

Abstract
DNA methylation is vital for proper chromatin structure and function in mammalian cells. Genetic removal of the enzymes that catalyze DNA methylation results in defective imprinting, transposon silencing, X chromosome dosage compensation, and genome stability. This epigenetic modification is interpreted by methyl-DNA binding domain (MBD) proteins. MBD proteins respond to methylated DNA by recruiting histone deacetylases (HDAC) and other transcription repression factors to the chromatin. The MBD2 protein is dispensable for animal viability, but it is implicated in the genesis of colon tumors. Here we report that the MBD2 protein is controlled by arginine methylation. We identify the protein arginine methyltransferase enzymes that catalyze this modification and show that arginine methylation inhibits the function of MBD2. Arginine methylation of MBD2 reduces MBD2-methyl-DNA complex formation, reduces MBD2-HDAC repression complex formation, and impairs the transcription repression function of MBD2 in cells. Our report provides a molecular description of a potential regulatory mechanism for an MBD protein family member. It is the first to demonstrate that protein arginine methyltransferases participate in the DNA methylation system of chromatin control.
The information contained within the DNA sequences of many organisms is augmented by epigenetic modifications of DNA and proteins bound to it. Methylation of cytosines in the context of CG dinucleotides is the predominant epigenetic modification of vertebrate genomes (27, 43). The majority of CG sites appears to be methylated in nonembryonic cells; only CG-rich segments located in gene control regions are generally unmethylated (7, 71).
Methylation is catalyzed postsynthetically by DNA methyltransferase (DNMT) enzymes (15, 27). DNMT1 is the major maintenance methyltransferase, and it ensures that newly synthesized DNA retains the methylation pattern of the template strand; DNMT3a and DNMT3b are de novo methyltransferases, setting up the methyl-CG landscape of the genome early in development. DNMT3L has no intrinsic enzyme activity, but it is essential for genome methylation, serving as a cofactor for DNMT3a and DNMT3b. DNMT2 has no detectable DNA methylation activity and was recently reclassified as a tRNA methyltransferase (28).
DNA methylation is vital for proper chromatin structure and function: genetic inactivation of each DNMT reveals its roles in X chromosome dosage compensation (3, 68), transposon silencing (12, 79), imprinting (13, 30, 37, 42, 53), and chromosome stability (20). These physiological phenomena have in common chromatin silencing.
At the molecular level, the methyl-CG mark can be attractive or repulsive to DNA binding factors that affect chromatin activity (43). The most thoroughly characterized set of factors that are attracted to methyl-CG is the methyl-DNA binding domain (MBD) protein family (31, 52, 59, 64). These proteins share a highly conserved MBD, which most of the five family members use to recognize methylated DNA (Fig. 1). Outside this region, the proteins are generally dissimilar at the amino acid (aa) level, although MBD1, MBD2, and methyl-CG-binding protein 2 (MeCP2) have in common a functionally homologous region termed the transcription repression domain (TRD). This region can recruit proteins that repress chromatin, including histone deacetylases (HDAC) and factors that regulate them, histone methyltransferases, and proteins with homology to ATP-dependent helicases (22, 41, 45, 63, 64, 75, 78, 83).
FIG. 1.

The mammalian MBD protein family. The MBDs (green) of each protein share both amino acid and functional homologies. The TRDs (red) share functional homology: they recruit histone deacetylase complexes to silence chromatin. The MBD of MBD2 overlaps the TRD and is indicated by a black line. The RG repeat domain in MBD2 is shown (yellow). The MBD of mammalian MBD3 has amino acid substitutions that do not allow it to recognize methylated DNA. MBD4 is a DNA mismatch repair enzyme (43).
Despite the fact that MBD1, MBD2, and MeCP2 share this ability to nucleate repression factors on methylated DNA, genetic analyses show that any molecular similarities between these proteins extend only loosely to biological function. The brain is the primary organ affected by the loss of each of these proteins, but the phenotypes are quite distinct: the loss of MBD1 compromises neurogenesis (84), MBD2 deficiency affects maternal behavior and the immune response to pathogens (32, 38, 39), and the loss of MeCP2 causes motor neuron dysfunction and other neurological symptoms (14, 29, 55).
