

Abstract
Schizophrenia and human leukocyte antigen (HLA) matching between couples or between mothers and offspring have independently been associated with prenatal/obstetric complications, including preeclampsia and low birth weight. Here, we report the results of a family-based candidate-gene study that brings together these two disparate lines of research by assessing maternal-fetal genotype matching at HLA-A, -B, and -DRB1 as a risk factor of schizophrenia. We used a conditional-likelihood modeling approach with a sample of 274 families that had at least one offspring with schizophrenia or a related spectrum disorder. A statistically significant HLA-B maternal-fetal genotype–matching effect on schizophrenia was demonstrated for female offspring (P=.01; parameter estimate 1.7 [95% confidence interval 1.22–2.49]). Because the matching effect could be associated with pregnancy complications rather than with schizophrenia per se, these findings are consistent with the neurodevelopmental hypothesis of schizophrenia and with accumulating evidence that the prenatal period is involved in the origins of this disease. Our approach demonstrates how genetic markers can be used to characterize the biology of prenatal risk factors of schizophrenia.
Schizophrenia is a major public health challenge because of its severity and prevalence. Several lines of converging evidence support the consensus that schizophrenia is a complex disorder with both genetic and environmental causes.1 Yet, the specific genetic and environmental processes that may combine to cause the disease have remained difficult to characterize. In a recent meta-analysis of 12 twin studies that used a multigroup twin model,1 evidence emerged of both a substantial additive genetic effect on schizophrenia (estimate of heritability 81% [95% CI 73%–90%]) and a common or shared environmental effect (estimate of heritability 11% [95% CI 3%–19%]). Although the intrauterine environments of twin pairs are not identical, they are more similar than are environments in later life; hence, the authors concluded that the presence of significant common-environment effects on risk of schizophrenia would most likely occur very early in life. This prediction is consistent with a neurodevelopmental etiology of schizophrenia2,3 and with the body of research on early risk factors of schizophrenia.4,5
Several reviews6,7 and a recent meta-analysis5 support the involvement of pregnancy complications—such as bleeding, diabetes, rhesus (Rh) incompatibility, preeclampsia, abnormal fetal growth and development, and delivery complications—as risk factors of schizophrenia. Furthermore, a recent study found that obstetric complications tended to cluster within families with schizophrenia, especially in multiply affected families,8 suggesting that the prenatal complications that increase susceptibility to schizophrenia may themselves have a genetic origin.9 One clear example of this is Rh incompatibility, since epidemiologic and genetic analyses have found evidence that the RHD locus (MIM 111680) is a risk factor of schizophrenia when maternal-fetal genotype (MFG) combinations produce Rh incompatibility.5,10–12
Rh incompatibility is an example of a maternal recognition of, or sensitization to, fetal cells—in this case, specifically to fetal red blood cells carrying an antigen of paternal origin that maternal red blood cells lack. Once sensitization has occurred, the maternal response involves the production of immunoglobulin G antibodies, which cross the placenta and lyse fetal red blood cells, producing hyperbilirubinemia and hypoxia in the fetus.13 Evidence from other lines of research suggests that these conditions can affect brain development in ways that increase the risk of schizophrenia.14–16 There is some evidence that susceptibility to schizophrenia as a function of Rh incompatibility pertains only to male offspring.12
It is generally accepted that there is maternal recognition of paternally derived fetal human leukocyte antigens (HLAs) during pregnancy, since maternal antibodies against these fetal antigens have been detected.17 Maternal recognition, or sensitization, likely occurs when fetal nucleated cells, which bear paternally derived HLAs that differ from the maternal HLAs, enter the maternal circulation system during pregnancy or delivery. However, because maternal antibodies to fetal antigens have been observed in a significant proportion of healthy pregnancies, there is some belief that maternal sensitization may be beneficial for implantation and maintenance of pregnancy and that lack of maternal sensitization may lead to adverse reproductive outcomes.17 This line of reasoning has given rise to a number of empirical studies evaluating the strength of the relationship between HLA matching and reproductive outcomes. Although results are inconsistent in the literature, there is some empirical evidence that HLA matching between couples or between mother and fetus—that is, situations in which maternal sensitization would not occur—increases the risk of fetal loss,18–21 preeclampsia,22–25 low birth weight,26–29 newborn encephalopathy, and seizures.30 The underlying biological mechanism(s) for poor reproductive outcomes are not yet known; however, an immunological-intolerance hypothesis posits that HLA similarity between mother and fetus fails to stimulate an adequate maternal immune response that is necessary for proper implantation and maintenance of pregnancy.20
The potential relevance of HLA matching to neurodevelopmental disorders has been recognized. Stubbs et al.31 hypothesized that parents of children with autism would be more likely to share HLAs than would parents of unaffected controls, in part because of increased rates of preeclampsia and spontaneous abortions in mothers of children with autism. The investigators found that parents of children with autism were significantly more likely to share at least one HLA-A, -B, or -C antigen in common, compared with parents of unaffected children.
