

Abstract
Histone modifications are associated with distinct transcription states and serve as heritable epigenetic markers for chromatin structure and function. While H3 K9 methylation defines condensed heterochromatin that is able to silence a nearby gene, how gene silencing within euchromatin regions is achieved remains elusive. We report here that histone H3 K4 methylation or K9/K14 acetylation defines distinct chromatin regions permissive or nonpermissive for transgene expression. A permissive chromatin region is enriched in H3 K4 methylation and H3 acetylation, while a nonpermissive region is poor in or depleted of these two histone modifications. The histone modification states of the permissive chromatin can spread to transgenic promoters. However, de novo histone H3 acetylation and H3 K4 methylation at a transgenic promoter in a nonpermissive chromatin region are stochastic, leading to variegated transgene expression. Moreover, nonpermissive chromatin progressively silences a transgene, an event that is accompanied by the reduction of H3 K4 methylation and H3 acetylation levels at the transgenic promoter. These repressive effects of nonpermissive chromatin cannot be completely countered by strong transcription activators, indicating the dominance of the chromatin effects. We therefore propose a model in which histone H3 acetylation and H3 K4 methylation localized to discrete sites in the mammalian genome mark distinct chromatin functions that dictate transgene expression or silencing.
In eukaryotic cells, chromatin either facilitates or hinders access of the DNA to transcription activators, thereby regulating gene expression. The basic unit of chromatin consists of an octamer of four core histones (H2A, H2B, H3, and H4) wrapped around 146 bp of DNA. While multiple lysine or arginine residues in the core histones, particularly H3 and H4, are subject to posttranslational modifications including methylation and acetylation, many of these modifications are associated with distinct transcription states (6, 45). For example, H3 hyperacetylation (H3ac) or methylation at lysine 4 (H3K4me) is often associated with active genes (40), whereas methylation of the same protein at lysine 9 (H3K9me) is generally found to exist in chromatin containing silent genes (25). Histone modifications may serve as docking sites for effectors that regulate chromatin structure or function and ultimately transcription (26). Indeed, H3 K4 methylation recruits chromatin-remodeling enzymes that lead to a relaxed or “open” chromatin structure permissive for transcription (41). Conversely, recruitment of heterochromatin protein 1 (HP1) by H3 K9 methylation results in the assembly of compact or “closed” chromatin around the DNA, leading to silenced gene expression (4, 22).
Interestingly, in addition to local control of transcription, histone modifications also serve as markers for distinct chromatin regions throughout the genome (13). In a genomic locus of fission yeast, H3 K9 methylation concentrates in a 20-kb silent heterochromatic region while H3 K4 methylation is localized to the surrounding euchromatin regions (31). Similarly, H3 K9 methylation in the mammalian genomes defines distinct heterochromatin or silent euchromatin regions where RNA polymerase II is excluded (33, 37). This distinct distribution of histone modifications thus defines discrete chromatin regions with a distinct structure. As a result, a gene localized to such distinct chromatin regions, either by gene arrangement or gene transfer, may be loaded into a distinct chromatin state, resulting in an unexpected transcription outcome varying from stable expression to silencing, a phenomenon referred to as position effect. Indeed, a gene translocated close to a heterochromatin region exhibits a mosaic pattern of expression (also referred to as position effect variegation, or PEV), due to spreading of the condensed chromatin state from the heterochromatin region to the gene location in a stochastic fashion (48). Variegated gene expression is also evident in model systems where HP1 is recruited to euchromatin via its fusion to a DNA-binding protein leading to enrichment of H3 K9 methylation and chromatin condensation (3, 43, 47).
In contrast to heterochromatin, euchromatin, characterized by prevalent gene expression, is depleted of H3 K9 methylation and other histone modifications associated with gene silencing (30, 33, 37). Nevertheless, translocated genes or transgenes are still frequently silenced in these regions. Such silencing is unlikely to be caused by PEV-like heterochromatinization, or large-scale tandem repetitive DNA that recruits proteins such as HP1 (42), since such DNA is rare in mammalian genome. These considerations prompted us to ask whether histone modifications other than H3 K9 methylation define distinct euchromatin regions where transgenes are differentially expressed. To address this, we have integrated various transgenes into separate but defined genomic locations utilizing FRT-mediated homologous recombination (50) and determined transgene expression and histone modification states in these locations. Here we report that, rather than H3 K9 methylation, high H3 acetylation (H3ac) and H3 K4 methylation (H3K4me) levels define distinct euchromatin regions allowing for stable expression of an integrated transgene. In contrast, integration into a region that is poor in or lacking H3ac and H3K4me confers mosaic expression and progressive silencing of the transgene. Further, when integrated into the nonpermissive regions, histone H3 associated with the transgenic promoter is subjected to de novo methylation and acetylation in a stochastic manner, providing a mechanism for variegated transgene expression. Our results also suggest that the reduction in the level of these modified histones at the trangenic promoters accounts for progressive silencing of transgenes in the nonpermissive locations. Thus, histone H3K4me and H3ac levels mark distinct chromatin regions that are permissive or nonpermissive for transgene expression.
MATERIALS AND METHODS
Cell culture and transfections.
All cells were routinely cultured in McCoy's 5A medium supplemented with 10% fetal bovine serum and antibiotics. Stable transfections were performed with poly-l-ornithine (Sigma, St. Louis, MO) as described previously (49), while Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used for transient transfections according to the manufacturer's recommendations.
FRT-directed homologous recombination.
We utilized a modified FLP recombination target (FRT)-directed homologous recombination system (50) to integrate various transgenes into defined genomic locations. First, human fibrosarcoma HT1080 cells were transfected with a plasmid pFRT/lacZeo harboring an FRT fragment. After being selected with 300 μg/ml Zeocin, the resistant clones were harvested and expanded, and clones integrated with a single copy of the FRT fragment were identified by Southern blotting analyses as described previously (50). These clones were designated the FRT lines, each representing a genomic location for subsequent transgene integrations. Second, the luciferase-encoding sequence in the pCM/Luc vector (50) was replaced with an enhanced green fluorescent protein (EGFP) coding sequence, and various promoters were subcloned into the upstream region of the reporter gene. We obtained plasmids harboring 1.7-kb MMP2 (36), 0.9-kb p16 (36), 1.2-kb TIMP3 (52), and 2.2-kb MMP9 promoters (all human origin) from E. N. Benveniste, E. Hara, J. Bennett, and M. Seiki, respectively. The cytomegalovirus (CMV) promoter and its 5′ deletions were cloned by PCR from the vector pcDNA5/FRT (Invitrogen, Carlsbad, CA). In addition, these promoters were subcloned into pGL3 for transient transfections to determine their strengths. Next, 1 μg of each of the plasmids harboring the EGFP gene driven by various promoters was cotransfected with 9 μg of pOG44, a plasmid encoding the Flp recombinase, into the FRT lines followed by selection of the transfected cells with 200 μg/ml hygromycin B. The obtained resistant clones were observed under a fluorescence microcope or pooled for fluorescence-activated cell sorter (FACS) analyses.
Chromosomal walking.