The essential functions of DNA methylation are underscored by the human pathologies inflicted when components of the methylation system are defective. ICF (immunodeficiency, centromeric instability, and facial anomaly) syndrome, a disorder characterized by chromosome instability and immunodeficiency, and Rett syndrome, a severe neurological disorder, are caused by DNMT3b and MeCP2 mutations, respectively (1, 46, 81). Furthermore, depletion of MBD2 confers resistance to intestinal tumors in mice. MBD2 is therefore an attractive target for a colorectal cancer therapeutic (6, 32, 73).
Identification of the regions of chromatin under the control of MBD proteins is expected to provide the bridge needed to connect these phenotypes with the molecular characteristics of the proteins. One class of MBD2 target in chromatin has been uncovered: helper T cells from MBD2-deficient mice are compromised in cytokine gene control (32, 38, 39). No global gene misregulation was detected in MeCP2 mutant animals by microarray analysis (77). However, relatively subtle misexpression of a number of genes in the absence of MeCP2 has been reported, including the brain-derived neurotrophic factor gene, imprinted DLX-5, UBE3A, and GABRB3 genes, and glucocorticoid-regulated genes (16, 36, 56, 65, 72). Precisely how misexpression of these genes relates to the phenotypes displayed by animals lacking each MBD protein is not yet clear.
Several puzzles are evident when the phenotypes resulting from the losses of DNMT and MBD proteins are considered in parallel. It may be that in some contexts, DNA methylation operates independently of MBD proteins, MBD proteins are functionally redundant, additional uncharacterized components are present, and/or MBD proteins participate in other activities. A subset of the MBD proteins can interact with certain classes of RNA molecules with high affinity, suggesting functions in addition to chromatin control (40). MeCP2 can modulate alternative splicing, supporting this possibility (82).
A full molecular understanding of the DNA methylation-mediated chromatin control system will illuminate the relationship between phenotypes resulting from the loss of DNMT and the loss of MBD protein. When and how each DNMT recognizes its target sites, when and how each MBD protein segregates between these methyl-CG sites, and how these events translate methyl-CG into altered chromatin configurations are areas of intense research efforts (43).
The DNA methylation system governs strikingly diverse biological activities and displays features indicative of complex regulation (16, 38, 56, 60, 74). The molecular mechanisms underlying these controls have yet to be uncovered. MBD2 was the first methyl-DNA-associated protein to be identified biochemically, and genome sequence analyses indicate that MBD2 is the ancestral family member: it is the sole MBD protein in invertebrate organisms (33). Here we report that mammalian MBD2 is controlled by methylation on arginine residues. We identified the protein arginine methyltransferases (PRMT) that catalyze the modifications in vivo and uncover the molecular and functional consequences of MBD2 modification: methylation reduces the affinity of MBD2 for methyl-CG DNA and for HDAC silencing complexes. Consistent with these molecular effects, we show that arginine methylation of MBD2 inhibits its function as a transcription repressor. MBD2 methylation can therefore act to silence a silencer.
MATERIALS AND METHODS
Plasmids.
