatry 11:5–8 10.1038/sj.mp.4001760 [DOI] [PubMed] [Google Scholar]
- 6.Chen J-J, Huang W, Gui J-P, Yang S, Zhou F-S, Xiong Q-G, Wu H-B, Cui Y, Gao M, Li W, Li J-X, Yan K-L, Yuan W-T, Xu S-J, Liu J-J, Zhang X-J (2005) A novel linkage to generalized vitiligo on 4q13-q21 identified in a genomewide linkage analysis of Chinese families. Am J Hum Genet 76:1057–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rademakers R, Cruts M, Sleegers K, Dermaut B, Theuns J, Aulchenko Y, Weckx S, De Pooter T, Van den Broeck M, Corsmit E, De Rijk P, Del-Favero J, van Swieten J, van Duijn CM, van Broeckhoven C (2005) Linkage and association studies identify a novel locus for Alzheimer disease at 7q36 in a Dutch population-based sample. Am J Hum Genet 77:643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roelfsema JH, White SJ, Ariyürek Y, Bartholdi D, Niedrist D, Papadia F, Bacino CA, den Dunnen JT, van Ommen G-J, Bruening MH, Hennekam RC, Peters DJM (2005) Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet 76:572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Risch N (2000) Searching for genetic determinants in the new millennium. Nature 405:847–856 10.1038/35015718 [DOI] [PubMed] [Google Scholar]
- 10.Mountain JL, Risch N (2004) Assessing genetic contributions to phenotypic differences among “racial” and “ethnic” groups. Nat Genet 36:S48–S53 10.1038/ng1456 [DOI] [PubMed] [Google Scholar]
- 11.Skol AD, Xiao R Boehnke M, Veterans Affairs Cooperative Study 366 Investigators (2005) An algorithm to construct genetically similar subsets of families with the use of self-reported ethnicity information. Am J Hum Genet 77:346–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang H, Quertermous T, Rodriguez B, Kardia SLR, Zhu X, Brown A, Pankow JS, Province MA, Hunt SC, Boerwinkle E, Schork NJ, Risch NJ (2005) Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. Am J Hum Genet 76:268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinha M, Larkin EK, Elston RC, Redline S (2006) Self reported race and genetic admixture. N Engl J Med 354:421–422 10.1056/NEJMc052515 [DOI] [PubMed] [Google Scholar]
- 14.Condit C, Templeton A, Bates BR, Bevan JL, Harris AT (2003) Attitudinal barriers to delivery of race-targeted pharmacogenomics among informed lay persons. Genet Med 5:385–392 [DOI] [PubMed] [Google Scholar]
- 15.Keita SO, Kittles RA, Royal CD, Bonney GE, Furbert-Harris P, Dunston GM, Rotimi CN (2004) Conceptualizing human variation. Nat Genet 36:S17–S20 10.1038/ng1455 [DOI] [PubMed] [Google Scholar]
- 16.Wang VO, Sue S (2005) In the eye of the storm. Am Psychol 60:37–45 10.1037/0003-066X.60.1.37 [DOI] [PubMed] [Google Scholar]
- 17.Parra EJ, Marcini A, Akey J, Martinson J, Batzer MA, Cooper R, Forrester T, Allison DB, Deka R, Ferrell RE, Shriver MD (1998) Estimating African American admixture proportions by use of population-specific alleles. Am J Hum Genet 63:1839–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuckerman M (1990) Some dubious premises in research and theory on racial differences: scientific, social, and ethical issues. Am Psychol 45:1297–1303 10.1037/0003-066X.45.12.1297 [DOI] [PubMed] [Google Scholar]
- 19.Schwartz RS (2001) Racial profiling in medical research. N Engl J Med 334:1392–1393 10.1056/NEJM200105033441810 [DOI] [PubMed] [Google Scholar]
- 20.Foster MW, Sharp RR (2002) Race, ethnicity, and genomics: social classifications as proxies of biological heterogeneity. Genome Res 12:844–850 10.1101/gr.99202 [DOI] [PubMed] [Google Scholar]
- 21.Jorde LB, Wooding SP (2004) Genetic variation, classification and “race”. Nat Genet 36:S28–S33 10.1038/ng1435 [DOI] [PubMed] [Google Scholar]
- 22.Tishkoff SA, Kidd KK (2004) Implications of biogeography of human populations for “race” and medicine. Nat Genet 36:S21–S27 10.1038/ng1438 [DOI] [PubMed] [Google Scholar]
- 23.Shields AE, Fortun M, Hammonds EM, King PA, Lerman C, Rapp R, Sullivan PF (2005) The use of race variables in genetic studies of complex traits and the goal of reducing health disparities. Am Psychol 60:77–103 10.1037/0003-066X.60.1.77 [DOI] [PubMed] [Google Scholar]
- 24.Rybicki BA, Harrington D, Major M, Simoff M, Popovich J Jr, Maliarik M, Iannuzzi MC (1996) Heterogeneity of familial risk in sarcoidosis. Genet Epidemiol 13:23–33 [DOI] [PubMed] [Google Scholar]
- 25.Rybicki BA, Iannuzzi MC, Frederick MM, Thompson BW, Rossman MD, Bresnitz EA, Terrin ML, Moller DR, Barnard J, Baughman RP, DePalo L, Hunninghake G, Johns C, Judson MA, Knatterud GL, McLennan G, Newman LS, Rabin DL, Rose C, Teirstein AS, Weinberger SE, Yeager H, Cherniack R, ACCESS Research Group (2001) Familial aggregation of sarcoidosis: a case-control etiologic study of sarcoidosis (ACCESS). Am J Respir Crit Care Med 164:2085–2091 [DOI] [PubMed] [Google Scholar]