The FRT integration sites were identified with the DNA Walking SpeedUp Premix kit (Seegene, Rockville, MD) according to the manufacturer's protocol. Briefly, genomic DNA was prepared from each of the FRT lines as PCR templates, and three nested primers specific to the upstream sequence of the FRT site in the pFRT/lacZeo plasmid were designed and used in conjunction with the ACP primers provided by the kit to amply the genomic DNA flanking the FRT integration site with PCR. The obtained DNAs were directly sequenced and contained chimeric plasmid and genomic sequences. The existence of the FRT upstream plasmid sequences indicated the walking specificity, while the genomic sequences were BLAST searched in the UCSC Human Genome Browser (19; http://www.genome.ucsc.edu/, May 2004 assembly) to identify the genomic locations.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed essentially as described previously (51) with modifications. Briefly, after cell lysis, the cross-linked chromatin was sonicated and then incubated with antibodies against modified histones or mixed-lineage leukemia protein (MLL) at 4°C overnight. We purchased antibodies against methylated H3 at K4 (05-791), acetylated H3 at K9/K14 (06-599), acetylated H4 at K4/K7/K11/K15 (06-598), dimethylated H3 at K9 (07-441), and trimethylated H3 at K9 (07-442) from Upstate (Charlottesville, VA) and antibodies against monomethylated (ab8895), dimethylated (ab7766), and trimethylated (ab8580) H3 at K4 and MLL (ab17959) from Abcam (Cambridge, MA). The immunocomplex was precipitated with protein A-agarose (Upstate, Charlottesville, VA), and the beads were washed as previously described, sequentially treated with 10 μg of RNase A (37°C for 30 min) and 75 μg of proteinase K (45°C for 4 h), and incubated at 65°C overnight to reverse cross-link of the chromatin. The DNA was recovered by phenol-chloroform extractions and coprecipitation with glycogen, and dissolved in 50 μl of Tris-EDTA (TE) buffer for real-time PCR assays.
Real-time PCR quantitation.
The amounts of DNA associated with modified histones were quantitated by real-time PCR as described previously (51). We obtained the flanking sequences of the integration sites by searching the UCSC Human Genome Browser and designed a series of primers to amplify 1, 2, 5, 8, 10, and 15 kb upstream and 2- and 9-kb regions downstream of each of the integration sites. We also designed primers to specifically amplify the regions of transgenic promoters but not the endogenous promoters to quantify the histone-modifying states of the transgenic promoters. To compare the real-time PCR data between ChIP assays, we normalized the modification levels against the levels in the promoter region of a housekeeping gene coding for GAPDH. Normalization in this way significantly reduced interassay variation arising from extended manipulations of chromatin during the experimental processes (see Fig. 7B and D).
FIG. 7.

Mosaic cell populations are distinct in their histone modification levels. (A and B) The cells containing the MMP2-EGFP cassette integrated at the F39 location were sorted twice to isolate subpopulations expressing [F39(+)MMP2] or not expressing [F39(−)MMP2] EGFP. After expansion for 4 days, a portion of cells was subjected to FACS analyses (A) while the remaining cells were subjected to ChIP assays as in Fig. 6B. The flanking primer pair corresponds to the primers amplifying the −1-kb region in Fig. 2. (C and D) The F59 cells were sorted and analyzed as for panels A and B.
RT-PCR.
To determine the expression levels of PLAUR, KIAA0329, or YTHD2, total RNA was prepared and reverse transcribed for reverse transcription-PCR (RT-PCR) as described previously (51). Primers were designed to amplify cDNA fragments spanning the last two exons of these genes. To determine PCR efficiencies (Ex) of each primer pair, cDNA was diluted 1:10, 1:100, 1:1000, and 1:10,000, and subjected to real-time PCR. The increase in the threshold cycle number (CT) after dilution (ΔCT) was plotted against the log change of cDNA amounts [log (dilution fold)], and the slope (k) of a linear regression equation for the curve was obtained and used for calculating PCR efficiency using the formula Ex = 10k − 1. To determine the relative expression levels of two genes, cDNA was subjected to real-time PCR with corresponding primer pairs, and CT values were obtained. The expression level (X2) of gene 2 relative to gene 1 (X1) was then calculated with the formula 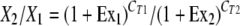 , in which Ex1/CT1 and Ex2/CT2 represent PCR efficiency/CT value for gene 1 and gene 2, respectively. The changes in gene expression before and after transgene integration were determined similarly, except that the gene expression level for each RNA sample was normalized against the β-actin expression level.
, in which Ex1/CT1 and Ex2/CT2 represent PCR efficiency/CT value for gene 1 and gene 2, respectively. The changes in gene expression before and after transgene integration were determined similarly, except that the gene expression level for each RNA sample was normalized against the β-actin expression level.
FACS analysis and cell sorting.
FACS analysis and cell sorting were performed using a Beckman Coulter instrument (EPICS XL-MCL). The percentages of EGFP-expressing cells were determined under standardized conditions using untransfected cells as the negative control. The levels of EGFP expression were defined as the mean fluorescence of 10,000 cells in the green channel.
RESULTS
Integration of transgenes in defined genomic locations through homologous recombination.
A prerequisite for studying the effects of the chromatin environment on transgene expression is the integration of the transgene into defined genomic locations. Towards this end, we have developed a homologous recombination system directing the insertion of a reporter expression cassette into given genomic locations (50). In this system, homologous recombination allows a transfected plasmid bearing an FRT fragment to “flip” into a genomic site harboring an FRT fragment (Fig. 1A). Therefore, once the FRT-integrated locus has been characterized, various transgenes can be introduced into the identical chromatin environment. This system allows us to study both the influence of the flanking chromatin on transgene expression and the effects of different promoters on position effects.
FIG. 1.

Integration of a transgene in a defined genomic location by homologous recombination. (A) Schematic showing that recombination between FRT fragments allows integration of an EGFP transgene driven by a promoter of interest at an FRT site within the genome. (B) HT1080 cells were transfected with an FRT-bearing vector. After selection in Zeocin-containing media, resistant clones were harvested and expanded. Genomic DNA was prepared, digested with HindIII or EcoRV (E) (for the F59 clone, digested with EcoRV plus BglII in the right panel), and subjected to Southern blotting with a probe spanning a plasmid region downstream of the FRT fragment. The four clones designated F29, F39, F55, and F59 contain a single copy of the FRT fragment (*). (C) Total RNA was prepared from HT1080 cells, reverse transcribed, and subjected to PCR to amplify fragments spanning the last two exons of the PLAUR, KIAA0329, and YTHD2 genes. A pair of β-actin primers was also included in the PCRs for loading control. (D) cDNA from HT1080 cells was subjected to real-time PCR. The relative expression levels were corrected for altered PCR efficiency among primer pairs. (E) cDNAs from HT1080 cells or F29, F39, F55, an F59 clones were subjected to real-time PCR quantitation for relative expression levels of PLAUR, KIAA0329, and YTHD2 and normalized to β-actin expression levels.
Thus, we developed several FRT clones derived from human fibrosarcoma HT1080 cells and for each clone confirmed the integration of a single copy of the FRT fragment by Southern blotting (Fig. 1B).
We further identified the FRT-integrated sites in these clones by chromosomal walking. We obtained and sequenced the DNAs flanking the integrated sites by sequential nested PCR with genomic DNAs isolated from these four FRT cells as templates and then BLAST searched the UCSC Human Genome Browser (19; http://www.genome.ucsc.edu/, May 2004 assembly) for the sequences. As shown in Table 1, the FRT fragment is localized in different genomic locations in these four cell clones. We refer to these genomic locations as F29, F55, F39, and F59 and the corresponding cell clone names are designated accordingly. Interestingly, none of these locations are close to the pericentromeres characterized by condensed heterochromatin. Rather, all of these locations are within gene-rich regions. In fact, a search of the Human Genome Browser indicated that with the exception of F55, all integration sites reside within or close to (distance of <15 kb) a known or predicted gene. Among them, the F29 site is located within exon 1 (upstream of start codon) of the PLAUR gene encoding the urokinase receptor, while the F39 site is within exon 2 but ∼13 kb downstream of the transcription start site of a predicted gene, KIAA0329 (Table 1). The expression levels of these three genes in HT1080 cells varied as is evident by RT-PCR (Fig. 1C). This difference was not caused by varied efficiencies of the PCRs, since real-time PCR quantitation corrected for PCR efficiency (Ex) for individual primer pairs (see Materials and Methods) revealed that the expression levels of KIAA0329 and YTHD2 were ∼3 and ∼57% of that of PLAUR, respectively (Fig. 1D). Interestingly, while integration of the FRT fragment did not alter the expression level of PLAUR or YTHD2 (possibly due to a bypass of any exogenous barrier to mRNA elongation), KIAA0329 expression was reduced by ∼40% (Fig. 1E), the latter possibly reflecting interference with mRNA elongation/splicing or a null allele.
