

Abstract
We examined the role of tumor necrosis factor (TNF-α) and its related signaling intermediates leading to apoptosis/proliferation in the peripheral blood mononuclear cells (PBMCs) of RA patients. The constitutive expression of mRNA for TNF-α receptors (TNFR-I and TNFR-II) and the adapter molecules, such as the TNF receptor-associated death domain protein (TRADD), Fas-associated death domain protein (FADD), receptor interacting protein (RIP), and TNF receptor-associated factor 2 (TRAF-2) were analyzed by reverse transcriptase-PCR (RT-PCR) in PBMCs from control and RA cases. PBMCs of RA patients showed a significant increase in TNF-α and TNFR-I expression as compared with that from control subjects along with significantly increased constitutive expression of TRADD, RIP, and TRAF-2 mRNA. There was a decrease in expression of FADD in RA patients, but the difference was not significant as compared to controls. These data suggested enhanced signaling by the TNFR-I-TRADD-RIP-TRAF-2 pathway and suppressed signaling by the TNFR-I-TRADD-FADD pathway in PBMCs of RA patients. However, the regulatory mechanisms for TNF-α induced signaling may not be explained only by these pathways.
INTRODUCTION
Abnormal proliferation and/or persistence of synoviocytes and inflammatory cells has long been described in inflammatory arthritis conditions, but only recently substantial attention has been drawn to the relevance of abnormal apoptotic processes in disease pathogenesis and treatment [1]. Rheumatoid arthritis (RA) is a chronic inflammatory disorder with autoimmune features that affects approximately 1% of the world's population. It is characterized by inflammation of synovial tissue and the formation of rheumatoid pannus, which is capable of eroding adjacent cartilage and bone causing subsequent joint destruction. Although the precise etiology of the disease is unknown, genetic and environmental factors seem to be involved in its pathogenesis [2]. Previous studies have indicated that the relative risk of developing the disease in siblings of affected individuals (λs) is 2–17 times higher as compared to the general population, suggesting the importance of genetic factors in rheumatoid arthritis [3]. Recently we reported an association of mannose binding lectin gene polymorphisms with the occurrence and disease progression [4]. The association of TNF-α microsatellite with susceptibility and progression of RA in our study population was found to be distinct from other populations [5].
A broad array of macrophage and fibroblast cytokines, including IL-1, IL-6, IL-15, IL-18, tumor necrosis factor (TNF-α), granulocyte-macrophage colony-stimulating factor (GM-CSF), various chemokines, and many others, is produced by rheumatoid synovium. Increased hyperplasia of the synovial membrane imposed by these proinflammatory cytokines has been suggested to play a crucial role in disease progression [6]. In rheumatoid arthritis, joint pathology has been shown to be associated with high IL1-β and TNF-αproduction and TNF antagonists have proven to be the most efficient therapy for RA thus far [6, 7].
TNF-α, a potent proinflammatory cytokine, is known to regulate cell survival, death, and/or growth depending upon the cell types [7]. The cytotoxic pathway involves interaction of death domain-containing adapter molecules and caspases leading to apoptosis, whereas the cell-protective pathway involves activation of transcription factors, including NF-kB [8]. TNF-α transduces its signals by binding with TNF receptors, TNFR-I and TNFR-II (Figure 1). These two receptors differ from each other, only by the presence of a conserved motif in the cytoplasmic tail called the death domain [9]. TNFR-I mediates most of the biological properties of TNF-α, such as programmed cell death and activation of NF-kB. Upon activation, the death domain serves as a docking site for the death domain-containing adaptor proteins such as TRADD (TNFR associated death domain) through homotypic death domain interaction [10]. The recruitment of caspase-8 or -10 via the complex of TRADD and FADD (Fas-associated death domain) leads to the activation of a caspase cascade resulting in apoptosis [8, 9]. Once TRADD binds to the TNFR-I, it can also lead to the recruitment of RIP (receptor interacting protein) and TRAF-2 (TNF receptor-associated factor 2). RIP is a death domain-containing kinase that is crucial for NF-kB activation [9]. TNFR-II lacks the death domain but the intracellular domain of this receptor contains a consensus motif that allows binding to TRAF-2 [11]. TRAF-2 activates both NF-kB and JNK (cJun N-terminal kinase) and mediates its antiapoptotic effect [12]. Therefore, TNFR-II is involved in the antiapoptotic effect of TNF-α, whereas TNFR-I involves both apoptotic and antiapoptotic signaling [8]. NF-kB has been implicated in linking inflammatory responses to an anti-inflammatory pathway [13]. NF-kB is a ubiquitous transcription factor that can be activated by proinflammatory agents, such as TNF-α, IL-1β, LPS, oxidative stress [14]. In quiescent cells, NF-kB is sequestered in the cytoplasm by a set of inhibitory molecules including I-kB. Upon stimulation by any proinflammatory molecule the I-kB undergoes signal-induced phosphorylation and subsequent degradation. Once I-kB undergoes degradation, NF-kB translocates inside the nucleus and regulates the transcription of various inflammation regulatory molecules [9] .
