

Abstract
Noncoding RNAs are recognized increasingly as important regulators of fundamental biological processes, such as gene expression and development, in eukaryotes. We report here the identification and functional characterization of the small noncoding human Y RNAs (hY RNAs) as novel factors for chromosomal DNA replication in a human cell-free system. In addition to protein fractions, hY RNAs are essential for the establishment of active chromosomal DNA replication forks in template nuclei isolated from late-G1-phase human cells. Specific degradation of hY RNAs leads to the inhibition of semiconservative DNA replication in late-G1-phase template nuclei. This inhibition is negated by resupplementation of hY RNAs. All four hY RNAs (hY1, hY3, hY4, and hY5) can functionally substitute for each other in this system. Mutagenesis of hY1 RNA showed that the binding site for Ro60 protein, which is required for Ro RNP assembly, is not essential for DNA replication. Degradation of hY1 RNA in asynchronously proliferating HeLa cells by RNA interference reduced the percentages of cells incorporating bromodeoxyuridine in vivo. These experiments implicate a functional role for hY RNAs in human chromosomal DNA replication.
In recent years, it has become apparent that noncoding RNAs are regulating many biological processes, from gene expression and chromatin dynamics to complex developmental programs (reviewed in references 2, 26, and 35). A fundamental process for which an involvement of noncoding RNAs has not been reported to date is the replication of chromosomal DNA in eukaryotes.
Chromosomal DNA replication is initiated at the G1-to-S phase transition of the cell division cycle. Regulators for this transition have been identified genetically and biochemically as proteins that interact with chromosomal DNA replication origins during G1 phase, directing the stepwise formation of preinitiation complexes (reviewed in references 1, 13, 25, 33, and 39). These protein factors are functionally conserved through evolution. The six-protein subunit origin recognition complex is assembled on origin DNA, from which Cdc6 and Cdt1 proteins recruit six minichromosome maintenance proteins (MCM2 to MCM7) to form a prereplicative complex, or replication license, in G1 phase. Conversion of this complex into active replication forks marks the entry into S phase, which is under the temporal and spatial control of S-phase cyclin-dependent kinase Cdk2 and Dbf4-dependent kinase Cdc7. Additional initiation proteins, including MCM10, Cdc45, GINS complex, Mus101 (Dbp11 and Cut5 in yeasts), and replication protein A (RPA) are recruited in this process to unwind origin DNA (1, 25, 39). Active DNA replication forks are established from there by the stepwise recruitment of DNA polymerase α/primase and the replicative DNA polymerases δ and ɛ, together with replication factor C and proliferating nuclear antigen (PCNA). This elaborate pathway has been worked out predominantly in the model systems of amphibian egg extracts and unicellular yeasts; later stages have been characterized in animal tumor virus systems. However, these systems do not necessarily reflect the cell cycle regulation in somatic cells of multicellular organisms.
To study the regulation of human chromosomal DNA replication directly at the molecular level, we have established and refined a human cell-free system that initiates semiconservative chromosomal DNA replication under cell cycle control (17, 19). Initiation-competent template nuclei are isolated from cultured human cells synchronized in the late G1 phase of the cell division cycle by the iron-chelating compound mimosine (18). These nuclei are licensed and competent to replicate but do not contain DNA replication forks. Active DNA replication forks are rapidly established in these nuclei upon the addition of a cytosolic extract from proliferating human cells (17, 23). DNA replication is confined to early replicating DNA sequences in this system, and origin specificity of initiation has been demonstrated for the early-firing lamin B2 origin (14). DNA replication does not depend on regulated nuclear transport in this system because it is efficiently detected in the absence of an intact nuclear envelope (17). The cytosolic extract therefore provides the source of soluble DNA replication factors required for establishment of active DNA replication forks, which can be purified and identified by straightforward fractionation and reconstitution experiments (37). We have recently published reports of the initial fractionation of the extract into three essential and nonredundant fractions and the purification of RPA and PCNA from two of these fractions (36, 37). In this article, we report the purification of a novel replication factor from the third essential fraction. Unexpectedly, we have identified this factor as a set of noncoding Y RNAs, which have not been reported as a constituent of any model system for DNA replication so far.
Human Y RNAs (hY RNAs) were originally found as the RNA component of soluble ribonucleoproteins (RNPs) termed Ro RNPs (11), which are detected by autoimmune sera of patients suffering from rheumatic diseases such as systemic lupus erythematosus or Sjögren's syndrome (reviewed in references 4 and 29). Ro RNPs consist of a Y RNA, which is associated with the autoimmune antigen proteins Ro60 (4) and La (45) and other less-well-characterized proteins (29). Four Y RNA species in humans have been described previously: hY1 (hY2 is a truncated form of hY1), hY3, hY4, and hY5 RNAs (11). Y RNAs have been found in all vertebrate species investigated so far (30). Orthologues have also been found in the nematode Caenorhabditis elegans (21, 42) and in a radiation-resistant prokaryote, Deinococcus radiodurans (3), but so far not in yeasts, plants, or insects. Even though functions of Ro60 and La proteins have recently been described as RNA stability, RNA quality control, and the resistance of cells to UV irradiation, no direct function has been demonstrated for Y RNAs in any organism to date (4, 29, 45). Against this background, our functional data now establish that hY RNAs have a function as essential factors for chromosomal DNA replication in human cell nuclei.
MATERIALS AND METHODS
Tissue culture, cell synchronization, preparation of template nuclei, and analysis of DNA replication reactions were performed as described preciously (see references 17, 34, 36, and 37 and references therein). In this study, nuclei were prepared from human HeLa cervix carcinoma cells, human EJ30 bladder carcinoma cells, or mouse NIH 3T3 fibroblast cells as specified, and cell extracts were prepared exclusively from HeLa cells.
Fractionation of cell extracts and purification of hY RNA.
Initial fractionation of human cytosolic S20 extract on Q Sepharose (Amersham Biosciences) was performed exactly as detailed before (37). Fraction QB was diluted in 50 mM Tris-HCl, pH 8.2, 1 mM EGTA, and 1 mM dithiothreitol (DTT) to adjust the KCl concentration to 150 mM and loaded onto arginine-Sepharose (Amersham Biosciences) equilibrated in 150 mM KCl, 50 mM Tris-HCl, pH 8.2, 1 mM EGTA, and 1 mM DTT. The material flowing through the column (termed ArFT) was collected. Bound material (termed ArE) was eluted in 350 mM KCl, 50 mM Tris-HCl, pH 8.2, 1 mM EGTA, and 1 mM DTT. The RNA present in fraction ArE was further purified by extraction with phenol-chloroform and precipitation in isopropanol (31). RNA was separated according to molecular mass on a Superdex-200 HR10/30 column (Amersham Biosciences) in TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). RNA was detectable between 7 ml and 20 ml of elution volume. RNA contained in each fraction was concentrated by ethanol precipitation and analyzed by agarose gel electrophoresis (31). RNA was visualized by staining with 1× SYBR gold (Molecular Probes). Inverted images are shown in the figures.
cDNA cloning and expression of recombinant human RNA.
For synthesis of cDNA clones from purified RNA preparations, RNA was first tailed at its 3′ end with poly(A) by using poly(A) polymerase (Amersham Biosciences). cDNA strands were primed by annealing a (dT)18 oligonucleotide (Sigma-Genosys) to the poly(A) tail and synthesized by SuperScript II RNase H− reverse transcriptase (Invitrogen). The RNA was subsequently digested with RNase H (Promega), and the single-stranded cDNA was purified by phenol extraction and ethanol precipitation (31). The 3′ ends of the purified single-stranded cDNA were extended with poly(dC), using terminal transferase and dCTP (New England Biolabs). Second-strand synthesis was primed by annealing a (dG)18 oligonucleotide (Sigma-Genosys) and synthesized by 30 cycles of PCR using (dT)18 and (dG)18 primers and the Expand high-fidelity PCR system (Roche). Double-stranded PCR products were cloned using the TOPO TA cloning kit (Invitrogen).