TABLE 1.
Genomic locations of the integrated FRT sites
| Site | Cytogenetic band | Genomic location (position)a | % GCb | Nearby gene:
|
|||
|---|---|---|---|---|---|---|---|
| Designation | Distance | Orientationc | Expressiond | ||||
| F29 | 19q13.31 | Chromosome 19 (48,866,117) | 49.53 | PLAUR | Within exon 1 | + | High |
| F55 | Xq23 | Chromosome X (113,493,021) | 33.39 | None | |||
| F39 | 14q32.32 | Chromosome 14 (101,912,842) | 44.13 | KIAA0329 | Within exon 2 | + | Low |
| F59 | 1p35.3 | Chromosome 1 (28,919,827) | 44.78 | YTHD2 | 3.6 kbe | − | Medium |
Based on the UCSC Human Genome Browser (May 2004 assembly).
Average of percents GC in a 5-base window of a 20-kb region spanning the integration site.
+, nearby gene has the same orientation as the transgene; −, nearby gene has the reverse orientation.
Expression levels were determined by quantitative real-time RT-PCR (see Fig 1C and D).
Distance to the 3- end of the YTHD2 gene.
Histone modifications define distinct chromatin regions for transgene integration.
Since posttranslationl modifications, particularly acetylation and methylation, of core histones may regulate chromatin structure and function, we quantified these modifications in the chromatin flanking the integration sites with ChIP assays followed by real-time PCR quantitation. We thus incubated the cross-linked chromatin with antibodies against methylated H3 at K4 (H3K4me), acetylated H3 at K9/K14 (H3ac), acetylated H4 (H4ac), and dimethylated (H3K9me2) or trimethylated H3 at K9 (H3K9me3), purified the immunoprecipitated DNAs, and determined the amounts of DNA associated with these modified histones with real-time PCR. For the five histone modifications examined, the first three represent markers of active gene expression (6) , while H3K9me2 and H3K9me3 are associated with silenced euchromatin and condensed heterochromatin, respectively (33, 37). Based on the UCSC Human Genome Browser, we designed a series of primers to amplify the DNA flanking the integration sites and spanning a region from 15 kb upstream (−15 kb) to 9 kb downstream (9 kb) (Fig. 2). Consistent with recent genome-wide analyses (7), both H3 K4 methylation and H3 acetylation were distributed unevenly in the human genome (Fig. 2A and B). Interestingly, the chromatin upstream of the integration site, at the F29 and F55 locations, was enriched in H3 K4 methylation (Fig. 2A) and H3 acetylation (Fig. 2B) compared with the F39 and the F59 locations. Of the four locations, H3 in the F39 location contained the smallest amount of K4 methylation and H3 acetylation was almost completely depleted from this location. In contrast, the H4 acetylation levels differed little among these four locations (Fig. 2C). The distribution of H3 K4 methylation correlated with that of H3 acetylation, consistent with the view that the K4 methyltransferase and acetylase are corecruited (44). We also examined the enrichment of di- or trimethylated H3 at lysine 9 at these genomic locations. The amounts of DNA precipitated by H3K9me3 and H3K9me2 antibodies were equal to or were only slightly higher than the DNA pulled down by normal immunoglobulin G (IgG) (Fig. 2D and E), indicating that these regions contain little of either of these two modified histones. More importantly, the levels of these K9-methylated histones differed little among these four locations (Fig. 2D and 2E). As a control, we determined the efficacy of these antibodies in immunoprecipitating chromatin in the pericentromeric region (42,121,930 to 42,121,983) of chromosome 10. The H3K9me2 and H3K9me3 antibodies pulled down substantially larger amounts of this DNA compared with normal IgG (Fig. 2F), suggesting that these ChIP experiments were working properly.
FIG. 2.

Histone modification states of chromatin regions at the FRT integration sites. Cross-linked chromatin derived from the four FRT clones was incubated with antibodies (1 μg each) against K4-methylated H3 (A), acetylated H3 (B), acetylated H4 (C), K9 dimethylated H3 (D), and K9 trimethylated H3 (E). The immunoprecipitated complexes were captured by protein A-agarose beads followed by sequential washes and treatments with RNase A and proteinase K. After cross-link reversal, the DNA was purified with phenol/choloroform, precipitated, dissolved in TE buffer, and subjected to real-time PCR assays. A series of primers amplifying various flanking regions were designed for each location based on the sequences retrieved from the UCSC Human Genome Browser. The approximate locations of DNA amplified by the primers are indicated (solid black box) below the genomic representation (line). The amounts of precipitated DNA (percent input) shown are corrected for interassay variation using the amounts of precipitated GAPDH promoter DNA. To confirm the utility of antibodies against K9 di- or trimethylated H3 in the ChIP assays, the DNA precipitated by these antibodies or normal IgG was also subjected to real-time PCR amplifying a fragment residing in the pericentromeric region of chromosome 10 (F).
Since the K4 residue of histone H3 can be subjected to either di- or trimethylation (H3K4me2 and H3K4me3), which is correlated with active transcription, we also determined the distributions of these two modified H3 histones in the four genomic locations. Similar to the total K4-methylated H3 described above, H3K4me2 and H3K4me3 were enriched in the regions upstream of the integration site within both the F29 and the F55 locations, while the corresponding regions within the F39 and the F59 locations contained lower levels of this modified H3 (Fig. 3A and B). Although the H3K4me3 levels across the F55 flanking region were lower than that in the corresponding region at F29, they were still higher than those of the F39 and F59 locations (Fig. 3B). Indeed, the latter two locations were almost completely depleted of H3K4me3 (Fig. 3B). Moreover, the F55 location contained high levels of H3K4me2 (Fig. 3A). Interestingly, the distributions of H3K4me and, in particular, H3K4me3 appeared to coincide with the chromatin binding of MLL, a histone methyltransferase (HMT) that catalyzes the addition of methyl groups to the K4 residue of H3 (27) (Fig. 3C). This is consistent with an earlier ChIP-on-chip analysis (14), suggesting that MLL might, at least in part, contribute to the maintenance of histone methylation states at the F29 and F55 locations between generations. Therefore, we conclude that these four genomic regions are characterized by their absence (or very low levels) of silent chromatin markers (H3K9me2 or H3K9me3) but marked by varying enrichments of H3 K4 methylation and acetylation (H3K4me or H3ac) often associated with active chromatin.
FIG. 3.

Histone K4 di- and trimethylation states of chromatin regions integrated at the FRT integration sites. The cross-linked chromatin from the FRT clones was incubated with antibodies (1 μg each) against K4 dimethylated H3 (A), trimethylated H3 (B), or MLL (C) and subjected to chromatin immunoprecipitation assays followed by real-time PCR as described for Fig. 2. The primers are indicated at the top of the graphs. The amounts of precipitated DNA (percent input) have been normalized for interassay variation as described in the legend to Fig. 2.
Variable expression of a transgene integrated at distinct chromatin regions.