- 26.Rybicki BA, Major M, Popovich J Jr, Maliarik MJ, Iannuzzi MC (1997) Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol 145:234–241 [DOI] [PubMed] [Google Scholar]
- 27.Teirstein AS, Siltzbach LE, Berger H (1976) Patterns of sarcoidosis in three population groups in New York City. Ann N Y Acad Sci 278:371–376 [DOI] [PubMed] [Google Scholar]
- 28.Yosida Y, Morimoto S, Hiramitsu S, Tsuboi N, Hirayama H, Itoh T (1997) Incidence of cardiac sarcoidosis in Japanese patients with high-degree atrioventricular block. Am Heart J 134:382–386 10.1016/S0002-8703(97)70070-X [DOI] [PubMed] [Google Scholar]
- 29.Grunewald J, Eklund A, Olerup O (2004) Human leukocyte antigen class I alleles and the disease course in sarcoidosis patients. Am J Respir Crit Care Med 169:696–702 10.1164/rccm.200303-459OC [DOI] [PubMed] [Google Scholar]
- 30.Israel HL, Gottlieb JE, Peters SP (1997) The importance of ethnicity in the diagnosis and prognosis of sarcoidosis. Chest 111:838–840 [DOI] [PubMed] [Google Scholar]
- 31.Iannuzzi MC (1998) Genetics of sarcoidosis. Monaldi Arch Chest Dis 53:609–613 [PubMed] [Google Scholar]
- 32.Rybicki BA, Maliarik MJ, Poisson LM, Iannuzzi MC (2004) Sarcoidosis and granuloma genes: a family-based study in African-Americans. Eur Respir J 24:251–257 10.1183/09031936.04.00005904 [DOI] [PubMed] [Google Scholar]
- 33.Schürmann M, Reichel P, Müller-Myhsok B, Schlaak M, Müller-Quernheim J, Schwinger E (2001) Results from a genome-wide search for predisposing genes in sarcoidosis. Am J Respir Crit Care Med 164:840–846 [DOI] [PubMed] [Google Scholar]
- 34.Iannuzzi MC, Iyengar SK, Gray-McGuire C, Elston RC, Baughman RP, Donohue JF, Hirst K, Judson MA, Kavuru MS, Maliarik MJ, Moller DR, Newman LS, Rabin DL, Rose CS, Rossman MD, Teirstein AS, Rybicki BA (2005) Genome-wide search for sarcoidosis susceptibility genes in African-Americans. Genes Immun 6:509–518 10.1038/sj.gene.6364235 [DOI] [PubMed] [Google Scholar]
- 35.Rybicki BA, Hirst K, Iyengar SK, Barnard JG, Judson MA, Rose CS, Donohue JF, Kavuru MS, Rabin DL, Rossman MD, Baughman RP, Elston RC, Maliarik MJ, Moller DR, Newman LS, Teirstein AS, Iannuzzi MC (2005) A Sarcoidosis Genetic Linkage Consortium: the Sarcoidosis Genetic Analysis (SAGA) study. Sarcoidosis Vasc Diffuse Lung Dis 22:115–122 [PubMed] [Google Scholar]
- 36.Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darvasi A, Shifman S (2005) The beauty of admixture. Nat Genet 37:118–119 10.1038/ng0205-118 [DOI] [PubMed] [Google Scholar]
- 39.Haseman JK, Elston R (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2:3–19 10.1007/BF01066731 [DOI] [PubMed] [Google Scholar]
- 40.Elston RC, Buxbaum S, Jacobs KB, Olson JM (2000) Haseman and Elston revisited. Genet Epidemiol 19:1–17 [DOI] [PubMed] [Google Scholar]
- 41.Shete S, Jacobs KB, Elston RC (2003) Adding further power to the Haseman and Elston method of detecting linkage in larger sibships: weighting sums and differences. Hum Hered 55:79–85 10.1159/000072312 [DOI] [PubMed] [Google Scholar]
- 42.Iyengar SK, Song D, Klein BEK, Klein R, Schick JH, Humphrey J, Millard C, Liptak R, Russo K, Jun G, Lee KE, Fijal B, Elston RC (2004) Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am J Hum Genet 74:20–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daniel WW (1999) Biostatistics: a foundation for analysis in the health sciences, 7th ed. John Wiley, New York, pp 252–253 [Google Scholar]
- 44.Falk CT (1997) Effect of genetic heterogeneity and assortative mating on linkage analysis: a simulation study. Am J Hum Genet 61:1169–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang T, Elston RC (2005) Two-level Haseman-Elston regression for general pedigree data analysis. Genet Epidemiol 29:12–22 10.1002/gepi.20075 [DOI] [PubMed] [Google Scholar]
- 46.Race, Ethnicity, and Genetics Working Group (2005) The use of racial, ethnic, and ancestral categories in research. Am J Hum Genet 77:519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shriver MD, Parra EJ, Dios S, Bonilla C, Norton H, Jovel C, Pfaff C, Jones C, Massac A, Cameron N, Baron A, Jackson T, Argyropoulos G, Jin L, Hoggart CJ, McKeigue PM, Kittles RA (2003) Skin pigmentation, biogeographical ancestry and admixture mapping. Hum Genet 112:387–399 [DOI] [PubMed] [Google Scholar]
- 48.Barnholtz-Sloan JS, Chakroborty R, Sellers TA, Schwartz AG (2005) Examining population stratification via individual ancestry estimates versus self-reported race. Cancer Epidemiol Biomarkers Prev 14:1545–1551 10.1158/1055-9965.EPI-04-0832 [DOI] [PubMed] [Google Scholar]
- 49.Cavalli-Sforza LL, Menozzi P, Piazza A (1994) The history and geography of human genes. Princeton University Press, Princeton, NJ [Google Scholar]