Full-length recombinant genes coding for all four human Y RNAs and their mutant derivatives, 5S rRNA, and U2 snRNA were generated by PCR amplification using either our isolated cDNA clones or synthetic templates (Sigma-Genosys). The forward primers contained a bacteriophage SP6 promoter sequence at their 5′ ends to initiate transcription at the first nucleotide. The reverse primers contained a restriction site at their 5′ ends, located immediately downstream of the last nucleotide of the cDNA (DraI for wild-type [wt] and mutant hY RNAs and 5S rRNA; SmaI for U2 snRNA). PCR products were cloned using the TOPO TA cloning kit (Invitrogen), and all constructs were confirmed by DNA sequencing. Transcription by SP6 RNA polymerase (Roche) was performed on templates cut with DraI or SmaI (Roche). RNA was purified by phenol extraction and ethanol precipitation and analyzed by denaturing polyacrylamide gels containing 8 M urea (31).
RNA degradation in vitro.
Cytosolic extracts were pretreated either with a final concentration of 0.3 mg/ml RNase A (Roche) or with 0.3 μM of single-stranded DNA oligonucleotides (detailed below) at room temperature for 1 h and then used immediately in DNA replication reactions. The oligonucleotides sequences were as follows: T3 sequencing primer, 5′-CGAAATTAACCCTCACTAAAGGGA; for the anti-hY1 loop, 5′-AAGGGGGGAAAGAGTAGAACAAGGA; for the anti-hY3 loop, 5′-GAGTGGAGAAGGAACAAAGAAATCT; for the anti-hY4 loop, 5′-GGGTTGTATACCAACTTTAGTGACA; and for the anti-hY5 loop, 5′-GGGAGACAATGTTAAATCAACTTAA.
RNA interference in vivo.
Small interfering RNAs (siRNAs) were chemically synthesized by using an Ambion Silencer siRNA construction kit as detailed previously (27). The following pairs of DNA oligonucleotides were synthesized (Sigma-Genosys) for direct generation of siRNAs in vitro: for firefly luciferase mRNA, lucA (AACTTACGCTGAGTACTTCGACCTGTCTC) and lucB (AATCGAAGTACTCAGCGTAAGCCTGTCTC); and for hY1 RNA, hY1a A (TTATCTCAATTGATTGTTCACCCTGTCTC) and hY1a B (GTGAACAATCAATTGAGATAACCTGTCTC) and hY1b A (TGTTCTACTCTTTCCCCCCTTCCTGTCTC), and hY1b B (AAGGGGGGAAAGAGTAGAACACCTGTCTC).
Transfections were performed with 10 nM siRNAs by using Lipofectamine 2000 reagent (Invitrogen) and OptiMEM (GibcoBRL), as specified by the suppliers.
Quantitative real-time RT-PCR.
cDNA was synthesized from total cytoplasmic RNA by using a set of primers complementary to the 3′ ends of all tested RNAs. For all comparisons, individual extracts were adjusted to contain identical protein amounts. The cDNA mix was used as the template for quantitative real-time reverse transcription-PCR (RT-PCR) on the iCycler iQ platform, using the iQ SYBR green supermix labeling kit (Bio-Rad) over 40 cycles and a hybridization temperature of 55°C. For RNA-specific cDNA amplification, the following primer sequences were used: for hY1, GGCTGGTCCGAAGGTAGTGA and GCAGTAGTGAGAAGGGGGGA; for hY3, GGCTGGTCCGAGTGCAGTGG and GAAGCAGTGGGAGTGGAGAA; for hY4, GGCTGGTCCGATGGTAGTGG and TTAGCAGTGGGGGGTTGTAT; for hY5, AGTTGGTCCGAGTGTTGTGG and AACAGCAAGCTAGTCAAGCG; for 5S, GTCTACGGCCATACCACCCT and AAAGCCTACAGCACCCGGTA; for 5.8S, CGACTCTTAGCGGTGGATCA and GGGCCGCAAGTGCGTTCGAA; for U2, TCGCTTCTCGGCCTTTTGGC and GGTGCACCGTTCCTGGAGGT; and for hypoxanthine ribosyltransferase (HPRT), AGATCCATTCCTATGACT and CATCTCCACCAATTACTT.
The relative amount (Ar) of RNA for each sample was calculated from the threshold cycle (CT) of each cDNA amplification by use of the following equation: 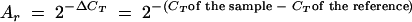 .
.
Reference RNAs were 5S rRNA or HPRT mRNA, as specified. Proportions of the relative amount of each RNA after treatment of a cytosolic extract with RNase A or DNA antisense oligonucleotides were normalized against the untreated control extract by use of the following equation: proportion of 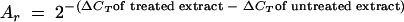 × 100%.
× 100%.
Proportions of the relative amounts of hY RNAs after RNA interference (RNAi) against hY1 RNA were normalized against the nontarget RNAi against luciferase mRNA by use of the following equation: proportion of 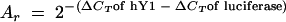 × 100%.
× 100%.
Data for each CT value were obtained in triplicate, and mean values from two to five independent experiments are presented.
RESULTS
Identification of small human RNAs as essential DNA replication factors.
In the experiments reported below, we used a well-characterized cell-free system derived from human cells to isolate and identify soluble factors that are functionally required for initiation and maintenance of chromosomal DNA replication forks in isolated human cell nuclei. In this system, a cytosolic extract from proliferating human cells initiates DNA replication and further supports bidirectional, semiconservative chromosomal DNA replication in about 50% of late-G1-phase template nuclei (14, 17, 23). In the absence of this extract, nuclear run-on DNA replication is observed in about 5% of nuclei that are present in the preparation as S-phase contaminants (17-19). Biochemical fractionation of the cytosolic extract has resulted in the functional identification of at least two fractions, termed QA and QB, each containing distinct replication factors (Fig. 1A) (37). Fraction QA contains RPA (37), whereas fraction QB contains PCNA and at least one additional essential replication factor (36).
FIG. 1.

Purification of RNA as a factor for human chromosomal DNA replication. (A) Schematic diagram of fractionation steps. (B) Representative fields of nuclei replicating in vitro. Template nuclei from late-G1-phase cells were incubated with combinations of the indicated fractions, and replicating nuclei were detected by confocal fluorescence microscopy as detailed in references 36 and 37. Nuclear DNA is visualized by propidium iodide (red signal). Replicated DNA is labeled by incorporation of digoxigenin-dUMP, which is detected by fluorescein-conjugated antidigoxigenin Fab fragments (green signal). A merged signal appears in yellow. (C) Quantitative analysis of replicating G1-phase nuclei in vitro. Mean values and standard deviations of the proportions of replicating nuclei from the indicated reactions of 12 to 22 independent experiments (n) are shown (see Materials and Methods). Protein amounts per experiment were 100 μg unfractionated S20 cytosolic extract, 15 μg QA, 35 μg QB, 8 μg ArFT, a 20-μl volume of concentrated ArE containing less than 0.1 μg protein or the equivalent RNA prepared from this volume as specified. Fractions were used as indicated. (D) Visualization of RNA purified from fraction ArE. The RNA present in fraction ArE was purified by phenol extraction and isopropanol precipitation, separated on a 2% neutral agarose gel, and visualized by staining with ethidium bromide (lane RNA). A ladder of multimeric 100-bp DNA fragments was used as a molecular weight marker (lane M). An inverted image of the fluorescent gel is shown.