To determine the effects of the distinct chromatin compositions on expression of an inserted gene, we constructed an EGFP expression cassette driven by a 1.7-kb MMP2 promoter and introduced it into these chromatin regions by FRT-mediated homologous recombination (Fig. 1A). To exclude the possible interference by the upstream transcription of the hygromycin B resistance gene, we inserted a synthesized polyadenylation signal (23) upstream of the promoter (Fig. 1A), a strategy that eliminates transcription noise (50). We selected the transfected cells with hygromycin B for 10 days and examined the resistant clones by fluorescence microscopy. EGFP expression varied greatly between these clones (Fig. 4A). All clones derived from transfections with the F29 and F55 cells strongly expressed EGFP (as clone 1 in Fig. 4A). In contrast, most of the clones derived from the F39 cells did not express EGFP (as clone 3 in Fig. 4A), while some of the F39 or F59 clones weakly expressed EGFP (as clone 2 in Fig. 4A). To determine the EGFP expression in the population, we randomly harvested and pooled 15 clones from each transfection, expanded the cells, and subjected them to FACS analyses. Clearly, when integrated into the chromatin regions (F29 and F55) marked by active histone modifications, EGFP was expressed in almost all of the cells (Fig. 4B). Conversely, mosaic EGFP expression was apparent when the transgene was localized in the chromatin regions (F39 and F59) that were poor in or lacking H3K4me and H3ac (Fig. 4B). While EGFP within the F59 region expressed in 59.7% of cells, it was silenced in the majority of the cells (83.2%) when integrated into the chromatin region (F39) that contained the lowest levels of H3K4me/H3ac (Fig. 4B). Therefore, the levels of H3 K4 methylation and H3 acetylation in the flanking chromatin not only were associated with transgene expression states, but also correlated with the extents of gene silencing. Furthermore, the mean fluorescence intensities that reflect the EGFP expression levels in each cell population also correlated well with the levels of the active chromatin markers in each genomic location (Fig. 4C). The difference in the EGFP expression in these cells was unlikely caused by variations in the levels of transcription factors, since the endogenous MMP2 levels, as determined by gelatin zymography (49), were constant among these cells (Fig. 4D). We conclude that histone modifications likely mark chromatin regions that are permissive or nonpermissive for transgene expression. Thus, a transgene localized in a nonpermissive chromatin region that is poor in or depleted of active histone modifications could be silenced.
FIG. 4.

Chromatin effects on expression of an EGFP reporter driven by the MMP2 promoter. (A) An EGFP expression cassette driven by the MMP2 promoter was introduced into the four FRT clones by homologous recombination. Resistant clones were observed under a fluorescence microscope (right panel) 12 days after transfection. Left, bright-field view. (B) Fifteen clones from each transfection were randomly picked and pooled, expanded, and subjected to FACS analyses for EGFP expression. The numbers inside each graph indicate the percentage of positive cells. The line inside each graph defines the EGFP-positive signal. Ctrl, control. (C) Mean fluorescence intensity for each integration site. (D) Conditioned medium from the pooled clones was harvested and subjected to gelatin zymography to determine the levels of endogenous MMP2 protein.
Promoter strength counters the chromatin effect.
Since the homologous system we have developed (50) allows the introduction of chimeric reporter constructs of choice into the same genomic location, we determined whether different promoters modulate the chromatin effect described above. We replaced the MMP2 promoter with an MMP9, p16, TIMP3, or CMV promoter and integrated the EGFP expression cassettes into the four defined chromatin regions. The strengths of these promoters range from weak to strong (MMP9 < p16 < MMP2 < TIMP3 < CMV) (Fig. 5A). Interestingly, the promoter strength dramatically affected EGFP expression (Fig. 5B). The activities of the MMP9 and p16 promoters were comparable to that of MMP2, and EGFP under the control of these two promoters was expressed or silenced following a similar pattern to that of the MMP2 promoter (Fig. 5B). Thus, EGFP expression driven by the MMP9 or p16 promoter occurred in the permissive chromatin regions (F29 and F55) marked by H3K4me and H3ac, while it was partly silenced when localized in the nonpermissive regions (F39 and F59) (Fig. 5B). However, when EGFP was driven by strong promoters (CMV or the TIMP3), gene silencing by the nonpermissive chromatin in the F39 and F59 locations was countered (Fig. 5B). While weaker than the CMV promoter, the TIMP3 promoter did not completely rescue the EGFP expression in the F39 location since there were still about 10% of cells that were silenced (Fig. 5B). This suggests that the strength of this promoter was insufficient to counter the strongest silencing effect of the chromatin in the F39 location. These results suggest that the expression state/level is determined by a balance between the negative and positive regulators (8) residing in both the flanking chromatin and the gene itself. To corroborate this assertion, we sequentially deleted the cis-elements in the CMV promoter and determined EGFP expression under the control of these deleted promoters. With the reduction of promoter strength (Fig. 5C), EGFP expression within the nonpermissive locations (F39 and F59) showed silencing (Fig. 5D) such that the silencing-countering effect was completely lost with the truncated CMV Δ2 promoter. Therefore, silencing of a transgene by the nonpermissive chromatin can be countered by transcription activators.
FIG. 5.

Promoter strength counters the repressive effect of chromatin. (A) The indicated promoters were subcloned into pGL3 vectors, and 0.4 μg of each plasmid was cotransfected with 0.001 μg of pRL-TK into HT1080 cells in a 24-well plate. After 24 h, the cells were harvested and cell lysates were subjected to luciferase assays. The numbers inside the graph indicate the mean relative luciferase activities. (B) The indicated promoters were subcloned into the EGFP reporter, and the plasmids were introduced into the four FRT clones by homologous recombination. Fifteen clones of each transfection were pooled, expanded, and subjected to FACS analyses. The number inside each graph indicates the percentage of GFP-positive cells. (C) Deletions of the CMV promoter were subcloned into pGL3. Each of the plasmids (0.4 μg) was cotransfected with 0.001 μg of pRL-TK into HT1080 cells and subjected to luciferase assays. (D) The indicated truncated promoters were subcloned into the EGFP reporter plasmid. The plasmids were introduced into the F55, F39, and F59 clones by homologous recombination. Fifteen clones from each transfection were pooled, expanded, and subjected to FACS analyses. The number inside each graph indicates the percentage of GFP-positive cells.
Mosaic cell populations are distinct in their H3K4me/H3ac levels.
Since histone modifications at the gene itself regulate transcription, we speculated that the aforementioned histone code defining the flanking region could direct histone modifications at the transgenic promoter. Therefore, we determined the H3K4me and H3ac levels at the integrated MMP2 promoter by ChIP and real-time PCR assays (primer location depicted in Fig. 6A). As expected, H3K4me or H3ac was enriched in the chromatin associated with the promoter within the permissive chromatin regions (F29 and F55), while the promoter that was integrated at the nonpermissive location (F39) showed lower levels of the modified histones (Fig. 6B). These results suggest that the permissive chromatin enriched in H3K4me or H3ac directs the modifications of histones at the transgene, while the nonpermissive chromatin depleted of H3K4me or H3ac provides for inefficient histone modifications at the promoter. However, we also observed that the H3K4me level at the promoters within the restrictive F59 location was only slightly lower than those (F29 and F55) within the permissive locations (Fig. 6B). Moreover, for the MMP2 promoter, the difference in histone modification levels between the different integration sites was far smaller than that between the corresponding flanking regions (compare Fig. 6B with Fig. 2A and B). These results suggest that the histone modification states of the local chromatin regions could not be faithfully propagated to the transgene. Similar results were obtained with the TIMP3 promoter (Fig. 6C), indicating that transcription cis-elements do not affect the de novo histone modifications associated with the promoters. (Unfortunately, we could not compare modified histone levels at the strongest [CMV] promoter, since this promoter showed an unacceptably high background in the ChIP assays.)