Constructs pcDNA3.1-FLAG-DEST, pDONR-221-MBD2, FLAG-MBD2, and FLAG-MBD2(−RG) (a mutant that removes aa 48 to 114) were described previously (40). Glutathione S-transferase (GST)-MBD2 (N-terminal and C-terminal proteins) and FLAG-MBD3 were kindly provided by Adrian Bird. To create pcDNA5/FRT/TO-FLAG-DEST, the original pcDNA5/FRT/TO plasmid (Invitrogen) was digested with PmeI to generate blunt ends and an oligonucleotide coding for the FLAG-tagged peptide sequence MDYKDDDK with an EcoRV site at the 3′ end was cloned into the plasmid. The RfA Gateway cassette was cloned into the EcoRV site to make it compatible for recombination cloning with the pDONR-221-DEST plasmid from the Gateway system (Invitrogen). The pOG44 plasmid was purchased from Invitrogen. For small interfering RNA (siRNA) plasmids, targeting siRNA sequences were designed based on the cDNA of human PRMT1 (GenBank accession no. NM_001536) and PRMT5 (GenBank accession no. NM_006109). Eight selected sequences from each gene were cloned into the pRetro.Super (pRS) vector (Oligoengine), used according to the supplier's instruction, and the same vector encoding siRNA against the p53 transcript was provided by Julian Downward. To create deletion mutants in the arginine-glycine (RG) domain of MBD2, pDNOR221-MBD2 was digested with FspI and SacII to delete aa 50 to 94 and double-stranded mutagenic oligonucleotides were ligated into the plasmid. For MBD2 aa 1 to 131, pDONR221-MBD2 was digested with BamHI and a mutagenic oligonucleotide was cloned into the plasmid to introduce a stop codon after residue 131 of the MBD2 protein. All MBD2 deletion mutants were cloned into the desired Gateway destination vectors by use of LR Clonase (Invitrogen) according to manufacturer's instruction. The pGal 1-94-DEST vector was created by digesting pGal aa 1 to 94 (a gift from Caroline Hill) with BamHI, blunting with T4 DNA polymerase, and cloning reading frame cassette B from the Gateway vector conversion system (Invitrogen) into the plasmid. pG5 DNA polymerase β was created by J. Millbrandt and provided by Adrian Bird. pRLTK was purchased from Promega.
Antibodies.
The following antibodies were obtained from the indicated sources: FLAG M2 and FLAG M2-agarose, Sigma; ASYM24, SYM10, PRMT1, and PRMT5, Upstate; MBD2, HDAC2, MTA2, and Gal4, Santa Cruz; HDAC1, Calbiochem; and RbAp46 and RbAp48, Oncogene. All commercial antibodies were used for Western blotting at dilutions recommended in the manufacturers' instructions. The hnRNP A1 (1:2,000) and methylosome protein 50 (MEP50) (1:2,000) antibodies were gifts from Gideon Dreyfuss.
SDS-PAGE, Western blotting, and fluorography.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting were performed as described previously (62). To visualize radiolabeled proteins by fluorography, gels were stained with Coomassie blue, destained and immersed in Amplify reagent (Amersham) for 15 min, and dried and exposed to Kodak BioMax MR film (Sigma) for at least 24 h before developing.
Recombinant protein production in bacteria.
GST-MBD2 N- and C-terminal proteins were overexpressed in BL21(DE3) cells and were purified as described previously (40).
Cell culture and transfection.
Human Burkitt's lymphoma B lymphocytes (Ramos), human embryonic kidney (293T), and human cervix adenocarcinoma (HeLa) cells were cultured at 37°C in Dulbecco's modified Eagle's medium supplemented with penicillin, streptomycin, and 10% fetal calf serum. Unless otherwise stated, cells (293T) were transfected at 30 to 40% confluence with 6 μg of plasmid DNA, 48 μl EC buffer, and 60 μl Effectene reagent (QIAGEN). All other transfections maintained the same ratio of DNA, EC buffer, and Effectene reagent, and cells were harvested around 48 h posttransfection.
Total cell lysate preparation.
All lysate and subsequent buffers for proteins contained 10 μg/ml each of leupeptin, aprotinin, pepstatin, 0.5 mM phenylmethylsulfonyl transferase, and 2 mM dithiothreitol, and all steps were carried out on ice or at 4°C. Cells were resuspended in RSB-800-Triton X-100 buffer (10 mM Tris, pH 7.4, 2.5 mM MgCl2, 800 mM NaCl, 0.5% Triton X-100) to a concentration of 107 cells/ml and sonicated (five times for 10 s with a 5-min interval between each sonication). The lysate was then rocked gently for at least 30 min, and insoluble proteins were removed by centrifugation at 14,000 × g for 20 min. The supernatant collected was used for immunoprecipitation or Western blotting. For purification of endogenous proteins (Ramos cells), the supernatant collected was further centrifuged at 100,000 × g for 30 min and passed through a 0.22-μm filter. We used Ramos cells because their MBD2 content is higher than those of other cell lines.
Recombinant proteins expressed in mammalian cells.