To purify this additional replication factor, we first separated fraction QB by anion exchange/affinity chromatography on arginine-Sepharose into two fractions, ArFT and ArE (Fig. 1A). Initiation and further elongation of chromosomal DNA replication were reconstituted in vitro when all three fractions (QA, ArFT, and ArE) were added together to template nuclei (Fig. 1B and C). Fraction ArFT contained the bulk of the proteins present in QB, and its further fractionation has led to the purification of PCNA (36). Surprisingly, we did not detect significant protein amounts in fraction ArE (data not shown). Instead, the activity of fraction ArE was resistant to phenol extraction and isopropanol precipitation (Fig. 1A), indicating that it is composed of nucleic acids rather than protein. Indeed, visualization after agarose gel electrophoresis revealed a discrete set of small nucleic acids (Fig. 1D). These nucleic acids were resistant to digestion with DNase I but sensitive to digestion with RNase A (data not shown), indicating that they are composed of RNA. Most importantly, this pure RNA preparation contains the DNA replication factor activity of fraction ArE (Fig. 1B and C). We conclude that small RNAs are functionally required for DNA replication in isolated nuclei from human late-G1-phase cells.
We have tested whether DNA replication in these nuclei can be reconstituted entirely by purified factors in vitro. Purified RPA (37), PCNA (36), and the small RNAs together do not increase the proportion of replicating nuclei (data not shown), indicating that additional, and so far not identified, soluble replication factors must be present in the unfractionated extract. In the subsequent reconstitution experiments, we have therefore used fractions QA and ArFT as sources for additional replication factors to supplement small RNAs.
Purification of noncoding hY RNAs as DNA replication factors.
To establish whether a discrete RNA is required for chromosomal DNA replication in vitro, we fractionated this RNA preparation further by gel filtration according to molecular mass (Fig. 2A). The replication factor activity was recovered as a discrete peak corresponding to fractions 40 to 42 (Fig. 2B). The active and inactive fractions contain different sets of small RNAs, indicating that the activity is mediated specifically by a discrete set of small RNAs rather than unspecifically by any small RNA.
FIG. 2.
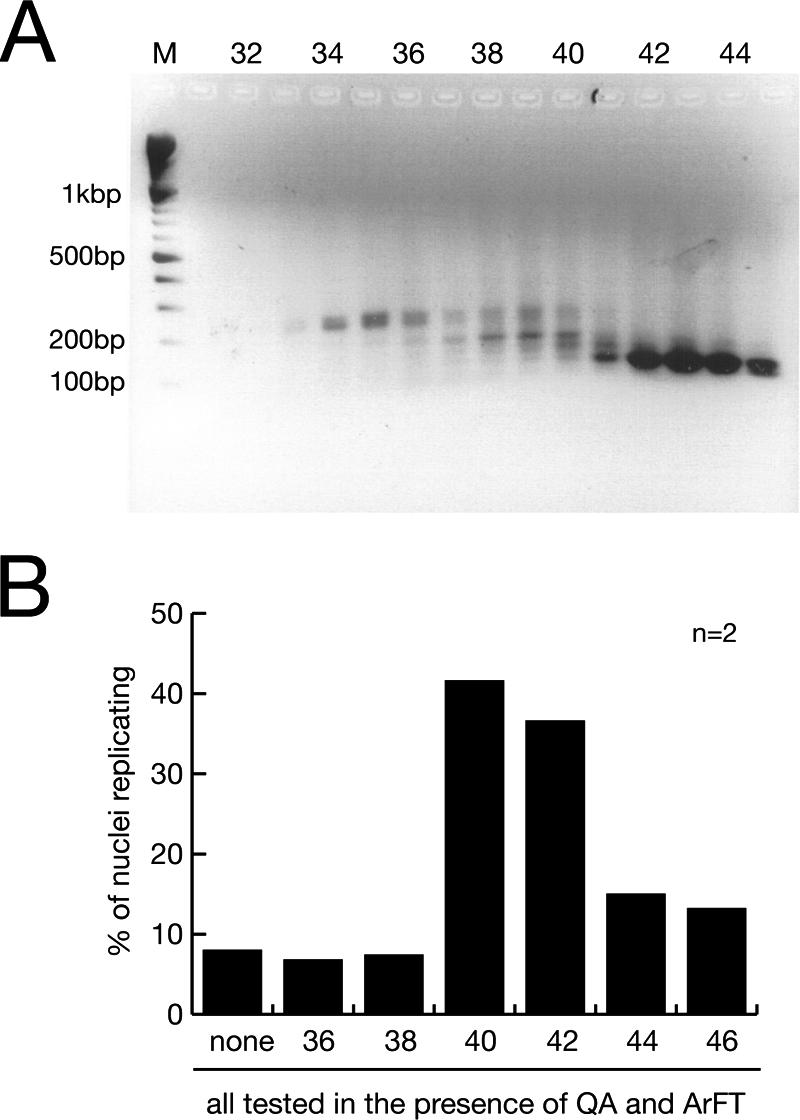
A discrete class of RNAs is essential for human chromosomal DNA replication. (A) Size fractionation of RNA prepared from ArE by gel filtration. Individual fractions were analyzed by agarose gel electrophoresis, and a 100-bp DNA ladder was used as a molecular weight marker (lane M). (B) Functional testing of the size-fractionated RNA. n, number of experiments. Template nuclei were incubated with the RNA purified from a constant volume of the indicated gel filtration fractions, supplemented with fractions QA and ArFT. Proportions of replicating nuclei were determined as described in the legend to Fig. 1.
We have therefore identified the RNA species present in the active fractions by cDNA cloning and sequencing. Out of a total of 19 obtained cDNA clones coding for defined short RNAs, we identified 4 independent clones (21% of the isolates) as 5S rRNA, 8 (42%) as U2 snRNA, 2 (10.5%) as hY4 RNA, and 5 (26.5%) as hY5 RNA. To establish which of these small RNAs is required for DNA replication, we tested them individually. Human Y RNAs are transcribed by RNA polymerase III and range in size from 80 to 110 nucleotides. They fold into a characteristic stem-loop structure based on their partially complementary 5′ and 3′ ends and share many conserved nucleotide sequences and structural elements (Fig. 3). We generated expression constructs containing the cDNAs for 5S rRNA, for U2 snRNA, and for all four hY RNAs (Fig. 4A), and we synthesized full-length recombinant RNAs from these templates by in vitro transcription (Fig. 4B). When assayed in the presence of fractions QA and ArFT, each of the four hY RNAs significantly increased the proportion of G1-phase nuclei replicating their chromosomal DNA over the background level obtained by fractions QA and ArFT alone (Fig. 4C). In contrast, 5S rRNA and U2 snRNA did not increase the proportion of replicating G1-phase nuclei in this system (Fig. 4C), and neither did a preparation of yeast tRNA (data not shown). We conclude that hY RNAs are specifically required for chromosomal DNA replication in human G1-phase nuclei in vitro.
FIG. 3.

Nucleotide (nt) sequences and predicted secondary structures of human Y RNAs (28, 41). Nucleotide sequences complementary to antisense DNA oligonucleotides (see below; see also Fig. 5) are highlighted by gray lines.
FIG. 4.

Human Y RNA is required for the reconstitution of chromosomal DNA replication. (A) Schematic drawing of the expression constructs for full-length human RNAs. (B) In vitro synthesis of full-length human wild-type RNA. Individual recombinant RNAs were synthesized in vitro from the constructs shown in panel A and visualized by SYBR green staining after denaturing gel electrophoresis. Multimeric 100-nucleotide (nt) RNA fragments were used for a molecular weight marker (lane M). (C) Functional reconstitution of chromosomal DNA replication with hY RNAs. n, number of experiments. Nuclei from late-G1-phase cells were incubated with fractions QA and ArFT, supplemented with 100 ng of the individual RNAs synthesized in vitro as indicated. Proportions of replicating nuclei were determined as described in the legend to Fig. 1.