FIG. 6.

Flanking chromatin affects histone modifications at transgenic promoters. (A) A primer pair (MMP) was designed to amplify a DNA fragment spanning the MMP2 promoter-EGFP coding sequence junction. The flanking primer pair corresponds to the primers amplifying the −1-kb region in Fig. 2. (B) The chromatin derived from cells containing the MMP2-EGFP cassette was incubated with antibodies (1 μg) against H3K4me or H3ac and subjected to ChIP assays followed by real-time PCR. The MMP primer described in panel A was used to amplify the integrated but not the endogenous MMP2 promoter. The amounts of immunoprecipitated DNA were normalized to the amounts of the GAPDH promoter and are presented as relative enrichments. (C) The chromatin derived from cells containing the TIMP3-EGFP cassette was subjected to ChIP assays as described in panel B. The primer pair used amplifies a region spanning the TIMP3 promoter-EGFP coding region junction. (D and E) The chromatin derived from the FRT cells and the MMP2-EGFP cells was incubated with antibodies against H3K4me (D) or anti-H3ac (E) and subjected to ChIP assays as described for Fig. 2.
Interestingly, insertion of the transgene had no significant effect on histone modifications in its local chromatin environment (Fig. 6D and E). This observation is consistent with the view that histone modifications are invariable between generations and heritable (20, 39).
A question surfaced as to why the EGFP gene integrated into the nonpermissive regions was silenced in a portion of cells given that the promoter itself was marked by active histones. One possibility is that the mosaic cell populations are subject to various levels of histone modifications at the transgenic promoter in a stochastic manner. To explore this possibility, we isolated an EGFP-positive population and an EGFP-silenced population by FACS from cells that harbor an integrated MMP2 promoter in the F39 genomic location and determined the H3K4me and H3ac levels by ChIP and real-time PCR assays. Due to rapid silencing of the transgene during the 4-day culturing expansion, we could only obtain a population with about 35% of EGFP-positive cells after two rounds of cell sorting (Fig. 7A). However, even compared with this unpurified cell population [F39(+)MMP2], both the promoter H3K4me and H3ac levels in the silenced cells [F39(−)MMP2] were much lower (Fig. 7B). As a control, we also determined the modified histone levels in the flanking sequence 1 kb upstream of the integration sites (Fig. 6A). No apparent difference was observed between these two subpopulations (Fig. 7B), suggesting that difference in the promoter histone modification levels was not due to random cell-cell variation. We repeated the experiments with the cells containing a transgene integrated at the F59 genomic location. Again, the levels of the active chromatin markers in the EGFP-positive cell population [F59(+)MMP2] were higher than that in the EGFP-negative cells [F59(−)MMP2] (Fig. 7D). However, the difference between the positive and negative cells was not as large as that in the F39 cells, probably due to impurity of the EGFP-negative cells (Fig. 7C) caused by reactivation of the transgene (11). These results suggest that integration of a transgene in nonpermissive chromatin regions poor in or depleted of active chromatin markers could generate cell subpopulations varying in the H3K4me or H3ac levels at the transgenic promoter. The subpopulation with higher H3K4me or H3ac levels expressed EGFP, while the one with lower modified histone levels was silenced. Therefore, varying levels of H3K4me or H3ac at the promoter are likely to contribute to mosaic or variegated expression of a transgene in the nonpermissive genomic locations.
Progressive silencing of the transgene is accompanied by reduced levels of the H3K4me and H3ac levels at the promoter.
Despite residing within regions poor in or depleted of H3 K4 methylation and H3 acetylation, the transgenic promoters initially contained relatively high levels of H3K4me and H3ac. One possible outcome from this imbalance could be the reduction of these modifications at the promoter resulting in progressive silencing of the transgene. To explore this possibility, we cultured the transgenic cells in the absence of hygromycin B in the culture medium for about 10 weeks and measured histone modifications and EGFP expression. Interestingly, FACS analyses indicated that the EGFP expression driven by either the MMP2 or the p16 promoter was progressively silenced in the nonpermissive locations (F39 and F59) but not in the permissive locations (F29 and F55) (Fig. 8A). Moreover, the silencing rate was higher at the F39 location than at the F59 locations (Fig. 8A). This is consistent with the observation in the cell-sorting experiments that the F39(+)MMP2 cells were silenced faster than the F59(+)MMP2 cells (Fig. 7A and C). Similar results were obtained when hygromycin B was present in the culture medium (data not shown), consistent with a previous report that expression cassettes in tandem respond to chromatin effects independently (10). We also determined the H3K4me and H3ac levels associated with the MMP2 promoter in cells that had already been cultured for 67 days and compared these levels to those at day 0. As expected, the H3K4me and H3ac levels at the promoter were higher in the F29 and F55 cells than those in the F39 and F59 cells at day 67 (Fig. 8B). In contrast to the levels at day 0, these levels correlated well with the expression states/levels of the EGFP gene (Fig. 8A). When compared to the cells at day 0, the H3K4me levels remained higher in the F29 and the F55 cells, but dropped rapidly in the F39 and F59 cells (Fig. 8C). Although the H3ac levels dropped in all of these four clones, they attenuated faster in the F39 and F59 cells than in the F29 and F55 cells. The final histone modification levels at the MMP2 promoter were comparable to those of the flanking chromatin regions, suggesting that a balance between the transgene and its local environment had been finally achieved, accompanied by progressive silencing of the transgene. On the other hand, the permissive chromatin could prevent the transgene from progressive silencing by preventing attenuation of the H3K4me levels at the transgenic promoter.
FIG. 8.

Progressive transgene silencing is accompanied by the reduction of modified histone levels. (A) The transgenic cells containing the MMP2 promoter and the p16 promoter were cultured for 67 days. The cells were periodically subjected to FACS analyses to determine EGFP expression. (B) Cells harboring the MMP2-EGFP cassette at the indicated genomic locus were cultured for 67 days. The chromatin derived from these cells was incubated with anti-H3K4me or anti-H3ac antibodies and subjected to ChIP assays followed by real-time PCR analyses. The amounts of precipitated DNA were normalized to the amounts of the GADPH promoter and are presented as relative enrichments. (C) Temporal changes in the H3K4me/H3ac levels between day 0 and day 67.
Since transcription activators could counter the effects of the nonpermissive chromatin on EGFP expression (Fig. 5), we determined the effects of strong promoters on progressive silencing. When driven by the TIMP3 promoter, the percentages of the EGFP-expressing cells remained constant in the F29, F55, and F59 locations but not the F39 location during the 10-week culture periods (Fig. 9A). On the other hand, the percentages of the EGFP-positive cells remained unchanged in all of these locations when the transgene was directed by the CMV promoter (Fig. 9A), suggesting that strong transcriptional activators prevent transgene silencing by a repressive chromatin environment. However, strong promoters failed to completely counter the adverse effects of the nonpermissive chromatin on the transgene expression, since the EGFP expression levels (as indicated by the mean fluorescence intensity) in the two nonpermissive locations were still progressively reduced (Fig. 9B). Furthermore, this reduction was accompanied by the reduction of H3 K4 methylation and H3 acetylation at the TIMP3 promoter (Fig. 9C and D). Therefore, strong transcription activators could not counter the reduction of H3 K4 methylation or H3 acetylation caused by the nonpermissive chromatin. The chromatin effect thus appears dominant over transcription activators in regulating transgene expression.
FIG. 9.