Figure 1.
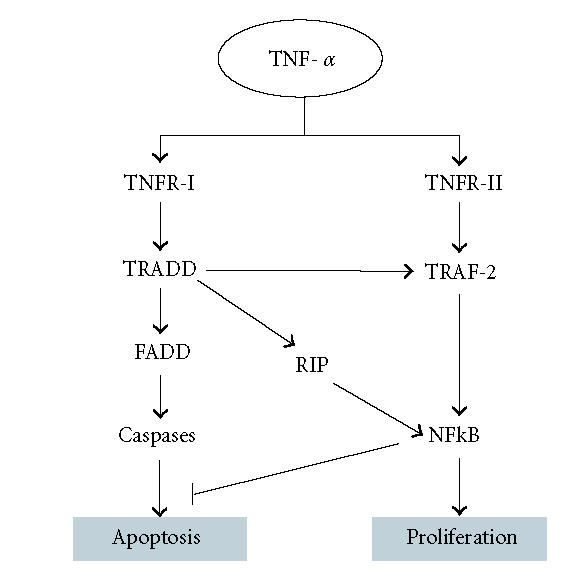
An overview of the TNF-α signaling pathway leading to apoptosis or proliferation of the cells.
In aged humans, the role of TNF-α induced apoptosis in decrease of T cells was examined by the expression of receptors for TNF-α by peripheral blood lymphocytes [15]. Increased constitutive expression of TNFR-I and TRADD and decreased expression of TNFR-II and TRAF-2 were observed in lymphocytes from aged as compared with young controls. These data suggest that increased TNF-α induced apoptosis plays a role in T-cell deficiency associated with human aging.
TNF-α was reported to be present at high concentrations in the blood and synovial fluid of RA patients. Therefore, differential expression and selective usage of TNF receptors may also work as a switch for TNF-α mediated signaling pathways on peripheral blood mononuclear cells (PBMCs) of RA patients. An increased expression of TNFR-II as compared to TNFR-I was also observed in synoviocytes as compared to peripheral blood mononuclear cells [9].
This study was designed to investigate the downstream signaling pathways of TNF-α in the PBMCs of RA patients. Expression of TNF receptors (TNFR-I and TNFR-II), TRADD, FADD, RIP, and TRAF-2 were analyzed to understand the mechanisms controlling apoptosis and proliferation in PBMCs of RA patients.
MATERIALS AND METHODS
Patients and controls
Blood samples from 27 patients diagnosed with rheumatoid arthritis were collected at the Out-Patient Department of Rheumatology, Army Hospital, Research and Referral, New Delhi, India, after a thorough investigation by the rheumatologist. All patients fulfilled the American College of Rheumatology classification criteria for the disease. The patients were diagnosed under the supervision of the rheumatologists using their medical records stating the disease duration, duration of morning stiffness, presence of extra-articular manifestations, presence of bone deformities, rheumatoid factor, and acute phase response (erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP)). The disease activity score using DAS-28 [16] disease score calculator was calculated on the basis of the number of tender joints involved, number of swollen joints, ESR, and visual analogue score (VAS) for general health as subjectively estimated by the patients. The patients had an active form of the disease but none of them were put on any disease modifying antirheumatic drugs (DMARDs). Informed consent was obtained from each patient and normal individual. Both the control and case individuals were age, sex, and ethnicity matched. Age in the control group was 40 ± 5 years and for the RA patients was 45 ± 6 years. There were 15 females and 12 males in our cohort of RA patients. The procedures followed were in accord with the ethical standards established by the ethics committee of the Institute and the Army Hospital. Samples from 30 healthy individuals served as controls.
The blood samples were collected in the sterile vacutainers with ACD-A buffer (Greiner Bio-One GmbH). Peripheral blood mononuclear cells (PBMCs) were immediately isolated from the blood samples using Histopaque-1077 (Sigma Chemicals, USA) by vendor recommended protocol. The PBMC samples were stored at −80°C without any freeze thawing till the samples were further processed for RNA isolation.