Next, we quantified the amounts of recombinant hY RNAs needed for the reconstitution of chromosomal DNA replication in the presence of fractions QA and ArFT. A dose-dependent increase in the proportion of replicating nuclei was observed upon the addition of increasing amounts of any of the four hY RNAs (data not shown). The maximal stimulation was reached when 30 to 100 ng of hY RNA was added to a standard 50-μl reaction volume; larger amounts of hY RNA did not increase the percentages of replicating nuclei further. These values translate into RNA concentrations ranging from 17 to 55 nM for hY1 to 22 to 74 nM for hY5 RNA. In comparison, the concentration of all hY RNAs present in a standard 50-μl replication reaction containing 10 μl of unfractionated S100 extract (mean ± standard deviation) is 25 ± 10 nM (Table 1), which is exactly within the concentration range obtained in fractionation and reconstitution experiments. Therefore, the quantitative requirement for pure hY RNAs to reconstitute chromosomal DNA replication in the fractionated system is within the physiological range of the system using unfractionated extract.
TABLE 1.
Abundance of hY RNAs in HeLa cell S100 extracts
| Sample RNA | Mean ± SD
|
||
|---|---|---|---|
| −ΔCTa | Relative RNA amount (103)b | RNA concnc | |
| HPRT mRNA | 0 | 1 | 0.8 pM |
| hY1 RNA | 14.1 ± 0.5 | 17.6 ± 7.3 | 13.4 ± 5.6 nM |
| hY3 RNA | 14.95 ± 0.36 | 31.7 ± 8.7 | 24.2 ± 6.6 nM |
| hY4 RNA | 13.6 ± 0.76 | 12.4 ± 8.6 | 9.5 ± 6.6 nM |
| hY5 RNA | 16.67 ± 0.47 | 104.3 ± 40.2 | 79.4 ± 30.7 nM |
| 5S rRNA | 17.26 ± 0.44 | 157 ± 56 | 120 ± 73.4 nM |
−ΔCT values were obtained as specified in Materials and Methods as averages from seven acquisition runs for each cDNA prepared from an S100 extract (as a reference, the protein concentration of the extract was 10 mg/ml). HPRT mRNA was used as reference RNA. Mean values ± standard deviations of the means from two independent experiments using two different S100 preparations are shown.
RNA amounts relative to the HPRT reference were calculated from the ΔCT values as specified in Materials and Methods.
The RNA concentration in the S100 extract was calculated from the relative RNA amounts by using calibration curves obtained by real-time PCR amplifications of defined amounts of hY cDNAs. The conversion factor of 0.763 × 10−3 nM was obtained as the average from three independent calibrations.
Depletion of hY RNAs inhibits chromosomal DNA replication in late-G1-phase nuclei.
The gain-of-function experiments reported above showed that hY RNAs are required for DNA replication in late-G1-phase nuclei in the presence of additional soluble protein factors. In the next set of experiments, we sought to confirm this requirement by loss-of-function and reconstitution experiments.
Human Y RNAs are expressed in human cells at estimated levels of less than 1 × 105 copies per HeLa cell, about 100-fold less abundant than ribosomal RNAs (11). We first used quantitative RT-PCR to determine the relative amounts of all four hY RNAs present in the cytosolic extract (Table 1). To standardize RNA levels, we used the very low copy number (estimated at 1 to 10 copies per cell) of HPRT mRNA, which has been reported to show a very low variation in expression levels among different human tissues and cell types (7, 40), as the reference. We found that all hY RNAs are present at 104- to 105-times-larger amounts than HPRT mRNA or at 1.5- to 13-fold-smaller amounts than ribosomal 5S rRNA in the extract (Table 1). Their relative abundance in the extract increased about sixfold, with decreasing size from hY1 to hY5 RNA, except that hY4 RNA showed the lowest relative abundance (about eightfold less abundant than hY5 RNA) (Table 1). These relative expression data are in good agreement with earlier data reported for hY RNA expression levels in HeLa cells (11, 30). In absolute terms, the concentrations of hY RNAs in the S100 extract ranged from 9.5 ± 6.6 nM for hY4 to 79.4 ± 30.7 nM for hY5 (Table 1).
We then degraded the RNA in the extract and investigated the consequences on DNA replication in isolated late-G1-phase nuclei (Fig. 5). After treatment with RNase A, the relative amounts of all tested RNAs present in the cytosolic extract were reduced by three to four orders of magnitude (Fig. 5A). Importantly, this treated cytosolic extract no longer increased the proportion of replicating G1-phase nuclei in vitro (Fig. 5B).
FIG. 5.

Degradation of hY RNAs in vitro inhibits chromosomal DNA replication in late-G1-phase template nuclei. n, number of experiments. (A) Specific degradation of hY RNA in vitro. S100 cytosolic extract from proliferating HeLa cells was treated with RNase A or with the DNA antisense oligonucleotides complementary to single-stranded domains of the hY RNAs as shown in Fig. 3. As a negative control, the standard bacteriophage T3 DNA sequencing primer was used. The proportions of the indicated relative RNA amounts remaining in the extract after the treatment were determined by quantitative RT-PCR, using 5S rRNA as the reference. (B) Y RNA degradation reduces the proportion of nuclei replicating in vitro. Late-G1-phase template nuclei were incubated in untreated and treated S100 cytosolic extract as indicated. Proportions of replicating nuclei were determined as described in the legend to Fig. 1. (C) Y RNA degradation reduces the amount of extract-dependent DNA synthesis in late-G1-phase nuclei in vitro. Nuclei were incubated in untreated and treated S100 cytosolic extract in the presence of [α-32P]dCTP as indicated. The incorporation of dNMPs into nascent chromosomal DNA under these conditions was quantitated by precipitation with trichloroacetic acid and scintillation counting. (D) Functional substitution of one depleted hY RNA in the extract by a different hY RNA. After depletion of either hY1 or hY3 RNA by antisense DNA oligonucleotides, the treated extract was supplemented with 100 ng of the indicated hY RNAs synthesized in vitro. Proportions of replicating nuclei were determined as described in the legend to Fig. 1.
Next, we targeted individual hY RNAs in the cytosolic extract for specific degradation by RNase H. We used single-stranded antisense DNA oligonucleotides complementary to hY RNA sequences at the 3′ side of the single-stranded loops (Fig. 3), which have been reported to direct specific degradation of hY RNA (24). The relative amounts of targeted hY1, hY3, and hY4 RNA species were specifically reduced about 300- to 500-fold by this treatment, whereas the relative amounts of untargeted RNAs remained unchanged (Fig. 5A). We were not able to detect a specific depletion of hY5 RNA by this approach (Fig. 5A), most likely because of the predominantly double-stranded nature of hY5 RNA, which would be refractory to antisense-mediated degradation (24). Similarly, we were not able to obtain depletion with antisense oligonucleotides directed against the conserved double-stranded stem of the hY RNAs (data not shown).
Importantly, after specific depletion of hY1 or hY3 RNA, the treated cytosolic extract no longer increased the proportions of replicating late-G1-phase nuclei significantly (Fig. 5B). Depletion of the least abundant hY4 RNA resulted in a partial inactivation of the replication activity of the extract (Fig. 5B). As a negative control, treatment with the nontarget bacteriophage T3 DNA sequencing primer neither influenced significantly the concentration of any RNA tested in the extract (Fig. 5A) nor reduced the proportion of G1-phase nuclei replicating in the extract (Fig. 5B).