Because risk of schizophrenia is associated with prenatal/obstetric complications, including preeclampsia and low birth weight, and because maternal-fetal HLA matching has been associated with these complications and with at least one other neurodevelopmental disorder, we hypothesized that maternal-fetal HLA matching may be a risk factor of schizophrenia. In this article, we examine the role of MFG matching at HLA-A (MIM 142800), -B (MIM 142830), and -DRB1 (MIM 142860) loci as a risk factor of schizophrenia.
Independent nuclear families in which DNA was available from at least one parent and one offspring affected with schizophrenia, schizoaffective psychosis disorder, or schizophrenia spectrum disorder (i.e., schizoid, schizotypal, and paranoid personality disorders; schizophreniform, delusional, and brief psychotic disorder; and psychosis not otherwise specified) were selected for the current study from a larger well-described and characterized Finnish schizophrenia sample.32–35 Probands in the larger study, born between 1940 and 1976, were identified through national health and population registers, and their first-degree relatives were recruited with permission from the proband. Two psychiatrists or psychiatric residents made independent DSM-IV36 best-estimate lifetime diagnoses for probands and their relatives, using all available inpatient and outpatient records. The research was approved by the Ministry of Social Affairs and Health in Finland and by the appropriate institutional review boards, and subjects provided informed consent.
DNA was extracted from blood in accordance with a standard procedure.37 HLA genotyping was performed by an HLA clinical diagnostic and reference laboratory with high inter- and intrasite reliability for DNA-based typing of class I38 and class II39 genes. The quality and quantity of the DNA was assessed by optical density and agarose-gel checks. We genotyped individuals, blind to their diagnostic status, for HLA-A, -B, and -DRB1 antigens by PCR amplification followed by hybridization with sequence-specific oligonucleotide probes, using Quick-Type HLA typing kits in accordance with the manufacturer’s protocol (Tepnel Lifecodes). A total of 30 A-locus, 47 B-locus, and 31 DRB1-locus probes were used to identify all World Health Organization (WHO)–designated HLA-A, -B, and -DRB1 alleles.40 The primers and probes used in the assay were validated before use, for sensitivity and specificity, on an HLA reference panel of DNA. With each batch of unknown samples, control samples of known genotype were evaluated to ensure accuracy. Negative controls (no DNA) were included in each assay, to monitor for PCR-amplicon contamination. After probe hybridization, membranes were scanned using Quick-Type HLA Scanning and Analysis software, and the assignment of the HLA typing was based on the reaction pattern, compared with patterns associated with published HLA gene sequences. The genotyping failure rate was ∼5% and was primarily due to insufficient quantities of DNA.
Individuals observed to have a single allele were treated as homozygous for that allele. Genotype error checking was performed using the mistyping option of Mendel v6.0,41,42 which flags genotypes requiring unlikely double recombination, as well as Mendelian transmission errors. A total of 13 individuals in 10 families with genotyping errors were excluded from the analyses. The per-locus genotyping error rate was 0.09%, 0.09%, and 0.9% for HLA-A, -B, and -DRB1, respectively. There was no significant evidence of violation of Hardy-Weinberg equilibrium at HLA-A (P=.28), -B (P=.21), or -DRB1 (P=.08) among the founder alleles when we used the Fisher's exact test implemented in Mendel v6.0.42
The current study sample is composed of 274 nuclear families with 484 affected offspring (n=372, 70, and 42 who received diagnoses of schizophrenia, schizoaffective psychosis disorder, and schizophrenia spectrum disorder, respectively). Sample sizes vary slightly by HLA locus, with genotypes available for both parents in 118 families for HLA-B and -DRB1 and in 117 families for HLA-A; in 115, 114, and 113 families for HLA-A, -DRB1, and -B, respectively, when the father’s genotype is missing; and in 41, 40, and 39 families for HLA-B, -DRB1, and -A, respectively, when the mother’s genotype is missing. The majority (90%) of missing parental genotypes were due to lack of availability of a DNA sample, and the remainder (⩽15 individuals per locus) were due to genotyping difficulties. All affected offspring with genotype data were included in the analyses. When available, genotype data for unaffected siblings were included, for the inference of parental genotypes when one parent's genotype was missing. The number of affected offspring per nuclear family ranged from one to six; 60% of families had two or more affected offspring, and 60% of affected offspring were male.