First-strand cDNA synthesis and gene specific polymerase chain reaction (PCR)
Total RNA was isolated from frozen PBMC samples by EZ-RNA isolation kit (Biological Industries) using vendor recommended protocol. The isolated RNA was dissolved in DEPC (diethyl pyrocarbonate, Sigma Chemicals, USA) treated deionized water and quantified. Two micrograms of the total RNA were used to initiate the first-strand cDNA synthesis using Advantage RT-for-PCR Kit (BD Clontech) by following vendor's recommended procedure. Control RNA and the primers for G3PDH are included in the kit and were used to validate the quality of cDNA synthesized for each reaction. Gene specific primers were designed using the DNASTAR software. Primer sequences specific for the TNF-α mediated signaling genes are presented in Table 1. The first-strand cDNA was diluted 1 : 5 with the deionized water and used for gene specific PCR reactions. The TNF-α mediated signaling genes were amplified with a DNA thermal cycler (MJ Research, Perkin Elmer) using the diluted cDNA, 25 mM MgCl2, 1.25 mM dNTP, 10 pM each of the forward and reverse primer and 2.5U of Taq DNA polymerase. After an initial denaturation at 94°C for 4 min, the amplification was conducted for 35 cycles at 94°C for 30 s, primer specific annealing temperature (Table 1) for 30 s, 72°C for 60 s and a final extension for 7 min at 72°C. The PCR product was visualized on a 1.2% agarose gel. The density of each band (PCR product) was measured using Image Scan and Analysis System (Alpha Innotech Corporation, USA) with Digidoc 1201 software.
Table 1.
Primer sequences specific for the TNF-α and TNF-α mediated signaling genes used for the amplification of cDNA prepared from the PBMCs of 27 RA patients and 30 healthy controls.
| Gene | Primer sequence | Amplicon length (bp) | Annealing temperature (°C) |
| TNF-α | 5′ CAGAGGGAAGAGTTCCCCAG 3′ | 325 | 60 |
| 5′ CCTTGGTCTGGTAGGAGACG 3′ | |||
| TNFR-I | 5′ ACCAAGTGCCACAAAGGAAC 3′ | 263 | 55 |
| 5′ CTGCAATTGAAGCACTGGAA 3′ | |||
| TNFR-II | 5′ GTT GGA CTG ATT GTG GGT GTG A 3′ | 454 | 60 |
| 5′ AGG GGC TGG AAT CTG TGT CTC 3′ | |||
| TRADD | 5′ GGTTCCTTCTGCGGCTATTGCTGA 3′ | 251 | 60 |
| 5′ TGAAACTGTAAGGGCTGGCTGTAA 3′ | |||
| FADD | 5′ CTGCCTTGGCAATTCTGTTATCAG 3′ | 267 | 60 |
| 5′ TGGCTGGGGTGGGGGTGGGGAGAC 3′ | |||
| RIP | 5′ TGGGAAAGCACTGGAAAAC 3′ | 200 | 55 |
| 5′ GTCGATCCTGGAACACTGGT 3′ | |||
| TRAF-2 | 5′ ACCAGCCCAGTCCTCAGATTTCAGA 3′ | 346 | 60 |
| 5′ CTAGGAATGCTCCCTTCTCTCTCCAG 3′ | |||
Statistical analysis
All data presented represent the mean and standard error (SE) for n determinations. Data analyses were performed using the World Wide Web available software. The density of PCR product for each sample was measured and the relative density mean (the value of the intensity of PCR product divided by the intensity for G3PDH) was calculated. All pairwise multiple comparisons were made using the Student t-test. The two tailed P values equal to or less than 0.05 were considered significant. Means ± SD (standard deviation) of the raw data used for normalization is presented in the results.
RESULTS
Expression of genes involved in the TNF-α mediated signaling pathway
We analyzed the constitutive expression of the genes that are involved in TNF-α mediated signaling in PBMCs of 30 normal healthy controls and 27 RA patients. We observed the following significant differences in the gene expression profile of the RA patients in comparison to the healthy controls.
Expression of TNF-α, TNFR-I, and TNFR-II genes
PBMCs from RA patients revealed a stronger expression of TNF-α and TNFR-I mRNA than those from the controls (Figure 2). There was a statistically significant difference (P < 0) between the expression of TNF-α in the RA patients (mean ± SD = 1.607 ± 0.39) with the healthy controls (mean ± SD = 1.0215 ± 0.224). An increase (P < .0003) in the expression of TNFR-I was observed in the RA patients (mean ± SD = 2.1289 ± 0.8117) as compared to the healthy controls (1.398 ± 0.5428, mean ± SD, Figure 2). However, we did not find an altered expression of TNFR-II gene in the PBMCs of the controls/cases in our cohort. The level of the gene expression in the controls (mean ± SD = 1 ± 0 ) was similar to that in the RA patients (mean ± SD = 1 ± 0) without any statistical significance (P = .692).