We next quantitated the amount of DNA synthesized in late-G1-phase template nuclei in the treated extracts by measuring the incorporation of radioactively labeled nucleotides into chromosomal DNA (Fig. 5C). Significantly, treatment with RNase A or the hY RNA-specific antisense DNA oligonucleotides reduced the amount of DNA synthesized in these nuclei to the levels observed in the complete absence of cytosolic extract. Treatment with the unspecific T3 sequencing primer led only to a limited reduction of DNA synthesis by about 20% (Fig. 5C). This suggests strongly that depletion of any hY RNA in the cytosolic extract inhibits the extract-dependent DNA synthesis in late-G1-phase template nuclei.
In the next experiments, we attempted to rescue the loss of function obtained through the specific depletion of one hY RNA by supplementing another nontargeted hY RNA. After the depletion of hY1 RNA, the percentage of late-G1-phase nuclei replicating in the cytosolic extract was restored to the level observed before the depletion upon the addition of hY3, hY4, or hY5 RNA (Fig. 5D). Conversely, after the depletion of hY3 RNA, the original levels of replicating nuclei were restored upon the addition of hY1 RNA (Fig. 5D). These experiments demonstrate that the four hY RNAs can functionally replace each other in this system.
We conclude from these experiments, taken together, that hY RNAs are essential factors for chromosomal DNA replication in human late-G1-phase nuclei.
Because these human late-G1-phase template nuclei have DNA damage as a result of the chemical synchronization procedure using mimosine (36, 38), we examined the requirement of Y RNAs for DNA replication in a different cell-free initiation system. We prepared template nuclei from mouse NIH 3T3 cells, which were synchronized in late G1 phase naturally by the release of contact-inhibited quiescent cells through subcultivation (34). Consistent with the results from published reports (5, 34), DNA synthesis is initiated in 25 to 30% of these nuclei upon incubation in cytosolic extract from human S-phase cells (Table 2). Importantly, RNase A treatment or specific depletion of hY3 RNA reduced the number of replicating nuclei to the background of contaminating S-phase nuclei. The addition of hY1 RNA to hY3 RNA-depleted extract restored efficient replication in vitro (Table 2). We conclude firstly that Y RNAs are conserved as essential chromosomal DNA replication factors between human and rodent replication systems and secondly that the dependency of DNA synthesis on Y RNA is neither unique to template nuclei having DNA damage nor a consequence of a particular synchronization protocol employed to obtain template nuclei.
TABLE 2.
Requirement of hY RNAs for DNA replication in late-G1-phase nuclei from mouse NIH 3T3 cells
| Replication in vitro witha: | % of nuclei replicatingb |
|---|---|
| Buffer (elongation) | 4.6 ± 4 |
| S cytosol (initiation and elongation) | 28.3 ± 4.1 |
| S cytosol plus: | |
| RNase A | 5.3 ± 3.6 |
| Nontarget DNA oligonucleotide | 24.2 ± 4.8 |
| Anti-hY3 DNA oligonucleotide | 3.3 ± 1.4 |
| Anti-hY3 oligonucleotide + hY1 RNA | 30.8 ± 5.1 |
Nuclei were prepared from mouse NIH 3T3 cells, synchronized in late G1 phase by a release from quiescence for 18 h (34). Semiconservative DNA replication is initiated in these nuclei upon incubation in cytosolic extract from S-phase cells in vitro (5, 34).
We used 300 μg of protein of S cytosol from HeLa cells and 300 ng of hY1 RNA per reaction as indicated in the text. Mean values ± standard deviations of the means from three to five independent experiments are shown.
Human Y RNAs are required for a reconstitution of semiconservative DNA replication.
Next, we analyzed the nature of Y RNA-dependent synthesis in template nuclei by density substitution experiments (Fig. 6). As reported previously (17), incubation of late-G1-phase template nuclei with unfractionated cytosolic extract led to the synthesis of hemisubstituted DNA products, demonstrating that semiconservative chromosomal DNA replication is initiated and maintained in these nuclei and that origins fire only once in this system (Fig. 6A). The addition of RNase A to this reaction led to a significant reduction of the amount of hemisubstituted DNA (Fig. 6A). Similarly, the addition of anti-hY1 oligonucleotides inhibited the synthesis of hemisubstituted DNA to the same extent, indicating that hY1 RNA is required for semiconservative DNA replication (Fig. 6A). Importantly, when we added recombinant hY3 RNA to reactions in which hY1 RNA was depleted by anti-hY1 oligonucleotides, we observed that the amount of hemisubstituted DNA was increased and that no additional products were synthesized with either fully substituted or less-than-hemisubstituted densities (Fig. 6B). Taken together, these density substitution experiments establish unambiguously that human Y RNAs are essential for semiconservative chromosomal DNA replication in this system and not for some other form of DNA synthesis like repair-associated DNA short-patch synthesis or rereplication from activated origins.
FIG. 6.

Human Y RNAs are required for semiconservative DNA replication. Late-G1-phase nuclei were incubated with S100 cytosolic extract in the presence of BrdU triphosphate and [α-32P]dCTP for 3 h, and purified DNA reaction products were analyzed by density gradient centrifugation and scintillation counting. (A) Specific degradation of hY RNA inhibits semiconservative DNA replication. Cytosolic extract from proliferating HeLa cells was left untreated, treated with RNase A, or treated with anti-hY1 oligonucleotides. Positions of unsubstituted (light-light [LL]), hemisubstituted (heavy-light [HL]), and fully substituted DNA (heavy-heavy [HH]) are indicated as determined by refractive indices. (B) Reconstitution of semiconservative DNA replication by hY RNA. After depletion of hY1 RNA by anti-hY1 oligonucleotides, the treated extract was supplemented with 100 ng of hY3 RNA synthesized in vitro. Representative results of one out of two independent experiments are shown (note that quantitative comparisons of the raw cpm values should not be drawn between the independent experiments of panels A and B).
Y RNAs are not priming DNA synthesis in vitro.
A possible explanation for how Y RNAs are involved in the establishment and further maintenance of chromosomal DNA replication forks is that they act directly as primers for DNA strand synthesis. Their conserved single-stranded 3′ polyuridine tail and unmodified 3′ hydroxyl end would be consistent with this hypothesis. We have addressed this possibility and used radioactive hY RNAs to initiate chromosomal DNA replication in vitro and analyzed the fate of these radioactive hY RNAs. We were not able to detect on denaturing urea gels any covalent extension of the input hY RNA by DNA synthesis during the in vitro reaction (data not shown). Furthermore, to block possible nucleotide chain extension, we capped the 3′ hydroxyl end of hY1 RNA with a 3′ deoxyuridine residue. This capped hY1 RNA initiated chromosomal DNA replication in the same percentages of late-G1-phase nuclei as the unmodified form (data not shown). We therefore conclude that hY RNAs do not act by priming DNA strand synthesis in this system, whether specifically or opportunistically.
Binding of hY1 RNA to Ro60 protein is not essential for DNA replication.
In all the organisms investigated, Y RNAs are efficiently bound by Ro60 protein to form Ro RNPs (4). Deletion of the homologous Ro60 protein in mice, C. elegans, and D. radiodurans resulted in viable organisms (3, 20, 46), indicating that Ro60 protein cannot be essential for chromosomal DNA replication per se. Therefore, the question arises whether Y RNAs exert their function during chromosomal DNA replication independently of Ro60 protein.