For purification of uncomplexed proteins, mammalian cell lysates were prepared as described above, but the RSB-800 buffer contained 0.5% Empigen BB (Calbiochem), in place of Triton X-100. Lysate prepared from cells (293T) transfected with plasmids encoding FLAG-tagged proteins was added to anti-FLAG M2 antibody-agarose beads (25-μl bead volume/plate of cells, washed with lysis buffer; Sigma). The slurry was rotated for at least 1 h, and beads were washed four times with the same lysis buffer and once with PRMT buffer consisting of 20 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 0.4 mM EDTA. The FLAG proteins were recovered from the agarose beads either by boiling in SDS sample buffer or by elution in buffer containing 20 mM Tris-HCl (pH 7.4), 100 mM NaCl, 2.5 mM MgCl2, and 0.4 mg/ml 3× FLAG peptide (Sigma) with a 1-h rotation. The eluate was collected by passing the slurry through a Micro Bio-Spin column (Bio-Rad). FLAG proteins were flash frozen in liquid nitrogen and stored at −80°C. Protein concentrations were determined by titration against bovine serum albumin (BSA) standards on Coomassie blue-stained gels. All FLAG-tagged proteins were >90% pure.
Immunopurification of endogenous MBD2 complex.
Anti-MBD2 antibody (10 μg) or control antibody was added to 2 ml of RSB-800-0.5% Triton X-100-total Ramos cell lysate and incubated overnight at 4°C. Protein G-Sepharose beads (50 μl with washed 1× phosphate-buffered saline [PBS]; Amersham) were added for at least 1 h to capture the antibody-protein complexes. Beads were washed four times with lysis buffer and twice with PRMT buffer. For Western blot analysis, protein complexes were eluted from the protein G-Sepharose beads by boiling in SDS sample buffer. To assay for copurifying PRMT activity, protein complexes on the beads were resuspended in 30 μl of PRMT buffer (see above) containing 2 μCi of S-adenosyl-l-[methyl-3H]methionine (Amersham), incubated at 37°C for 90 min, and analyzed by SDS-PAGE, Western blotting, and fluorography.
In vitro methylation assay.
GST-MBD2 proteins (0.5 μg) were incubated with 2 μCi of S-adenosyl-l-[methyl-3H]methionine (Amersham) and total Ramos cell lysate (15 μg total protein). The lysate was pretreated in the absence or presence of 0.1 mM S-(5′-adenosyl)-l-homocysteine (Sigma) at 37°C for 20 min. Reactions were carried out at 37°C for 1.5 h in buffer containing 20 mM Tris-HCl, pH 8, 200 mM NaCl, 0.4 mM EDTA, 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, and 10 μg/ml each of leupeptin, aprotinin, and pepstatin. Reactions were analyzed by SDS-PAGE and fluorography.
In vivo methylation assay.
Cells (293T) were transfected with FLAG protein expression plasmids for 48 h and were subjected to in vivo methylation as described previously (49, 54). To increase the unmethylated arginine content of cellular proteins, cells were treated with 6 μM periodate-oxidized adenosine (AdOx; Sigma) immediately after transfection and AdOx-containing medium was replaced with medium without AdOx 30 min prior to the in vivo labeling assay. Labeled cells were lysed with RSB-800 and 0.5% Empigen BB; FLAG proteins were purified by immunoprecipitation as described previously, recovered from the FLAG M2-agarose by a boiling in SDS sample buffer, and analyzed by SDS-PAGE and fluorography.
Flp-In T-REx stable cell lines.
The plasmid pcDNA5/FRT/TO-FLAG-DEST (1 μg) containing either MBD2 or MBD2(−RG41) was cotransfected with pOG44 (9 μg) into Flp-In T-REx 293T cells (Invitrogen). Posttransfection (48 h), the cells were selected with medium as described above but supplemented with 2.5 μg/ml blasticidin (Invitrogen) and 50 μg/ml hygromycin (Invitrogen) for one week. Selected cells were induced with 1 to 25 ng/ml tetracycline (Invitrogen) and analyzed for the expression of FLAG-MBD2 by Western blot titration against endogenous MBD2 protein in HeLa cells. Lysates that express FLAG-MBD2 at endogenous levels were used for immunoprecipitation and analyzed by SDS-PAGE and Western blotting.
siRNA stable cell lines.