We first conducted tests of NPL separately for HLA-A, -B, and -DRB1, using Mendel v6.042 to determine whether additional parameters for high-risk allelic effects should be included in the models examining the role of MFG matching. Single-point linkage analyses did not yield strong significant evidence of linkage in the families (additive pairs NPL statistic for HLA-A P=.09, -B P=.11, and -DRB1 P=.18). Alternative NPL statistics (recessive blocks, additive all, and dominant blocks) resulted in equivalent or larger P values. Given the weakly significant linkage findings, particularly for HLA-A and -B, we next tested for association. Multiallelic transmission/disequilibrium tests did not yield significant evidence of an association with a specific HLA-A, -B, or -DRB1 allele that could be acting through the affected offspring’s genotype to increase susceptibility to schizophrenia (P=.83, P=.48, and P=.21, respectively). Repeating these analyses by grouping together alleles with similar antigen specificity (according to the WHO assignments40) did not produce substantively different results. Because these analyses did not provide evidence of the role of a high-risk allele, subsequent analyses focused only on investigating whether matching MFG combinations would increase risk of schizophrenia.
To test hypotheses of MFG matching at these loci, the multiple-sibling extension of the MFG test43 was modified to handle multiallelic loci and missing parental genotypes.44 This likelihood approach evaluates the distribution of parental and affected-offspring genotypes conditioned on the offspring’s affection status and fits the likelihood i=1NPr(Gi1, Gi0, Gim, Gif | Di1), where Gi1 and Gi0 are genotypes for the ni affected offspring and mi unaffected offspring, respectively, in family i=1,…,N; Gim and Gif are genotypes for the mother and father, respectively; and D is affection status. Phenotypes of the unaffected offspring are not used in the likelihood calculation. With the use of the Bayes theorem and the assumption that siblings’ phenotypes are independent conditional on the genotypes, this likelihood can be parameterized in terms of penetrance functions and population mating-type frequencies.43,45 We adopt a log-linear model for the penetrances
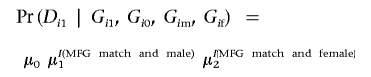 |
where I(.) is the indicator function. The parameter μ0 is the baseline risk, which ultimately cancels out the likelihood. The parameters μ1 and μ2 represent relative risks due to MFG matching when a matched affected offspring is male or female, respectively, compared with a nonmatched affected offspring. The parameters μ0, μ1, and μ2 are all positive. No assumption of Hardy-Weinberg equilibrium is necessary. We included data from families with incomplete parental genotypes in the likelihood calculation, by assuming that the genotypes are missing at random and summing over all possible genotypes for the parent.44
HLA loci are highly polymorphic. However, it is well known from organ transplantation work that there are subsets of alleles with similar antigen specificity, meaning that they recognize and produce an immune response to the same antigens. Because the immune hypothesis is based on the idea of maternal sensitization, or lack thereof, HLA-A, -B, and -DRB1 alleles with similar antigen specificity were grouped together according to the WHO assignments,40 for analyses to test hypotheses about the effect of MFG matching on susceptibility to schizophrenia. The WHO grouping preserves the biological hypothesis of maternal immunological intolerance and reduces the number of “alleles” in the analyses.
Consistent with previous research reported elsewhere,46 we further hypothesized that MFG matching increases risk of schizophrenia through a general allele-matching phenomenon rather than through specific allele combinations. Hence, we define parental mating types in the model using the parental allele identity-by-state (IBS) status under the assumption that the parental mating is symmetric with regard to genotypes.46 For example, consider a locus with six alleles, numbered 1–6. In the IBS schema, families with maternal genotype 1/3 and paternal genotype 3/4 are in the same mating type as are families with maternal genotype 2/4 and paternal genotype 4/5. In each case, both parents are heterozygous; IBS=1. The IBS schema has the additional attractive feature of reducing the number of mating types, which are considered nuisance parameters, to be estimated.