Ro binds to a conserved bulged RNA helix motif in the stem of the Y RNAs (10, 32). The specific binding of Ro60 to Y3 RNA is abolished when the conserved bulged single cytidine residue in the 5′ section of the stem is deleted, when the conserved bulged loop in the 3′ section of the stem is deleted, or when nucleotides are swapped between the two strands of the stem (10). We generated the homologous mutant forms of hY1 RNA (Fig. 7A) and synthesized them in vitro (Fig. 7B). Control experiments confirmed the loss of specific binding of recombinant human Ro60 protein to these mutant hY1 RNAs (data not shown). Importantly, all three of these mutants increased the proportion of late-G1-phase nuclei replicating in the presence of protein fractions QA and ArFT to the same values as the wt form of hY1 RNA (Fig. 7C).
FIG. 7.

The conserved binding site for Ro60 protein on hY1 RNA is not essential for chromosomal DNA replication. scram, scrambled nucleotide sequence mutant. (A) Nucleotide sequences and predicted structures of wild-type (hY1wt) and mutant hY1 RNAs. Nucleotide reference numbers for the RNAs are indicated by gray numbers. Structure predictions were performed using the RNAfold algorithm (http://rna.tbi.univie.ac.at) (12). Two alternative and interconvertible structural configurations of the wt stem are shown on the left (hY1wt and hY1wt*). The stems of three mutants deficient in binding to Ro60 protein (10) are shown in the middle [hY1ΔC8, hY1Δ(G96-C99):U, and hY1swap(4-7)]. The mutated binding sites for Ro60 are boxed. A scrambled nucleotide sequence mutant that maintains the overall predicted structure is shown on the left (hY1scram). The connecting wt loop is not drawn for some mutants. (B) In vitro synthesis of full-length mutant hY1 RNAs. Individual recombinant RNAs are visualized after denaturing gel electrophoresis as detailed in the legend to Fig. 3B. nt, nucleotides. (C) Functional reconstitution of chromosomal DNA replication with mutant hY1 RNAs. n, number of experiments. Nuclei from late-G1-phase cells were incubated in fractions QA and ArFT, supplemented with 100 ng of the individual mutant hY1 RNAs as indicated. Proportions of replicating nuclei were determined as described in the legend to Fig. 1C.
For a negative control, we also synthesized a mutant hY1 RNA in which the nucleotide sequence was randomized (scrambled) but the overall secondary structure was preserved (Fig. 7A and B). Importantly, this mutant did not increase the proportion of late-G1-phase nuclei replicating in the presence of fractions QA and ArFT (Fig. 7C), suggesting strongly that nucleotide sequence information is a functional determinant of hY1 RNA. Taken together, we conclude that binding to Ro60 protein is not essential for the DNA replication factor activity of hY RNAs.
hY RNAs are required for DNA replication in vivo.
Finally, we investigated whether hY RNAs have a role in chromosomal DNA replication in cultured human cells in vivo. We used RNA interference (8) to degrade hY1 RNA and analyzed the consequences on DNA replication by determining the percentages of S-phase cells by bromodeoxyuridine (BrdU) incorporation (Fig. 8).
FIG. 8.

Degradation of hY1 RNA in human cells by RNA interference in vivo leads to an inhibition of DNA replication. n, number of experiments. (A) Generation of siRNAs specific for hY1 RNA. The nucleotide sequences complementary to the two siRNAs (termed a and b) are indicated by gray lines. (B) Quantification of RNA levels after RNAi against hY1 RNA. At 48 h after transfection of asynchronously proliferating HeLa cells with the indicated siRNAs, the expression levels of hY1 RNA and 5.8S rRNA, relative to 5S rRNA, were determined by quantitative real-time RT-PCR. Relative RNA amounts were normalized against transfection with a nontarget siRNA specific for firefly luciferase mRNA. (C) Quantification of replicating S-phase cells after RNAi. At the indicated times after transfection with the indicated siRNAs, cells were pulse labeled for 1 h with BrdU, and percentages of BrdU-positive cells were determined by immunofluorescence microscopy. Mean values and standard deviations of three to five independent experiments are shown as indicated.
To target hY1 RNA for specific degradation, we synthesized two nonoverlapping 21-nucleotide siRNAs specific for the unique loop domain of hY1 RNA (Fig. 8A). After transfection of asynchronously proliferating HeLa cells with these siRNAs, we determined the relative expression levels of hY1 RNA by quantitative real-time PCR (Fig. 8B). We observed that the expression of the nontargeted reporter 5.8S rRNA, a control, stayed within a 90 to 110% bracket compared to the normalized expression of 5S rRNA. Importantly, transfection with either of the two siRNAs against hY1 RNA reduced the expression levels of hY1 RNA 5- to 10-fold, demonstrating that RNAi against the small noncoding hY1 RNA is feasible in human cells.
After transfection with either of the two siRNAs against hY1 RNA the proportion of S-phase cells incorporating BrdU was reduced significantly by a factor of three over a 48-h time course (Fig. 8C). As control, transfection with a siRNA against nontarget firefly luciferase mRNA did not reduce the percentages of S-phase cells significantly. These data support a functional role of hY RNAs for chromosomal DNA replication in proliferating human cells in vivo.
DISCUSSION
The data presented in this paper are relevant for the fields of DNA replication and noncoding RNA function. First, we have identified a novel factor for chromosomal DNA replication in human cell nuclei. In gain-of-function experiments using a fractionation and reconstitution approach, we have discovered a requirement of hY RNAs for chromosomal DNA replication in vitro. Conversely, in loss-of-function experiments using specific degradation of hY RNAs, we have observed an inhibition of chromosomal DNA replication in vitro and in vivo. The addition of pure recombinant hY RNAs to human cell extracts after hY RNA degradation in vitro led to the reconstitution of semiconservative DNA replication in the template nuclei. Second, through these experiments, we have also provided the first direct demonstration of a biological function for Y RNAs since their discovery in the early 1980s.
We have employed a cell-free system from human cells to purify essential DNA replication factors from a cytosolic extract. Chromosomal DNA replication, which is initiated in isolated late-G1-phase nuclei upon incubation in the extract, reflects all canonical properties observed in vivo, namely: it is semiconservative (17), dependent on S-phase-specific cyclin-dependent protein kinases (17, 22), site specific and temporally controlled (14), and bidirectional (23). Fractionation and reconstitution experiments of the cytosolic extract have resulted in the purification of RPA (37), PCNA (36), and now hY RNAs as essential soluble factors required for chromosomal DNA replication in this system.
The template nuclei employed in these studies are prepared from human cells synchronized in the late G1 phase of the cell division cycle, which do not contain active DNA replication forks (14, 17, 18). Therefore, the DNA replication factors purified through fractionation and reconstitution experiments contain at least one soluble activity which is essential for the initiation step of chromosomal DNA replication. Previously purified factors RPA and PCNA are directly involved in the recruitment of replicative DNA polymerases to activated chromosomal origins of DNA replication in other eukaryotic model systems (1, 25, 39). We have therefore interpreted them as initiation factors for human chromosomal DNA replication (36, 37). However, in the light of lacking precedents, we cannot infer the precise interface of hY RNA with the DNA replication process at molecular resolution. Therefore, we prefer at present to describe the hY RNAs more broadly as DNA replication factors, because they may be required for either the initiation step leading to the establishment of active replication forks or for elongation steps during DNA replication fork progression following their establishment in vitro or even for both steps.