Cells (293T) were transfected with empty pRetro.Super puromycin-selectable plasmid or the same plasmid generating siRNA against p53, PRMT1, or PRMT5 transcripts. Posttransfection (48 h), cells were washed once with PBS, and the medium was supplemented with 2 μg/ml puromycin (Sigma). The cells were maintained in selective medium for one week (with fresh medium every two days) until colonies formed. Colonies were picked from the plates, and positive clones were determined by Western blotting of cell lysate with normalized protein levels. Stable clones with the highest protein knockdown levels were those transfected with constructs that generate siRNA of 5′-AGATTACTACTTTGACTCC(TT)-3′ and 5′-AGCTTTCTCCATGGATTCG(TT)-3′ for PRMT1 and PRMT5, respectively (parentheses indicate bases that are not present in the sequences of PRMT1 and PRMT5 and that were added in the sequence design because TT bases at the 3′ end are essential for the function of the siRNA).
HDAC assay.
Biotinylated histone H4 peptide was acetylated with [3H]acetyl-coenzyme A (Amersham) by using reagents and instructions provided in the histone deacetylase kit (Upstate). After labeling and coupling to streptavidin-agarose, the labeled histone H4 peptide was quantified by scintillation counting in Ultima Gold LLT (Perkin Elmer) scintillation fluid. Histone H4 peptide (30,000 to 40,000 cpm) was incubated with FLAG-MBD2 complex (30 to 300 ng, purified from cells as described in the legend to Fig. 5C). Sodium butyrate (250 mM) was included as the control for unspecific release of [3H]acetate. Reaction mixtures were incubated, stopped, and assayed for [3H]acetate release according to the manufacturer's instructions.
FIG. 5.

Depletion of PRMTs reduces the methylarginine content of MBD2 and increases its affinity for the HDAC complex. (A) Human (293T) cells were transfected with empty plasmid (pRS) or plasmids containing sequences that would generate siRNA against the transcripts for p53, PRMT1 (P1), or PRMT5 (P5). Transfected cells were selected with puromycin to generate stable clones. Total cell lysate from each clone was analyzed by Western blotting with the indicated antibodies. As loading controls, lysates were normalized by Bradford and anti-Ran antibody Western blotting. (B and C) The stable cell lines in panel A were transiently transfected with plasmids expressing FLAG-MBD2 or FLAG-MBD2(−RG) proteins. The proteins shown in panels B and C are from a single purification, prepared as described in the legend to Fig. 3B and analyzed by Western blotting with the indicated antibodies. Data shown are representative of results obtained from at least three independent experiments. (D) FLAG-MBD2 complexes prepared as described for panel C were incubated with 3H-acetylated histone H4 peptide at 37°C for 3 h with the indicated amount of protein (ng) and in the absence or presence of the HDAC inhibitor sodium butyrate (250 mM). Release of [3H]acetate (in cpm) from the histone H4 peptide was determined by scintillation counting.
Methyl-CG DNA interaction assay.
5′-GATCJGAJGAJGAJGAJGAJGAJGAJGAJGAJGAJGAJGA-3′ DNAs (sense and antisense) were synthesized (J represents cytosine or methylcytosine, and a 5′ biotin label was coupled to the first base). This DNA sequence is identical to that of GAM12 (40, 52). DNA was resuspended in annealing buffer (30 mM HEPES-KOH, pH 7.4, 100 mM potassium acetate, and 2 mM magnesium acetate), and annealed by incubating 4 min at 95°C, 10 min at 75°C, and 1 h equilibration to room temperature. Streptavidin-agarose beads (50 μl; Sigma) were blocked 1 h with 500 μl of 3% BSA in PBS. The beads were washed three times with 1 ml of DNA wash buffer (40 mM HEPES-KOH, pH 7.5, 350 mM KCl, and 0.01% NP-40) and incubated with 1 μg DNA in binding buffer (40 mM HEPES-KOH, pH 7.5, 100 mM KCl, 6 mM MgCl2) rotating overnight. Streptavidin-agarose-DNA was washed four times in DNA wash buffer and incubated for 2.5 h with 5 μg of FLAG-MBD2 (Empigen purified) diluted in 250 μl of 30 mM HEPES-KOH, pH 7.5, 100 mM NaCl, 5% glycerol, 0.25 mg/ml tRNA, and 0.5% Triton X-100. Protein was sequentially eluted from the beads for 10 min with wash buffers (40 mM HEPES-KOH, pH 7.5, 2 mM EDTA, 0.5% Triton X-100) containing increasing NaCl concentrations. Eluted protein was recovered by ethanol-methanol-acetone precipitation.