For each locus, MFG matching was defined in terms of allele matching between the mother and offspring. Specifically, mother and offspring match if the offspring’s alleles are identical to the maternal alleles (e.g., mother 1/1 and offspring 1/1 or mother 1/2 and offspring 1/2) or if the offspring’s alleles are a subset of the maternal alleles (e.g., mother 3/4 and offspring 3/3). In each of these cases, the antigens produced by the offspring’s alleles would not be perceived by the mother as different from her own during pregnancy, and maternal sensitization would not occur.
We evaluate the role of MFG matching by placing different constraints on the relative risk parameters. Under a null hypothesis—that MFG matching does not increase susceptibility to schizophrenia—one would expect that the distribution of parent and affected-offspring genotypes would simply be based on the allele frequencies in the population. The null model is evaluated by setting μ1 and μ2 to the null value of 1, whereas placing no constraints on μ1 and μ2 allows the MFG-matching risk to be estimated separately for male and female offspring.
The maximum-likelihood estimates of the MFG-matching parameters and the mating-type parameters were obtained by solving score equations of the log likelihood, and the SEs of the estimates were derived through the observed information matrix.47 Hypothesis testing was performed by a likelihood-ratio test whose statistic follows asymptotically a χ2 distribution with degrees of freedom equal to the difference in the number of parameters estimated in nested models.
We tested the primary hypothesis of an HLA-A, -B, or -DRB1 MFG-matching effect for males and females, by comparing the model in which μ1 and μ2 are estimated with a model in which μ1=μ2=1. There was no significant evidence of an HLA-A (χ2=2.29, 2 df; P=.32) or -DRB1 (χ2=3.34, 2 df; P=.19) MFG-matching effect on schizophrenia. In contrast, this model comparison yielded significant evidence of an HLA-B MFG-matching effect on schizophrenia (χ2=9.1, 2 df; P=.01) (see table 1 for comparison of model 0 with model 1), with parameter estimates of 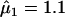 (95% CI 0.76–1.6) and
(95% CI 0.76–1.6) and 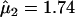 (95% CI 1.22–2.49). Inspection of μ1 and μ2 parameter estimates and their 95% CIs reveals that HLA-B MFG matching produces a higher risk of schizophrenia for female offspring than for male offspring (table 1). We note that, in this analysis, there were 185 families that could be assigned to a specific mating type. Of those, 61 families were informative for MFG matching and yielded 72 instances (35 females and 37 males) of MFG matching, compared with 37 instances (12 females and 25 males) of mismatching between mothers and their affected offspring. The rate of matching between affected daughters and their mothers was higher than that between affected sons and their mothers (74.5% and 59.7%, respectively), which is consistent with the estimates of the matching parameters. Although this comparison does not take into account the effects of dependencies among siblings and missing parental genotype data (whereas the statistical analysis does), it provides an intuitive feel for the increased frequency of MFG matching at the HLA-B locus among affected female offspring.
(95% CI 1.22–2.49). Inspection of μ1 and μ2 parameter estimates and their 95% CIs reveals that HLA-B MFG matching produces a higher risk of schizophrenia for female offspring than for male offspring (table 1). We note that, in this analysis, there were 185 families that could be assigned to a specific mating type. Of those, 61 families were informative for MFG matching and yielded 72 instances (35 females and 37 males) of MFG matching, compared with 37 instances (12 females and 25 males) of mismatching between mothers and their affected offspring. The rate of matching between affected daughters and their mothers was higher than that between affected sons and their mothers (74.5% and 59.7%, respectively), which is consistent with the estimates of the matching parameters. Although this comparison does not take into account the effects of dependencies among siblings and missing parental genotype data (whereas the statistical analysis does), it provides an intuitive feel for the increased frequency of MFG matching at the HLA-B locus among affected female offspring.
Table 1. .
Model Comparisons and Estimates of Relative Risks of Schizophrenia Due to HLA-B MFG Matching
| Model |
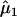 (95% CI)a (95% CI)a
|
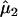 (95% CI)b (95% CI)b
|
Log Likelihood |
| 0 | =1 | =1 | −1,018.17 |
| 1 | 1.11 (0.76–1.61) | 1.74 (1.22–2.49) | −1,013.63 |
| 2 | =1 | 1.69 (1.20–2.38) | −1,013.79 |
Relative risk for sons who match at HLA-B with their mother.