The template nuclei used in the fractionation and reconstitution have DNA strand breaks as result of the cell synchronization process using mimosine (38). As a result of a regulated DNA damage response, sites of these breaks have already recruited repair proteins in vivo, and their presence does not prevent initiation of DNA replication in the same nuclei (36, 38). DNA strand breaks were detected by single-DNA-fiber analysis not at actual initiation sites of chromosomal DNA replication but at random positions further downstream (23). Importantly, the data from our in vitro experiments using late-G1-phase template nuclei from naturally synchronized mouse cells (Table 2) and in vivo experiments using asynchronously proliferating human cells (Fig. 8) also showed a requirement of hY RNAs for chromosomal DNA replication. We therefore conclude that hY RNAs are not required specifically for DNA replication in the context of preexisting DNA breaks and an activated DNA damage response in mimosine-treated cell nuclei but are for chromosomal DNA replication in a wider context.
Our data presented here have identified a requirement of Y RNAs for chromosomal DNA replication in vertebrate cell nuclei in vitro and in vivo. The next major step forward will be the identification of the molecular mechanism(s) by which Y RNAs regulate replication and whether this regulation is achieved directly or indirectly. To guide future research, some principal observations should be considered. For instance, hY RNAs are relatively abundant (Table 1) (11, 30). This suggests strongly a stoichiometric or structural requirement for hY RNAs during DNA replication fork assembly and/or maintenance rather than a catalytic function.
The four hY RNAs show functional redundancy as DNA replication factors in vitro. Each of the four hY RNAs can individually increase the proportion of G1-phase nuclei replicating in the presence of additional protein factors (Fig. 4). Considering the nucleotide sequences and predicted structures of wild-type and mutant Y RNAs (Fig. 3 and 7), we would like to propose that nucleotide sequences which are conserved between the four hY RNAs are likely candidates for functional determinants of hY RNAs in DNA replication, making the individual hY RNAs functionally redundant with each other. Importantly, we can already rule out the conserved binding site for Ro60 protein (10, 32) as an essential functional determinant because its disruption does not inhibit the replication factor activity of hY1 RNA (Fig. 7). Future detailed mutagenesis will be required to identify the essential nucleotide sequence elements which may constitute functional determinants for the replication factor activity of Y RNAs.
Our data suggest that the functional redundancy of hY RNAs is complex. Depletion of any hY RNA from the HeLa cell extract inhibits the replication activity of the treated extract, despite the presence of the other three nontargeted hY RNAs, and functionality is restored experimentally upon increasing to the maximum the amount of a nontargeted hY RNA (Fig. 5; Table 1). One possible explanation is that the overall amount of all hY RNAs present in the system, regardless of their identity, therefore constitutes the replication activity. However, the functional redundancy is likely to be more complex because of our quantitative data. Depletion of the least abundant hY4 RNA reduced the amount of overall DNA synthesis as efficiently as depletion of the more abundant hY1 or hY3 RNA, or as unspecific depletion by RNase A (Fig. 5B). This observation could be explained in a hypothetical model, in which Y RNAs would form multimeric complexes with each other before becoming active DNA replication factors in vitro. Any hY RNA would be able to form an active complex containing more than one Y RNA molecule, and disruption of any Y RNA in a formed complex would inactivate the replication factor activity of entire complex. The addition of any exogenous Y RNA after RNA degradation would lead again to the formation of new active complexes. Although suitable as a speculative conceptual framework to explain our observations, future experiments would be required to test its validity.
Despite the functional redundancy of the hY RNAs, we cannot exclude subtle differences in the contribution of individual hY RNAs to chromosomal DNA replication. In late-G1-phase template nuclei, such differences could become apparent at the level of individual origin specification, individual origin activation, or individual replication fork elongation rates.
An emerging principle for noncoding RNA functionality is that the RNAs associate with proteins to form functional RNP particles (reviewed in references 2, 26, and 35). A noncoding RNA moiety in the RNP particles often serves as a specificity device to direct a RNP-associated function to a target nucleic acid via strand hybridization, for example, as characterized for snRNA in mRNA splice site recognition (44) or snoRNA in guiding RNA modification (16). Therefore, a key question is the identification of potential target nucleic acids and/or proteins which specifically interact with hY RNA during DNA replication. It is well conceivable in this context that hY RNAs either bind to and activate established DNA replication proteins or bind to and inhibit inhibitor proteins of DNA replication, thereby acting as anti-inhibitors.
Finally, a potentially relevant molecular link between hY RNAs and DNA replication proteins can be taken from the literature. On one hand, the abundant nuclear protein nucleolin has been found to interact with the polypyrimidine loop domains of hY1 and hY3 RNAs (9; S. N. Klinge and T. Krude, unpublished observations). On the other hand, nucleolin interacts specifically after genotoxic stress with the essential DNA replication protein RPA to prevent the initiation of DNA replication (6, 15, 43). This link may point towards a possible function of hY RNAs as anti-inhibitors, perhaps interfering with the replication-inhibiting interaction of nucleolin with RPA. However, hY4 and hY5 RNAs are only weakly associated or not at all associated with nucleolin (9; S. N. Klinge and T. Krude, unpublished observations), and we have observed a requirement of Y RNAs for DNA replication in the absence of genotoxic stress in vitro and in vivo (Fig. 8; Table 2). Therefore, it is unlikely that this molecular link would be sufficient to explain the requirement of hY RNAs for human chromosomal DNA replication. Future research will therefore be directed at investigating whether hY RNAs may in fact associate with other nucleic acids and at identifying which proteins interact with hY RNAs directly and indirectly under conditions that support chromosomal DNA replication.
Acknowledgments
We thank Isabel M. Palacios and Sebastian N. Klinge for a critical reading of the manuscript.
This work was supported by Cancer Research UK (project grants C1471/A2612 and C1471/A4635) and the Human Frontier Science Program Organization (research grant RGP0375). Timothy J. Gardiner is supported by a Research Studentship from the MRC.