Strong promoters do not prevent reduction of the modified histone levels. (A) The transgenic cells bearing the TIMP3 promoter- and the CMV promoter-EGFP cassettes were cultured for 67 days. The cells were periodically subjected to FACS analyses to determine the EGFP expression. (B) The relative mean fluorescence intensity (in fluorescent light units [FLU]) representing the relative EGFP expression level was calculated. (C) The TIMP3 cells were cultured for 67 days and subjected to ChIP assays as for Fig. 6C. (D) Temporal changes in the H3K4me/H3ac levels between day 0 and day 67.
DISCUSSION
Frequent silencing of transgenes constitutes a major barrier for gene therapy and transgenic studies. While it is widely accepted that the local chromatin environment dictates the transcription outcome of a transgene, the determinants residing in the chromatin, particularly euchromatin, remain elusive. We have demonstrated herein that H3 K4 methylation and K9/K14 acetylation levels demark distinct euchromatin regions as permissive or nonpermissive for transgene expression. Moreover, these local histone modifications direct parallel histone changes at the transgenic promoter, thereby controlling transgene expression at a given chromosomal location. Thus, we advance a model for transgene position effect in which transgene silencing reflects the depletion of H3K4me/H3ac at a specified chromatin region.
These findings could very well have ramifications beyond the control of transgene expression. Recent genome-wide analyses of histone modifications have revealed that H3K4me and H3ac distribute in punctate sites in the genomes and are likely to mark active chromatin regions (7, 24, 35, 38). While many of these sites coincide with transcription starts (7, 24, 35, 38), some localize to intra- or intergenic regions spanning several kilobases of DNA. We have shown that such a region allows transgenes to be expressed, highlighting a possibility that these regions might be “open” for transcriptional machinery and contribute to abundant intergenic transcription (16, 17) in eukaryotic cells.
A distinct feature of the nonpermissive chromatin regions we have defined here is that although these regions are poor in or depleted of active chromatin markers like H3K4me or H3ac, they also lack heterochromatin makers such as H3K9me2 and H3K9me3. This suggests that the mechanism underlying transgene position effects evident in these euchromatin regions is distinct from PEV. In PEV, heterochromatin-assembling factors including HP1 are recruited and promote condensation of the chromatin (3). On the other hand, our findings argue that the euchromatin associated with the nonpermissive integration sites is “closed” simply due to a lack of H3K4me/H3ac which relaxes chromatin.
However, transgenes within these nonpermissive regions were not silenced in the entire cell population. Instead, both EGFP-expressing and silent clones were generated when a transgene was integrated into a defined nonpermissive location (Fig. 4A), suggesting that transgene expression states are stochastically determined at the time of transgene integration. Intriguingly, the cell subpopulation that expresses EGFP contained higher levels of H3K4me/H3ac at the transgene promoter than the silenced cells, underlining the importance of H3K4me/H3ac in maintaining the “open” chromatin structure for gene expression.
While the mechanism for histone modifications on the de novo-synthesized nucleosomes associated with a transgene is unknown, it may be similar to the mechanism for maintaining chromatin states during cell division. In one of these models, unmodified histones deposited randomly at the newly replicating DNA are targeted by histone methyltransferases/acetylases recruited via effectors (including HP1) binding to the parental modified histone residues (39). A similar model would predict spreading of methylated/acetylated histone states from the chromatin regions to the transgenic promoter. Thus, a promoter within the permissive regions enriched for H3K4me or H3ac would acquire the information for modifications on the newly synthesized nucleosomes. In line with this model, we found that MLL, a well-characterized histone methyltransferase (14, 27), was enriched in the permissive but not the nonpermissive regions (Fig. 3C).
However, this model cannot explain the enrichment of H3K4me/H3ac at the transgenic promoter under nonpermissive conditions. Instead, the histones at the promoter under these conditions appeared to be modified stochastically. Stochasticity results in all-or-none (binary) states when an effector is limiting (18, 21). Since nonpermissive chromatin regions contained little H3K4me or H3ac, the local histone-modifying enzymes such as MLL (Fig. 3C) recruited by these chromatin markers could be limiting. Thus, these enzymes would stochastically load on the newly-synthesized nucleosomes, resulting in nucleosomes varying in their methylation and acetylation levels (Fig. 10). Alternatively, H3K4me or H3ac may direct active chromatin to a nuclear compartment that contains high levels of methyltransferases and acetylases, a notion consistent with the observations of discrete nuclear distributions of modified histones or histone-modifying enzymes (5, 9, 15, 37). As a result, transgenic DNA within a permissive chromatin domain enters a compartment rich in histone-modifying enzymes, the latter efficiently loading on the newly synthesized nucleosomes to modify the histone tails. In contrast, transgenes within the H3K4me/H3ac-poor region are directed to a nuclear compartment depleted of modifying enzymes, resulting in stochastic histone modifications in the de novo-synthesized nucleosomes (Fig. 10).
FIG. 10.

A model for variegated transgene position effect. Histone-modifying enzymes (e.g., HMT as depicted) recruited by modified histone residues (H3K4me or H3ac) (solid circle) in the flanking chromatin (nucleosomes in gray) randomly load on the newly synthesized nucleosomes (in white), resulting in parallel modifications of histone residues at the transgenic promoter while transgene expression is facilitated. In a nonpermissive chromatin region, however, lack of H3K4me or H3ac results in limited number of local histone-modifying enzymes (A). As a result, stochastic loading of these enzymes generates cell subpopulations that differ in their de novo histone modification levels associated with the transgenic promoter (B), resulting in mosaic (variegated) transgene expression (C). Alternatively, histone modifications in the flanking chromatin may direct the transgene to distinct nuclear compartments rich in or lacking histone-modifying enzymes that modify the transgene-associated histones by a similar mechanism.
This model would also explain the reduction of histone methylation/acetylation during progressive silencing of the transgene within nonpermissive chromatin regions. Despite initial high H3K4me/H3ac levels at its promoter, the transgene localizes to a nuclear compartment deficient in histone-modifying enzymes, with the low enzyme levels insufficient to maintain histone modifications after cell division. This may culminate in dilution of H3K4me/H3ac and ultimately equalization of the modified levels between the transgenic promoters and the local chromatin. Indeed, EGFP was silenced faster at the F39 location than at the F59 location, consistent with the lower level of H3K4me/H3ac at this location (Fig. 8A). Furthermore, histone modifications at the MMP2 promoter approximated the nearby chromatin regions after about 10 weeks (Fig. 8B).
Interestingly, strong transcription activators failed to prevent the long-term reduction of H3K4Me/H3Ac levels (Fig. 9), arguing against the possibility that transcription activators recruit chromatin-modifying enzymes directly to the transgenic promoter (34) where active chromatin markers serve as transcription “memory” (29). We also think it unlikely that DNA methylation establishes progressive gene silencing, since the EGFP expression was subjected to silencing regardless of the CpG dinucleotide density in the transgenic promoters (Fig. 8 and 9). Of these four promoters, p16 and TIMP3 contain a well-characterized CpG island (32, 52). While DNA methylation might occur for these promoters, it is probably an event subsequent to the reduction of H3K4me levels during transgene silencing, as suggested in a previous report (28).
Since dimethyl K4 sometimes localizes to sites separate from that of trimethyl K4 and the latter correlates better with transcription starts (7), it was proposed that dimethyl K4 functions differently from trimethyl K4, perhaps by marking functional elements in the DNA (7). However, we found no difference in these two types of methylations in defining chromatin regions for transgene expression. While the F29 domain was enriched in both di- and trimethyl K4, the F55 domain was preferentially enriched in dimethyl K4 (Fig. 3) and both regions were permissive for gene expression. Indeed, it would appear that the overall K4 methylation level rather than the amount of dimethyl or trimethyl K4 defines a permissive chromatin region for transgene expression.