Relative risk for daughters who match at HLA-B with their mother.
Because the coverage on μ1 includes the null value of 1, we next examined whether the effect was limited to females by comparing the model where μ1 and μ2 are unconstrained (which allows males and females to be at different risk) with the model where μ1=1 (model 1 and model 2, respectively, in table 1). We found that the unconstrained model did not fit significantly better than the model with μ1=1 (χ2=0.32, 1 df; P=.57), which indicates that, in this sample, only females are significantly affected by HLA-B MFG matching.
This article provides evidence that the HLA-B locus increases schizophrenia risk for female offspring through an MFG-incompatibility mechanism, in which risk is increased when mothers and their daughters match for alleles at that locus. Further studies are needed to elucidate the mechanism through which HLA-B matching increases risk of schizophrenia. However, because HLA matching has been found in some studies to increase the risk of prenatal complications, the presence of an HLA-B MFG-matching effect suggests that susceptibility to schizophrenia may be increased by an adverse prenatal environment, because the matching effect could be associated with pregnancy complications rather than with schizophrenia per se. These findings are consistent with the neurodevelopmental hypothesis of schizophrenia and with evidence that the prenatal period is involved in the origins of this disease. Furthermore, our results suggest that females are more vulnerable to the effects of HLA-B MFG matching on schizophrenia than are males. Although the explanation for this differential sex phenomenon is not known, a recent study demonstrated that, with preeclampsia (a condition that has been associated with HLA-B matching) with preterm delivery, female fetuses were more likely to survive than were male fetuses.48 Thus, the effects of preeclampsia on schizophrenia may be more likely to be observed among females because fewer males are likely to survive the effects of preeclampsia.
We note that the estimates presented in this article may be underestimates and that the true effects may be larger. An assumption of the MFG test is that the probability of an affected individual surviving to clinical detection does not depend on the HLA genotypes of the parents and offspring,49 and this assumption may be violated when loci potentially reducing fetal viability are assessed, as in the case of HLA loci. The effect of this violation would be a reduction in the number of mother-offspring matches in the sample, which would underestimate the true MFG-matching effect. Analyses that incorporate genotype data on phenotype-unknown siblings would enable fetal viability to be estimated and would lead to a more accurate estimate of the MFG-matching effect.45
Until recently, the investigation of prenatal risk factors has been based on data extracted from medical records. However, growing evidence that prenatal risk factors of schizophrenia may, in fact, have a genetic origin motivates the use of a genetic approach to identify and further study the effects of these risk factors. Identifying genes that contribute to an adverse prenatal environment that increases risk of schizophrenia may serve to (i) increase power to detect schizophrenia susceptibility genes, (ii) foster research into the study of how susceptibility genes and prenatal environment act separately and together to produce schizophrenia, and (iii) produce tailored risk assessments for individuals with personal or family histories of schizophrenia.
Acknowledgments
We thank the families that participated in this research and two anonymous reviewers for their constructive criticisms on the early version of this article. This research was supported in part by United States Public Health Service grants MH066001 and GM53275, by The Center of Excellence in Disease Genetics of the Academy of Finland, and by Biocentrum Helsinki, Finland.