REFERENCES
- 1.Arias, E. E., and J. C. Walter. 2004. Initiation of DNA replication in xenopus egg extracts. Front. Biosci. 9:3029-3045. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein, E., and C. D. Allis. 2005. RNA meets chromatin. Genes Dev. 19:1635-1655. [DOI] [PubMed] [Google Scholar]
- 3.Chen, X., A. M. Quinn, and S. L. Wolin. 2000. Ro ribonucleoproteins contribute to the resistance of Deinococcus radiodurans to ultraviolet irradiation. Genes Dev. 14:777-782. [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, X., and S. L. Wolin. 2004. The Ro 60 kDa autoantigen: insights into cellular function and role in autoimmunity. J. Mol. Med. 82:232-239. [DOI] [PubMed] [Google Scholar]
- 5.Coverley, D., H. Laman, and R. A. Laskey. 2002. Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat. Cell Biol. 4:523-528. [DOI] [PubMed] [Google Scholar]
- 6.Daniely, Y., and J. A. Borowiec. 2000. Formation of a complex between nucleolin and replication protein A after cell stress prevents initiation of DNA replication. J. Cell Biol. 149:799-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Kok, J. B., R. W. Roelofs, B. A. Giesendorf, J. L. Pennings, E. T. Waas, T. Feuth, D. W. Swinkels, and P. N. Span. 2005. Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab. Investig. 85:154-159. [DOI] [PubMed] [Google Scholar]
- 8.Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498. [DOI] [PubMed] [Google Scholar]
- 9.Fouraux, M. A., P. Bouvet, S. Verkaart, W. J. van Venrooij, and G. J. Pruijn. 2002. Nucleolin associates with a subset of the human Ro ribonucleoprotein complexes. J. Mol. Biol. 320:475-488. [DOI] [PubMed] [Google Scholar]
- 10.Green, C. D., K. S. Long, H. Shi, and S. L. Wolin. 1998. Binding of the 60-kDa Ro autoantigen to Y RNAs: evidence for recognition in the major groove of a conserved helix. RNA 4:750-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendrick, J. P., S. L. Wolin, J. Rinke, M. R. Lerner, and J. A. Steitz. 1981. Ro small cytoplasmic ribonucleoproteins are a subclass of La ribonucleoproteins: further characterization of the Ro and La small ribonucleoproteins from uninfected mammalian cells. Mol. Cell. Biol. 1:1138-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofacker, I. L. 2003. Vienna RNA secondary structure server. Nucleic Acids Res. 31:3429-3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kearsey, S. E., and S. Cotterill. 2003. Enigmatic variations: divergent modes of regulating eukaryotic DNA replication. Mol. Cell 12:1067-1075. [DOI] [PubMed] [Google Scholar]
- 14.Keller, C., O. Hyrien, R. Knippers, and T. Krude. 2002. Site-specific and temporally controlled initiation of DNA replication in a human cell-free system. Nucleic Acids Res. 30:2114-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim, K., D. D. Dimitrova, K. M. Carta, A. Saxena, M. Daras, and J. A. Borowiec. 2005. Novel checkpoint response to genotoxic stress mediated by nucleolin-replication protein a complex formation. Mol. Cell. Biol. 25:2463-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiss, T. 2002. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell 109:145-148. [DOI] [PubMed] [Google Scholar]
- 17.Krude, T. 2000. Initiation of human DNA replication in vitro using nuclei from cells arrested at an initiation-competent state. J. Biol. Chem. 275:13699-13707. [DOI] [PubMed] [Google Scholar]
- 18.Krude, T. 1999. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp. Cell Res. 247:148-159. [DOI] [PubMed] [Google Scholar]
- 19.Krude, T., M. Jackman, J. Pines, and R. A. Laskey. 1997. Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell 88:109-119. [DOI] [PubMed] [Google Scholar]
- 20.Labbe, J. C., S. Hekimi, and L. A. Rokeach. 1999. The levels of the RoRNP-associated Y RNA are dependent upon the presence of ROP-1, the Caenorhabditis elegans Ro60 protein. Genetics 151:143-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Labbe, J. C., M. Jannatipour, and L. A. Rokeach. 1995. The Caenorhabditis elegans rop-1 gene encodes the homologue of the human 60-kDa Ro autoantigen. Gene 167:227-231. [DOI] [PubMed] [Google Scholar]
- 22.Laman, H., D. Coverley, T. Krude, R. Laskey, and N. Jones. 2001. Viral cyclin-cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol. Cell. Biol. 21:624-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marheineke, K., O. Hyrien, and T. Krude. 2005. Visualization of bidirectional initiation of chromosomal DNA replication in a human cell free system. Nucleic Acids Res. 33:6931-6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matera, A. G., M. R. Frey, K. Margelot, and S. L. Wolin. 1995. A perinucleolar compartment contains several RNA polymerase III transcripts as well as the polypyrimidine tract-binding protein, hnRNP I. J. Cell Biol. 129:1181-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendez, J., and B. Stillman. 2003. Perpetuating the double helix: molecular machines at eukaryotic DNA replication origins. Bioessays 25:1158-1167. [DOI] [PubMed] [Google Scholar]
- 26.Michel, U. 2002. Non-coding ribonucleic acids—a class of their own? Int. Rev. Cytol 218:143-219. [DOI] [PubMed] [Google Scholar]
- 27.Nabatiyan, A., and T. Krude. 2004. Silencing of chromatin assembly factor 1 in human cells leads to cell death and loss of chromatin assembly during DNA synthesis. Mol. Cell. Biol. 24:2853-2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Brien, C. A., K. Margelot, and S. L. Wolin. 1993. Xenopus Ro ribonucleoproteins: members of an evolutionarily conserved class of cytoplasmic ribonucleoproteins. Proc. Natl. Acad. Sci. USA 90:7250-7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pruijn, G. J., F. H. Simons, and W. J. van Venrooij. 1997. Intracellular localization and nucleocytoplasmic transport of Ro RNP components. Eur. J. Cell Biol. 74:123-132. [PubMed] [Google Scholar]
- 30.Pruijn, G. J., P. A. Wingens, S. L. Peters, J. P. Thijssen, and W. J. van Venrooij. 1993. Ro RNP associated Y RNAs are highly conserved among mammals. Biochim. Biophys. Acta 1216:395-401. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 32.Stein, A. J., G. Fuchs, C. Fu, S. L. Wolin, and K. M. Reinisch. 2005. Structural insights into RNA quality control: the Ro autoantigen binds misfolded RNAs via its central cavity. Cell 121:529-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stillman, B. 2005. Origin recognition and the chromosome cycle. FEBS Lett. 579:877-884. [DOI] [PubMed] [Google Scholar]
- 34.Stoeber, K., A. D. Mills, Y. Kubota, K. Marheineke, T. Krude, P. Romanowski, R. A. Laskey, and G. H. Williams. 1998. Cdc6 causes premature entry into S phase in a mammalian cell-free system. EMBO J. 17:7219-7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Storz, G., and K. M. Wassarman. 2005. An abundance of RNA regulators. Annu. Rev. Biochem. 74:199-217. [DOI] [PubMed] [Google Scholar]
- 36.Szüts, D., C. Christov, L. Kitching, and T. Krude. 2005. Distinct populations of human PCNA are required for initiation of chromosomal DNA replication and concurrent DNA repair. Exp. Cell Res. 311:240-250. [DOI] [PubMed] [Google Scholar]
- 37.Szüts, D., L. Kitching, C. Christov, A. Budd, S. Peak-Chew, and T. Krude. 2003. RPA is an initiation factor for human chromosomal DNA replication. Nucleic Acids Res. 31:1725-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szüts, D., and T. Krude. 2004. Cell cycle arrest at the initiation step of human chromosomal DNA replication causes DNA damage. J. Cell Sci. 117:4897-4908. [DOI] [PubMed] [Google Scholar]
- 39.Takeda, D. Y., and A. Dutta. 2005. DNA replication and progression through S phase. Oncogene 24:2827-2843. [DOI] [PubMed] [Google Scholar]
- 40.Vandesompele, J., K. De Preter, F. Pattyn, B. Poppe, N. Van Roy, A. De Paepe, and F. Speleman. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Gelder, C. W., J. P. Thijssen, E. C. Klaassen, C. Sturchler, A. Krol, W. J. van Venrooij, and G. J. Pruijn. 1994. Common structural features of the Ro RNP associated hY1 and hY5 RNAs. Nucleic Acids Res. 22:2498-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Horn, D. J., D. Eisenberg, C. A. O'Brien, and S. L. Wolin. 1995. Caenorhabditis elegans embryos contain only one major species of Ro RNP. RNA 1:293-303. [PMC free article] [PubMed] [Google Scholar]
- 43.Wang, Y., J. Guan, H. Wang, D. Leeper, and G. Iliakis. 2001. Regulation of DNA replication after heat shock by replication protein a-nucleolin interactions. J. Biol. Chem. 276:20579-20588. [DOI] [PubMed] [Google Scholar]
- 44.Will, C. L., and R. Lührmann. 2001. Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 13:290-301. [DOI] [PubMed] [Google Scholar]
- 45.Wolin, S. L., and T. Cedervall. 2002. The La protein. Annu. Rev. Biochem. 71:375-403. [DOI] [PubMed] [Google Scholar]
- 46.Xue, D., H. Shi, J. D. Smith, X. Chen, D. A. Noe, T. Cedervall, D. D. Yang, E. Eynon, D. E. Brash, M. Kashgarian, R. A. Flavell, and S. L. Wolin. 2003. A lupus-like syndrome develops in mice lacking the Ro 60-kDa protein, a major lupus autoantigen. Proc. Natl. Acad. Sci. USA 100:7503-7508. [DOI] [PMC free article] [PubMed] [Google Scholar]