Transcription repression assay.
pG5 DNA polymerase β reporter plasmid (2 μg) encoding the firefly luciferase protein and 5 ng of control plasmid encoding the Renilla luciferase protein (pRL-TK) were cotransfected with 45 to 405 ng (293T) or 5 to 45 ng (siRNA clones) of Gal4-MBD2 expression plasmid. Empty pGal vector was added to make the DNA mix 2.45 μg (293T) or 2.06 μg (siRNA clones) in total. Cells (293T and siRNA clones) were transfected at 70% confluence in six-well plates. Luciferase activity was measured 48 h posttransfection using the dual-luciferase assay kit (Promega) according to the manufacturer's instructions. Relative luciferase units (RLU) were expressed as a ratio of firefly activity to Renilla activity, and the results for each sample are representative of those for three independent transfections. Total protein was obtained by precipitating the cell extracts with ethanol-methanol-acetone. These were resuspended in SDS sample buffer and analyzed by Western blotting.
RESULTS
MBD2 can be methylated in vitro.
The mammalian MBD2 protein contains a long stretch of RG repeats within its N terminus. Proteins with RG repeats are commonly methylated on arginine residues within this motif (26, 57). This prompted us to test whether MBD2 can be posttranslationally modified by methylation in vitro. MBD2 N-terminal and C-terminal halves were purified and subjected to an in vitro methylation assay using mammalian total cell lysate as a source of protein methyltransferase (PMT) enzymes and [3H]S-adenosylmethionine ([3H]SAM) as a methyl donor. The assay was performed in the absence or presence of S-adenosyl-homocysteine (SAH), an inhibitor of all methyltransferases (61). Reaction mixtures were analyzed by SDS-PAGE, Coomassie blue staining, and fluorography (Fig. 2). The N terminus of MBD2, containing the RG repeats, was robustly labeled, and the C terminus was not. The methyltransferase inhibitor SAH abolished labeling of MBD2. Therefore, PMT enzymes present in the mammalian cell lysate can methylate the N-terminal RG-containing domain of MBD2.
FIG. 2.
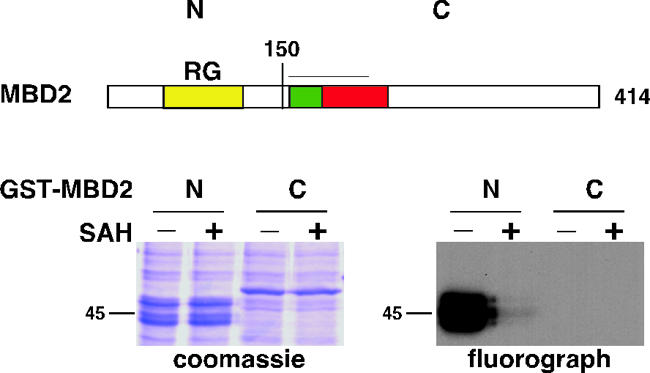
MBD2 is methylated in vitro. Recombinant GST-MBD2 N terminus (aa 1 to 150) and GST-MBD2 C terminus (aa 151 to 414) expressed in and purified from bacteria were incubated with mammalian total cell lysate and [3H]SAM, in the absence or presence of the methyltransferase inhibitor SAH, for 90 min. The reaction mixtures were analyzed by SDS-PAGE, Coomassie blue staining, and fluorography. For an explanation of the MBD2 structure presented, see the legend to Fig. 1.
MBD2 is methylated in vivo.