Web Resource
The URL for data presented herein is as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for RHD, HLA-A, HLA-B, and HLA-DRB1)
References
- 1.Sullivan PF, Kendler KS, Neale MC (2003) Schizophrenia as a complex trait. Arch Gen Psychiatry 60:1187–1192 10.1001/archpsyc.60.12.1187 [DOI] [PubMed] [Google Scholar]
- 2.Marenco S, Weinberger DR (2000) The neurodevelopmental hypothesis of schizophrenia: following a trail of evidence from cradle to grave. Dev Psychopathol 12:501–527 10.1017/S0954579400003138 [DOI] [PubMed] [Google Scholar]
- 3.McGrath JJ, Feron FP, Burne TH, Mackay-Sim A, Eyles DW (2003) The neurodevelopmental hypothesis of schizophrenia: a review of recent developments. Ann Med 35:86–93 10.1080/07853890310010005 [DOI] [PubMed] [Google Scholar]
- 4.Dalman C, Allebeck P, Cullberg J, Grunewald C, Koster M (1999) Obstetric complications and the risk of schizophrenia: a longitudinal study of a national birth cohort. Arch Gen Psychiatry 56:234–240 10.1001/archpsyc.56.3.234 [DOI] [PubMed] [Google Scholar]
- 5.Cannon M, Jones PB, Murray RM (2002) Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 159:1080–1092 10.1176/appi.ajp.159.7.1080 [DOI] [PubMed] [Google Scholar]
- 6.McNeil TF, Cantor-Graae E, Ismail B (2000) Obstetric complications and congenital malformation in schizophrenia. Brain Res Rev 31:166–178 10.1016/S0165-0173(99)00034-X [DOI] [PubMed] [Google Scholar]
- 7.Mueser KT, McGurk SR (2004) Schizophrenia. Lancet 363:2063–2072 10.1016/S0140-6736(04)16458-1 [DOI] [PubMed] [Google Scholar]
- 8.Walshe M, McDonald C, Taylor M, Zhao J, Sham P, Grech A, Schulze K, Bramon E, Murray RM (2005) Obstetric complications in patients with schizophrenia and their unaffected siblings. Eur Psychiatry 20:28–34 10.1016/j.eurpsy.2004.07.007 [DOI] [PubMed] [Google Scholar]
- 9.Preti A (2005) Obstetric complications, genetics and schizophrenia. Eur Psychiatry 20:354–357 10.1016/j.eurpsy.2005.03.008 [DOI] [PubMed] [Google Scholar]
- 10.Palmer CGS, Turunen JA, Sinsheimer JS, Minassian S, Paunio T, Lonnqvist J, Peltonen L, Woodward JA (2002) RHD maternal-fetal genotype incompatibility increases schizophrenia susceptibility. Am J Hum Genet 71:1312–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollister JM, Laing P, Mednick SA (1996) Rhesus incompatibility as a risk factor for schizophrenia in male adults. Arch Gen Psychiatry 53:19–24 [DOI] [PubMed] [Google Scholar]
- 12.Insel BJ, Brown AS, Bresnahan MA, Schaefer CA, Susser ES (2005) Maternal-fetal blood incompatibility and the risk of schizophrenia in offspring. Schizophr Res 80:331–342 10.1016/j.schres.2005.06.005 [DOI] [PubMed] [Google Scholar]
- 13.Guyton AC (1981) Textbook of medical physiology. W. B. Saunders, Philadelphia, pp 86–88 [Google Scholar]
- 14.Moises HW, Zoega T, Gottesman II (2002) The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. BMC Psychiatry 2:8 10.1186/1471-244X-2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stefanis N, Frangou S, Yakeley J, Sharma T, O’Connell P, Morgan K, Sigmudsson T, Taylor M, Murray R (1999) Hippocampal volume reduction in schizophrenia: effects of genetic risk and pregnancy and birth complications. Biol Psychiatry 46:697–702 10.1016/S0006-3223(99)00089-X [DOI] [PubMed] [Google Scholar]
- 16.van Erp TGM, Saley PA, Rosso IM, Huttunen M, Lonnqvist J, Pirkola T, Salonen O, Valanne L, Poutanen VP, Standertskjold-Nordenstam CG, Cannon TD (2002) Contributions of genetic risk and fetal hypoxia to hippocampal volume in patients with schizophrenia or schizoaffective disorders, their unaffected siblings, and healthy unrelated volunteers. Am J Psychiatry 159:1514–1520 10.1176/appi.ajp.159.9.1514 [DOI] [PubMed] [Google Scholar]