Taken together, our studies suggest a novel mechanism for transgene position effects whereby active histone modifications (H3K4me/H3ac) define chromatin regions that specify the expression state or level of a transgene. Although distinct from PEV, this mechanism can unify with the latter in invoking a generalized role of histone modifications in defining chromatin regions permissive/restrictive for transgene expression. Thus, H3 K9 methylation defines a heterochromatin region where a transgene is silenced or stochastically silenced if the transgene is localized proximal to this region. Additionally, H3K4me or H3ac defines a euchromatin region allowing transgene expression, whereas variegated transgene expression occurs in a euchromatin region deficient in these markers. Therefore, our findings underline the importance of the chromatin environment in determining gene expression.
Other models including repetitive DNA (42) or transcriptional interference from the flanking sequence (12) have been proposed to explain transgene position effects in euchromatin. Repetitive DNAs recruit heterochromatin assembly factors and result in condensation of the chromatin (42). However, large-scale repetitive DNA is very rare in the mammalian genome, and in fact, the four genomic locations we investigated did not enrich in H3K9me2 or H3K9me3, markers for heterochromatinization. Likewise, a role for transcriptional interference in the position effects we observed here is less likely since the inclusion of an additional strong polyadenylation signal sequence upstream of the transgenic promoters (Fig. 1A) would likely abolish the transcriptional interference from the flanking DNA (50).
As expected (1, 46), strong promoters presumably recruiting strong transcription activators countered the silencing effects of the nonpermissive chromatin. This observation supports the “site-exposure model” that proposes a dynamic equilibrium between a “closed” and a “open” chromatin state that allows strong transcription activators access to the DNA, thereby shifting the balance between negative and positive regulators toward activated transcription (1, 2). According to this model, the chromatin state is not altered by transcription activators, consistent with our observations that the strong TIMP3 and the weak MMP2 promoters shared similar H3 acetylation and K4 methylation patterns. Importantly, neither the reduction of modified histone levels at the promoter nor the reduction of the EGFP expression levels in the transgenic cells was abolished by the TIMP3 promoter localized to the nonpermissive regions, suggesting that the ultimate balance was shifted back toward negative regulation. These findings could very well explain the frequent progressive loss over time of expression of stably transfected genes driven by strong promoters, suggesting that repressive chromatin effects dominate over trans activation.
In summary, we have demonstrated that the level of H3K4me/H3ac defines a chromatin region distinct in its ability to either permit or restrict transgene expression. Further, we propose that variegated transgene expression is a function of stochastic H3K4 methylation and/or H3 acetylation at the transgenic promoter dictated in turn by the histone code in the flanking chromatin region.
Acknowledgments
This work was supported by R01 grants CA58311 and DE 10845 to DB.
We are grateful to E. N. Benveniste, E. Hara, J. Bennett, and M. Seiki for the generous gifts of the plasmids containing the promoters.
REFERENCES
- 1.Ahmad, K., and S. Henikoff. 2001. Modulation of a transcription factor counteracts heterochromatic gene silencing in Drosophila. Cell 104:839-847. [DOI] [PubMed] [Google Scholar]
- 2.Ahmad, K., and S. Henikoff. 2002. Epigenetic consequences of nucleosome dynamics. Cell 111:281-284. [DOI] [PubMed] [Google Scholar]
- 3.Ayyanathan, K., M. S. Lechner, P. Bell, G. G. Maul, D. C. Schultz, Y. Yamada, K. Tanaka, K. Torigoe, and F. J. Rauscher III. 2003. Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 17:1855-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannister, A. J., P. Zegerman, J. F. Partridge, E. A. Miska, J. O. Thomas, R. C. Allshire, and T. Kouzarides. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120-124. [DOI] [PubMed] [Google Scholar]
- 5.Bartova, E., J. Pachernik, A. Harnicarova, A. Kovarik, M. Kovarikova, J. Hofmanova, M. Skalnikova, M. Kozubek, and S. Kozubek. 2005. Nuclear levels and patterns of histone H3 modification and HP1 proteins after inhibition of histone deacetylases. J. Cell Sci. 118:5035-5046. [DOI] [PubMed] [Google Scholar]
- 6.Berger, S. L. 2002. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12:142-148. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein, B. E., M. Kamal, K. Lindblad-Toh, S. Bekiranov, D. K. Bailey, D. J. Huebert, S. McMahon, E. K. Karlsson, E. J. Kulbokas, T. R. Gingeras, S. L. Schreiber, and E. S. Lander. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120:169-181. [DOI] [PubMed] [Google Scholar]
- 8.Dillon, N., and R. Festenstein. 2002. Unravelling heterochromatin: competition between positive and negative factors regulates accessibility. Trends Genet. 18:252-258. [DOI] [PubMed] [Google Scholar]
- 9.Esteve, P.-O., D. Patnaik, H. G. Chin, J. Benner, M. A. Teitell, and S. Pradhan. 2005. Functional analysis of the N- and C-terminus of mammalian G9a histone H3 methyltransferase. Nucleic Acids Res. 33:3211-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eszterhas, S. K., E. E. Bouhassira, D. I. K. Martin, and S. Fiering. 2002. Transcriptional interference by independently regulated genes occurs in any relative arrangement of the genes and is influenced by chromosomal integration position. Mol. Cell. Biol. 22:469-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng, Y.-Q., R. Alami, and E. E. Bouhassira. 1999. Enhancer-dependent transcriptional oscillations in mouse erythroleukemia cells. Mol. Cell. Biol. 19:4907-4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng, Y.-Q., R. Warin, T. Li, E. Olivier, A. Besse, A. Lobell, H. Fu, C. M. Lin, M. I. Aladjem, and E. E. Bouhassira. 2005. The human β-globin locus control region can silence as well as activate gene expression. Mol. Cell. Biol. 25:3864-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischle, W., Y. Wang, and C. D. Allis. 2003. Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 15:172-183. [DOI] [PubMed] [Google Scholar]
- 14.Guenther, M. G., R. G. Jenner, B. Chevalier, T. Nakamura, C. M. Croce, E. Canaani, and R. A. Young. 2005. Global and Hox-specific roles for the MLL1 methyltransferase. Proc. Natl. Acad. Sci. USA 102:8603-8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh, J. J.-D., P. Ernst, H. Erdjument-Bromage, P. Tempst, and S. J. Korsmeyer. 2003. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol. Cell. Biol. 23:186-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huttenhofer, A., P. Shattner, and N. Polacek. 2005. Non-coding RNAs: hope or hype? Trends Genet. 21:289-297. [DOI] [PubMed] [Google Scholar]
- 17.Johnson, J. M., S. Edwards, D. Shoemaker, and E. E. Shadt. 2005. Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 21:93-102. [DOI] [PubMed] [Google Scholar]
- 18.Kaern, M., T. C. Elston, W. J. Blakes, and J. J. Collins. 2005. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6:451-464. [DOI] [PubMed] [Google Scholar]