Figure 2.

(a) Agarose gel (1.2%) showing gene expression profile of TNF-α, TNFR-I, and TNFR-II from PBMCs of five representative healthy controls and RA patients. G3PDH gene was used for estimating the relative density of gene specific expression. (b) Relative density mean (density of gene specific product/density of housekeeping gene G3PDH) of 30 healthy controls and 27 RA patients for the expression of the genes.
Expression of TRADD and FADD genes
The representative RT-PCR gels demonstrating the expression of TRADD and FADD genes from control and RA cases are shown in Figure 3. As evident, there was a statistically significant increase in the expression of TRADD gene (P < 0) in the RA patients (mean ± SD = 1.919 ± 0.581) in comparison to the normal healthy controls (mean ± SD = 1.312 ± 0.2885 ). However, the FADD gene was observed to be equally expressed in both the cases and the controls (patients mean ± SD = 1.497 ± 0.782, controls mean ± SD = 1.7816 ± 0.360).
Figure 3.

(a) Gene expression profile of TRADD and FADD from PBMCs of representative healthy controls and RA patients. (b) Relative density mean (density of gene specific product/density of housekeeping gene G3PDH) of 30 healthy controls and 27 RA patients for the expression of TRADD and FADD genes.
Expression of RIP and TRAF-2 genes
On analyzing the expression of RIP and TRAF-2 genes in the controls and RA patients (Figure 4), we observed an increase in the expression of both the RIP (P = 0.0123) and TRAF-2 genes (P = 0) in RA patients (RIP: mean ± SD = 2.9799 ± 1.4946, TRAF2: mean ± SD = 1.942 ± 0.564) as compared to the healthy controls (RIP: mean ± SD = 2.0966 ± 0.9343 TRAF2: mean ± SD = 1.1944 ± 0.582).
Figure 4.

(a) Agarose gel (1.2%) showing gene expression profile of RIP and TRAF-2 from PBMCs of five representative healthy controls and RA patients. (b) Graph showing the relative density mean of 30 healthy controls and 27 RA patients for the expression of RIP and TRAF-2 genes.
DISCUSSION
Rheumatoid arthritis (RA), an autoimmune disease, is characterized by chronic inflammatory symptoms leading to damage of synovial tissue and joints. Proinflammatory cytokines like TNF-α reportedly contributes to the pathogenesis of rheumatoid arthritis [17]. In support of such reports we also observed an overexpression of TNF-α gene in the PBMCs of RA patients as compared to the healthy controls. TNF-α is associated with various cell signaling systems via two types of cell surface receptors, TNFR-I (p55) and TNFR-II (p75) [8, 10]. TNFR-I is involved in both proapoptotic and antiapoptotic signaling via the interaction with TRADD-FADD and TRADD-RIP-TRAF-2, respectively, [10]. Both TNFR-I and Fas-mediated apoptotic pathways use FADD as a common conduit [18] for the activation of apoptotic signaling. In contrast, TNFR-II is involved in an antiapoptotic effect of TNF-α via TRAF-2 [8].
The present study, for the first time, demonstrates that the PBMCs of RA patients showed an overexpression of TNFR-I and TRADD genes in comparison to the healthy controls; however, only a mild increase in TNFR-II expression was observed. Overexpression of TRADD results in apoptotic cell death via TNF-α induced activation of caspases as reported in many cell lines [19]. We observed a similar expression of the FADD gene among the RA patients and the healthy controls. This result indicates that the TNF-α-TRADD/FADD induced apoptotic pathway is not being followed in the PBMCs of the RA patients.
We observed a constitutive expression of TNFR-II gene in both the controls and the RA patients without any statistical significance. This nullifies any significant overexpression of the TNFR-II mediated proliferative/antiapoptotic pathway in the RA patients. However the TRAF-2 and RIP genes in the PBMCs were significantly over expressed among the RA patients. This demonstrates a significant overexpression of the antiapoptotic pathway in the PBMCs of the RA patients in contrast to the healthy individuals.
Our observations of increased TRAF-2/RIP expression in PBMCs suggest that an increased TRAF-2/RIP may result in TNF-α induced proliferation via NF-kB-dependent pathway.