- 17.Ober C (1998) HLA and pregnancy: the paradox of the fetal allograft. Am J Hum Genet 62:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ober C, Hyslop T, Elias S, Weitkamp LR, Hauck WW (1998) Human leukocyte antigen matching and fetal loss: results of a 10 year prospective study. Hum Reprod 13:33–38 10.1093/humrep/13.1.33 [DOI] [PubMed] [Google Scholar]
- 19.Unander AM, Olding LB (1983) Habitual abortion: parental sharing of HLA antigens, absence of maternal blocking antibody, and suppression of maternal lymphocytes. Am J Reprod Immunol 4:171–178 [DOI] [PubMed] [Google Scholar]
- 20.Beydoun H, Saftlas AF (2005) Association of human leucocyte antigen sharing with recurrent spontaneous abortions. Tissue Antigens 65:123–135 10.1111/j.1399-0039.2005.00367.x [DOI] [PubMed] [Google Scholar]
- 21.Ober CL, Martin AO, Simpson JL, Hauck WW, Amos DB, Kostyu DD, Fotino M, Allen FH (1983) Shared HLA antigens and reproductive performance among Hutterites. Am J Hum Genet 35:994–1004 [PMC free article] [PubMed] [Google Scholar]
- 22.Fujisawa S (1985) HLA antigens-antibodies system and its association with severe toxemia of pregnancy. Nippon Sanka Fujinka Gakkai Zasshi 37:124–130 [PubMed] [Google Scholar]
- 23.Bolis PF, Martinetti BM, La Fianza A, Franchi M, Cuccia Belevedere M (1987) Immunogenetic aspects of pre-eclampsia. Biol Res Pregnancy Perinatol 8:42–45 [PubMed] [Google Scholar]
- 24.Schneider K, Knutson F, Tamsen L, Sjoberg O (1994) HLA antigen sharing in preeclampsia. Gynecol Obstet Invest 37:87–90 [DOI] [PubMed] [Google Scholar]
- 25.de Luca Brunori I, Battini L, Simonelli M, Clemente F, Brunori E, Mariotti ML, Genazzani AR (2000) Increased HLA-DR homozygosity associated with pre-eclampsia. Hum Reprod 15:1807–1812 10.1093/humrep/15.8.1807 [DOI] [PubMed] [Google Scholar]
- 26.Larizza D, Martinetti M, Dugoujon JM, Tinelli C, Calcaterra V, Cuccia M, Salvaneschi L, Severi F (2002) Parental GM and HLA genotypes and reduced birth weight in patients with Turner’s syndrome. J Pediatr Endocrinol Metab 15:1183–1190 [DOI] [PubMed] [Google Scholar]
- 27.Ober C, Simpson JL, Ward M, Radvany RM, Andersen R, Elias S, Sabbagha R (1987) Prenatal effects of maternal-fetal HLA compatibility. Am J Reprod Immunol Microbiol 15:141–149 [PubMed] [Google Scholar]
- 28.Verp MS, Sibul M, Billstrand C, Bellen G, Hsu M, Ober C (1993) Maternal-fetal histocompatibility in intrauterine growth retarded and normal weight babies. Am J Reprod Immunol 29:195–198 [DOI] [PubMed] [Google Scholar]
- 29.Reznikoff-Etievant MF, Bonneau JC, Alcalay D, Cavelier B, Toure C, Lobet R, Netter A (1991) HLA antigen-sharing in couples with repeated spontaneous abortions and the birthweight of babies in successful pregnancies. Am J Reprod Immunol 25:25–27 [DOI] [PubMed] [Google Scholar]
- 30.Cowan LD, Hudson L, Bobele G, Chancellor I, Baker J (1994) Maternal-fetal HLA sharing and risk of newborn encephalopathy and seizures: a pilot study. J Child Neurol 9:173–177 [DOI] [PubMed] [Google Scholar]
- 31.Stubbs EG, Ritvo ER, Mason-Brothers A (1985) Autism and shared parental HLA antigens. J Am Acad Child Psychiatry 24:182–185 [DOI] [PubMed] [Google Scholar]
- 32.Ekelund J, Lichtermann D, Hovatta I, Ellonen P, Suvisaari J, Terwilliger JD, Juvonen H, Varilo T, Arajarvi R, Kokko-Sahin M-L, Lonnqvist J, Peltonen L (2000) Genome-wide scan for schizophrenia in the Finnish population: evidence for a locus on chromosome 7q22. Hum Mol Genet 9:1049–1057 10.1093/hmg/9.7.1049 [DOI] [PubMed] [Google Scholar]
- 33.Ekelund J, Hovatta I, Parker A, Paunio T, Varilo T, Martin R, Suhonen J, Ellonen P, Chan G, Sinsheimer JS, Sobel E, Juvonen H, Arajarvi R, Partonen T, Suvisaari J, Lonnqvist J, Meyer J, Peltonen L (2001) Chromosome 1 loci in Finnish schizophrenia families. Hum Mol Genet 10:1611–1617 10.1093/hmg/10.15.1611 [DOI] [PubMed] [Google Scholar]