- 19.Kent, W. J., C. W. Sugnet, T. S. Fuery, K. M. Roskin, T. H. Pringle, A. M. Zahler, and D. Haussler. 2002. The human genome browser at UCSC. Genome Res. 12:996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kouskouti, A., and I. Talianidis. 2005. Histone modifications defining active genes persist after transcriptional and mitotic in activation. EMBO J. 24:347-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurakin, A. 2005. Stochastic cell. IUBMB Life 57:59-63. [DOI] [PubMed] [Google Scholar]
- 22.Lachner, M., D. O'Carroll, S. Rea, K. Mechtler, and T. Jeuwein. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410:116-120. [DOI] [PubMed] [Google Scholar]
- 23.Levitt, N., D. Briggs, A. Gil, and N. J. Proudfoot. 1989. Definition of an efficient synthetic poly(A) site. Gene Dev. 3:1019-1025. [DOI] [PubMed] [Google Scholar]
- 24.Liang, G., J. C. Lin, V. Wei, C. Yoo, J. C. Cheng, C. T. Nguyen, D. J. Weisenberger, G. Egger, D. Takai, F. A. Gonzales, and P. A. Jones. 2004. Distinct localization of histone H3 acetylaton and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. USA 101:7357-7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Litt, M. D., M. Simpson, M. Gaszner, C. D. Allis, and G. Felsenfeld. 2001. Correlation between histone lysine methylation and developmental changes at the chicken β-globin locus. Science 293:2453-2455. [DOI] [PubMed] [Google Scholar]
- 26.Martin, C., and Y. Zhang. 2005. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell. Biol. 6:838-849. [DOI] [PubMed] [Google Scholar]
- 27.Milne, T. A., S. D. Briggs, H. W. Brock, M. E. Martin, D. Gibbs, C. D. Allis, and J. L. Hess. 2002. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 10:1107-1117. [DOI] [PubMed] [Google Scholar]
- 28.Mutskov, V., and G. Felsenfeld. 2004. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J. 23:138-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng, H. H., F. Robert, R. A. Young, and K. Struhl. 2003. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 11:709-719. [DOI] [PubMed] [Google Scholar]
- 30.Nishioka, K., J. C. Rice, K. Sarma, H. Erdjument-Bromage, J. Werner, Y. Wang, S. Chuikov, P. Valenzuela, P. Tempst, R. Steward, J. T. Lis, C. D. Allis, and D. Reinberg. 2002. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone 4 and is associated with silent chromatin. Mol. Cell 9:1201-1213. [DOI] [PubMed] [Google Scholar]
- 31.Noma, K., C. D. Allis, and S. T. Grewal. 2001. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293:1150-1155. [DOI] [PubMed] [Google Scholar]
- 32.Ohtani, N., Z. Zebedee, T. J. Huot, J. A. Stinson, M. Sugimoto, Y. Ohashi, A. D. Sharrocks, G. Peters, and E. Hara. 2001. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 409:1067-1070. [DOI] [PubMed] [Google Scholar]
- 33.Peters, A. H. F. M., S. Kubicek, K. Mechtler, R. J. O'Sullivan, A. A. H. A. Derijck, L. Perez-Burgos, A. Kohlmaier, S. Opravil, M. Tachibana, Y. Shinkai, J. H. A. Martens, and T. Jenuwein. 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12:1577-1589. [DOI] [PubMed] [Google Scholar]
- 34.Peters, A. H. F. M., and D. Schubeler. 2005. Methylation of histones: playing memory with DNA. Curr. Opin. Cell Biol. 17:230-238. [DOI] [PubMed] [Google Scholar]
- 35.Pokholok, D. K., C. T. Harbison, S. Levine, M. Cole, N. M. Hannett, T. I. Lee, G. W. Bell, K. Walker, P. A. Rolfe, E. Herbolsheimer, J. Zeitlinger, F. Levitter, D. K. Gifford, and R. A. Young. 2005. Genome-wide map of mucleosome acetylation and methylation in yeast. Cell 122:517-527. [DOI] [PubMed] [Google Scholar]
- 36.Qin, H., Y. Sun, and E. N. Benveniste. 1999. The transcription factors Sp1, Sp3, and AP-2 are required for constitutive matrix metalloproteinase-2 gene expression in astroglioma cells. J. Biol. Chem. 274:29130-29137. [DOI] [PubMed] [Google Scholar]
- 37.Rice, J. C., S. D. Briggs, B. Ueberheide, C. M. Barber, J. Shabanowitz, D. F. Hunt, Y. Shinkai, and C. D. Allis. 2003. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 12:1591-1598. [DOI] [PubMed] [Google Scholar]
- 38.Roh, T. Y., S. Cuddapah, and K. Zhao. 2005. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 19:542-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santoro, R., and F. De Lucia. 2005. Many players, one goal: how chromatin states are inherited during cell division. Biochem. Cell Biol. 83:332-343. [DOI] [PubMed] [Google Scholar]
- 40.Santos-Rosa, H., R. Schneider, A. J. Bannister, J. Sherriff, B. E. Bernstein, N. C. T. Emre, S. L. Schreiber, J. Mellor, and T. Kouzarides. 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419:407-411. [DOI] [PubMed] [Google Scholar]
- 41.Santos-Rosa, H., R. Schneider, B. E. Bernstein, N. Karabetsou, A. Morillon, C. Weise, S. L. Schreiber, J. Mellor, and T. Kouzarides. 2006. Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol. Cell 12:1325-1332. [DOI] [PubMed] [Google Scholar]
- 42.Saveliev, A., C. Everett, T. Sharpe, Z. Webster, and R. Festenstein. 2003. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 422:909-913. [DOI] [PubMed] [Google Scholar]
- 43.Seum, C., M. Delattre, A. Spierer, and P. Spierer. 2001. Ectopic HP1 promotes chromosome loops and variegated silencing in Drosophila. EMBO J. 20:812-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith, S. T., S. Petruk, Y. Sedkov, E. Cho, S. Tillib, E. Canaani, and A. Mazo. 2004. Modulation of heat shock gene expression by the TAC1 chromatin-modifying complex. Nat. Cell Biol. 6:162-167. [DOI] [PubMed] [Google Scholar]
- 45.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Nature 403:41-45. [DOI] [PubMed] [Google Scholar]
- 46.Sutter, N. B., D. Scalzo, S. Fiering, M. Groudine, and D. I. K. Martin. 2003. Chromatin insulation by a transcriptional activator. Proc. Natl. Acad. Sci. USA 100:1105-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verschure, P. J., I. van der Kraan, W. de Leeuw, J. van der Vlag, A. E. Carpenter, A. S. Belmont, and R. van Driel. 2005. In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol. Cell. Biol. 25:4552-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wakimoto, B. T. 1998. Beyond the nucleosome: epigenetic aspects of position-effect variegation in Drosophila. Cell 93:321-324. [DOI] [PubMed] [Google Scholar]
- 49.Yan, C., W. Heng, and D. D. Boyd. 2002. ATF3 represses 72-kDa type IV collagenase (MMP-2) expression by antagonizing p53-dependent trans-activation of the collagenase promoter. J. Biol. Chem. 277:10804-10812. [DOI] [PubMed] [Google Scholar]
- 50.Yan, C., H. Wang, B. Aggarwal, and D. D. Boyd. 2004. A novel homologous recombination system to study 92 kDa type IV collagenase transcription demonstrates that the NF-κB motif drives the transition from a repressed to an activated state of gene expression. FASEB J. 18:540-541. [DOI] [PubMed] [Google Scholar]
- 51.Yan, C., H. Wang, Y. Toh, and D. D. Boyd. 2003. Repression of 92-kDa type IV collagenase expression by MTA1 is mediated through direct interactions with the promoter via a mechanism which is both dependent on and independent of histone deacetylation. J. Biol. Chem. 278:2309-2316. [DOI] [PubMed] [Google Scholar]
- 52.Zeng, Y., R. C. Rosborough, Y. Li, A. R. Gupta, and J. Bennett. 1998. Temporal and spatial regulation of gene expression mediated by the promoter for the human tissue inhibitor of metalloproteinases-3 (TIMP-3)-encoding gene. Dev. Dyn. 211:228-237. [DOI] [PubMed] [Google Scholar]