To address whether MBD2 is methylated in vivo, cells expressing MBD2 were subjected to an in vivo methylation assay (49, 54). In this assay, cells are incubated with either [35S]methionine (radiolabel incorporated into proteins during protein synthesis) or l-[methyl-3H]methionine (radiolabel incorporated during protein synthesis or after cellular conversion of methionine to SAM; it can be posttranslationally incorporated by PMTs). Human (293T) cells were transiently transfected with FLAG-MBD2 or FLAG-MBD2(−RG) and labeled with each type of methionine in the absence or presence of the protein synthesis inhibitors cycloheximide and chloramphenicol (CX/CP). FLAG-MBD2 was isolated from cell lysates by immunoprecipitation and analyzed. The radiolabel from [35S]methionine was incorporated into MBD2 proteins in the absence of CX/CP as expected, and the incorporation was reduced to a very low level in the presence of CX/CP, showing that protein translation was effectively inhibited (Fig. 3A). Radiolabel from l-[methyl-3H]methionine was also incorporated into MBD2, and this label transfer was immune to the presence of protein synthesis inhibitors, indicating that protein methyltransferases had methylated MBD2 in these cells (Fig. 3A). When MBD2 lacking the RG domain was analyzed, no label from l-[methyl-3H]methionine was incorporated when protein translation was blocked (Fig. 3A). We conclude that FLAG-MBD2 can be methylated in cells and that this methylation requires an intact RG domain.
FIG. 3.

MBD2 is methylated on arginine residues in vivo. (A) Human (293T) cells were transiently transfected with plasmids expressing the FLAG-MBD2 or FLAG-MBD2(−RG) proteins. The cells were incubated with either [35S]methionine or l-[methyl-3H]methionine (analyzed by fluorography [fluoro]), in the absence or presence of protein synthesis inhibitors CX/CP. FLAG proteins were purified from total cell lysate by immunoprecipitation with anti-FLAG antibody. Proteins were detached from the antibody resin by boiling in SDS sample buffer and analyzed as described in the legend to Fig. 2. The asterisk indicates the antibody heavy chain. (B) Human (293T) cells were transiently transfected with plasmids expressing the indicated FLAG proteins. FLAG proteins were immunopurified from total cell lysate as described for panel A and eluted from the antibody resin with FLAG peptide. The purified FLAG proteins were normalized by titration against a BSA standard by SDS-PAGE and Coomassie blue staining (not shown). Normalized FLAG proteins were analyzed by Western blotting with the indicated antibodies. (C) Human (293T) Flp-In T-REx cells were used to generate stable cell lines expressing FLAG-MBD2 or FLAG-MBD2(−RG41) proteins driven by a tetracycline-inducible promoter [the MBD2(−RG41) mutant is shown in Fig. 7A]. Total cell lysate from tetracycline-induced cells and total cell lysate from HeLa cells were analyzed by Western blotting with anti-MBD2 antibody. The arrows indicate the tetracycline concentrations used in panel D. As loading controls, lysates were normalized by Bradford and anti-Ran antibody Western blotting. (D) Stable cells induced with the tetracycline concentrations indicated in panel C were used to immunopurify MBD2 as described for panel A. Total cell lysate (input) and immunoprecipitated (IP) MBD2 proteins were analyzed by Western blotting with the indicated antibodies. The asterisk indicates the antibody heavy chain.
MBD2 is methylated on arginine residues in vivo.
Proteins can be posttranslationally methylated on arginine, lysine, and histidine residues (26). Since the MBD2 methylation observed in vitro and in vivo depends on the presence of its RG domain, the arginines within this region are good candidates for the modification. To date, nine PRMT enzymes in mammalian cells have been characterized (5, 17). The PRMTs fall into two groups according to their catalytic end products. Both groups catalyze the formation of monomethylarginine. Type I enzymes can then produce asymmetric dimethylarginine, whereas type II enzymes produce symmetric dimethylarginine. PRMT1, PRMT3, PRMT4, PRMT6, and PRMT8 are type I enzymes, while PRMT5, PRMT7, and PRMT9 belong to the type II group. No enzymatic activity has yet been reported for PRMT2 (5, 50).