- 34.Hovatta I, Varilo T, Suvisaari J, Terwilliger JD, Ollikainen V, Arajarvi R, Juvonen H, Kokko-Sahin M-L, Vaisanen L, Mannila H, Lonnqvist J, Peltonen L (1999) A genomewide screen for schizophrenia genes in an isolated Finnish subpopulation, suggesting multiple susceptibility loci. Am J Hum Genet 65:1114–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paunio T, Ekelund J, Varilo T, Parker A, Hovatta I, Turunen JA, Rinard K, Foti A, Terwilliger JD, Juvonen H, Suvisaari J, Arajarvi R, Suokas J, Partonen T, Lonnqvist J, Meyer J, Peltonen L (2001) Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum Mol Genet 10:3037–3048 10.1093/hmg/10.26.3037 [DOI] [PubMed] [Google Scholar]
- 36.American Psychiatric Association (1994) Diagnostic and statistical manual, 4th ed. American Psychiatric Association, Washington, DC [Google Scholar]
- 37.Blin N, Stafford DW (1976) A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res 3:2303–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muramoto J, Cecka JM, Reed EF (2004) Class I DNA typing proficiency approaches 99% consensus: summary of the 10th year of the International HLA DNA Exchange. Hum Immunol Suppl 65:580 [Google Scholar]
- 39.Lau M, Locke AF, Park MS, Cecka JM, Reed EF (2005) The International Cell Exchange: analysis of class II polymorphism. Hum Immunol Suppl 66:29 10.1016/j.humimm.2005.08.051 [DOI] [Google Scholar]
- 40.Schreuder GMT, Hurley CK, Marsh SGE, Lau M, Maiers M, Kollman C, Noreen HJ (2001) The HLA dictionary 2001: a summary of HLA-A, -B, -C, -DRB1/3/4/5 and -DQB1 alleles and their association with serologically defined HLA-A, -B, -C, -DR, and -DQ antigens. Eur J Immunogenet 28:565–596 10.1046/j.0960-7420.2001.00284.x [DOI] [PubMed] [Google Scholar]
- 41.Sobel E, Papp JC, Lange K (2002) Detection and integration of genotyping errors in statistical genetics. Am J Hum Genet 70:496–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lange K, Cantor RM, Horvath S, Perola M, Sabatti C, Sinsheimer JS, Sobel E (2001) MENDEL version 4.0: a complete package for the exact genetic analysis of discrete traits in pedigree and population data sets. Am J Hum Genet Suppl 69:A1886 [Google Scholar]
- 43.Kraft P, Palmer CGS, Woodward JA, Turunen JA, Minassian S, Paunio T, Lonnqvist J, Peltonen L, Sinsheimer JS (2004) RHD maternal-fetal genotype incompatibility and schizophrenia: extending the MFG test to include multiple siblings and birth order. Eur J Hum Genet 12:192–198 10.1038/sj.ejhg.5201129 [DOI] [PubMed] [Google Scholar]
- 44.Hsieh HJ (2005) Assessing maternal, offspring and maternal-fetal genotype incompatibility effects at a highly-polymorphic locus. PhD dissertation, University of California, Los Angeles [Google Scholar]
- 45.Hsieh HJ, Palmer CGS, Harney S, Newton JL, Wordsworth P, Brown MA, Sinsheimer JS (2006) The v-MFG test: investigating maternal, offspring, and maternal-fetal genetic incompatibility effects on disease and viability. Genet Epidemiol 30:333–347 10.1002/gepi.20148 [DOI] [PubMed] [Google Scholar]
- 46.Sinsheimer JS, Palmer CGS, Woodward JA (2003) Detecting genotype combinations that increase risk for disease: the maternal-fetal genotype incompatibility test. Genetic Epidemiology 24:1–13 10.1002/gepi.10211 [DOI] [PubMed] [Google Scholar]
- 47.Ferguson TS (1996) A course in large sample theory. Chapman & Hall/CRC, London [Google Scholar]
- 48.Vatten LJ, Skjaerven R (2004) Offspring sex and pregnancy outcome by length of gestation. Early Hum Dev 76:47–54 10.1016/j.earlhumdev.2003.10.006 [DOI] [PubMed] [Google Scholar]
- 49.Umbach DM, Weinberg CR (2000) The use of case-parent trials to study joint effects of genotype and exposure. Am J Hum Genet 66:251–261 [DOI] [PMC free article] [PubMed] [Google Scholar]