In conclusion, PBMCs of the RA patients are observed to be more susceptible to TNF-α induced proliferation as compared to control subjects and are associated with a decreased expression of molecules (FADD) involved in apoptotic pathways. Increased expression of TNF-α in the PBMCs and increased susceptibility of these cells to TNF-α induced antiapoptosis may play a role in the pathogenesis of the disease.
The present study therefore supports the notion that the changes in the expression of TNF-α associated downstream molecules are more complicated than the expression of TNF-α/TNF receptors. This is probably caused by the complex regulatory mechanisms including cross talks with other signaling pathways that function in association with TNF-α mediated signaling pathways.
ACKNOWLEDGMENTS
The authors wish to thank the Council of Scientific and Industrial Research, India, for the financial assistance and Mr. A. N. Shirkey of Army Hospital, Department of Rheumatology, for his constant support during collection of blood samples from the patients.
References
- 1.Peng SL. Fas (CD95)-related apoptosis and rheumatoid arthritis. Rheumatology. 2006;45(1):26–30. doi: 10.1093/rheumatology/kei113. 2005 September 13: [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 2.Kim-Howard XR, Staudt L, James JA. Update in rheumatoid arthritis therapy. The Journal of the Oklahoma State Medical Association. 2005;98(2):53–62. [PubMed] [Google Scholar]
- 3.Suzuki A, Yamada R, Chang X, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nature Genetics. 2003;34(4):395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 4.Gupta B, Agrawal C, Raghav SK, et al. Association of mannose-binding lectin gene (MBL2) polymorphisms with rheumatoid arthritis in an Indian cohort of case-control samples. Journal of Human Genetics. 2005;50(11):583–591. doi: 10.1007/s10038-005-0299-8. [DOI] [PubMed] [Google Scholar]
- 5.Agrawal C, Raghav SK, Gupta B, et al. Tumor necrosis factor-alpha microsatellite polymorphism association with rheumatoid arthritis in Indian patients. Archives of Medical Research. 2005;36(5):555–559. doi: 10.1016/j.arcmed.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 6.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 7.Tracey KJ, Cerami A. Tumor necrosis factor, other cytokines and disease. Annual Review of Cell Biology. 1993;9:317–343. doi: 10.1146/annurev.cb.09.110193.001533. [DOI] [PubMed] [Google Scholar]
- 8.Sawanobori M, Yamaguchi S, Hasegawa M, et al. Expression of TNF receptors and related signaling molecules in the bone marrow from patients with myelodysplastic syndromes. Leukemia Research. 2003;27(7):583–591. doi: 10.1016/s0145-2126(02)00095-4. [DOI] [PubMed] [Google Scholar]
- 9.Youn J, Kim HY, Park JH, et al. Regulation of TNF-α-mediated hyperplasia through TNF receptors, TRAFs, and NF-κB in synoviocytes obtained from patients with rheumatoid arthritis. Immunology Letters. 2002;83(2):85–93. doi: 10.1016/s0165-2478(02)00079-2. [DOI] [PubMed] [Google Scholar]
- 10.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81(4):495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 11.Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995;269(5229):1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 12.Reinhard C, Shamoon B, Shyamala V, Williams LT. Tumor necrosis factor alpha-induced activation of c-jun N-terminal kinase is mediated by TRAF2. The EMBO Journal. 1997;16(5):1080–1092. doi: 10.1093/emboj/16.5.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miagkov AV, Kovalenko DV, Brown CE, et al. NF-kappaB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(23):13859–13864. doi: 10.1073/pnas.95.23.13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annual Review of Immunology. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 15.Aggarwal S, Gollapudi S, Gupta S. Increased TNF-alpha-induced apoptosis in lymphocytes from aged humans: changes in TNF-alpha receptor expression and activation of caspases. Journal of Immunology. 1999;162(4):2154–2161. [PubMed] [Google Scholar]
- 16.Prevoo ML, van 't Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis and Rheumatism. 1995;38(1):44–48. doi: 10.1002/art.1780380107. [DOI] [PubMed] [Google Scholar]
- 17.Brennan FM, Maini RN, Feldmann M. TNF alpha—a pivotal role in rheumatoid arthritis? British Journal of Rheumatology. 1992;31(5):293–298. doi: 10.1093/rheumatology/31.5.293. [DOI] [PubMed] [Google Scholar]
- 18.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 19.Park A, Baichwal VR. Systematic mutational analysis of the death domain of the tumor necrosis factor receptor 1-associated protein TRADD. The Journal of Biological Chemistry. 1996;271(16):9858–9862. doi: 10.1074/jbc.271.16.9858. [DOI] [PubMed] [Google Scholar]