To assess whether the methyl groups that are posttranslationally deposited on MBD2 are attached to arginines, we used antibodies that recognize dimethylarginine within peptides and proteins (10, 18). The ASYM24 antibody specifically recognizes asymmetric dimethylarginines, and the SYM10 antibody recognizes symmetric dimethylarginines. FLAG-MBD2 was transiently expressed in and immunopurified from human cells as in the in vivo methylation assay. Both ASYM24 and SYM10 recognized purified MBD2, while MBD2 lacking the RG domain was not recognized by these antibodies (Fig. 3B). The MBD3 protein, which has 70% amino acid sequence identity to MBD2 (31), was not recognized by either of these antibodies. These experiments show that transiently overexpressed MBD2 is asymmetrically and symmetrically dimethylated on arginines in cells. To confirm unequivocally that MBD2 is methylated in vivo, we generated a stable cell line that expresses tetracycline-inducible FLAG-MBD2 protein at levels comparable to those of endogenous MBD2 (Fig. 3C). We also generated a stable cell line that expresses FLAG-MBD2 with an RG domain deleted [MBD2(−RG41)]. This is a smaller deletion than the mutant (−RG), but all RG are removed (see Fig. 7A). FLAG-MBD2 was immunopurified from these stable cell lines and analyzed in the same way as the transiently expressed proteins. The tagged MBD2 protein expressed at endogenous MBD2 levels is dimethylated on arginines in cells. The RG domain mutant is not methylated (Fig. 3D).
FIG. 7.

Hypermethylated MBD2 mutants have reduced transcription repression activity. (A) The RG domain of MBD2. The RG domain deletion mutants we generated are shown. Mutant MBD2(−RG41) has all 14 R residues in the context of RG deleted. Mutants MBD2(−RG29) and MBD2(−RG23) have eight of the R residues in the context of RG deleted. Black lines indicate the deleted segments. WT, wild type. (B) Human (293T) cells were transiently transfected with FLAG-MBD2 or the FLAG-MBD2 RG domain mutants shown in panel A; the proteins were prepared as described in the legend to Fig. 3B and analyzed by SDS-PAGE, Coomassie blue staining, and Western blotting with the indicated antibodies. Input represents pooled lysate from all transfections. (C) Human (293T) cells were transiently transfected with FLAG-MBD2 or the FLAG-MBD2 hypermethylated mutants shown in panel B and were incubated in the absence or presence of AdOx. The AdOx was removed, and cells were then incubated with l-[methyl-3H]methionine and CX/CP as described in the legend to Fig. 3A. FLAG proteins were prepared and normalized as described in the legend to Fig. 3B and were analyzed by SDS-PAGE, Coomassie blue staining, and fluorography (fluoro). (D) Purified FLAG-MBD2 protein complexes from the preparation described above for panel B were analyzed by Western blotting with the indicated antibodies. Input represents pooled lysate from all transfections. (E) Human (293T) cells were transiently cotransfected with the indicated plasmids expressing Gal4-MBD2 proteins and the pG5 DNA polymerase β firefly luciferase and pRL-TK Renilla luciferase reporter plasmids. Luciferase readings were measured after 48 h, and RLU values are expressed as firefly/Renilla ratios. Each bar value represents an average of three independent transfections. Gal4-MBD2 proteins were quantified by Western blotting with the indicated antibodies. The anti-Gal4 antibody is relatively insensitive, detecting only the highly expressed Gal4-MBD2(1-131) protein (lane 7) and a ∼60-kDa nonspecific protein (asterisk) that migrates very similarly to Gal4-MBD2(−RG23). Therefore, anti-MBD2 antibody was used to detect Gal4-MBD2 and Gal4-MBD2(−RG23). This antibody cannot detect Gal4-MBD2(1-131) because the epitope lies outside the portion of MBD2 from aa 1 to 131 (lane 7). Anti-Ran antibody was used to ensure equivalent total protein loading. WT, wild type. (F) The stable cell lines shown in Fig. 5A were transiently cotransfected with reporter plasmids as described for panel E. RLU measurements were obtained as described for panel E and were normalized using MBD2(1-131). WT, wild type; P1, PRMT1; P5, PRMT5.
We conclude that MBD2 is a bona fide PRMT substrate that is arginine dimethylated in vivo, both asymmetrically and symmetrically. The MBD2 RG domain is essential for these modifications.
MBD2 was detected in a large-scale proteomics study using the SYM11 antibody to immunoprecipitate candidate methylated proteins in cell extracts (11). SYM11 recognizes symmetrically dimethylated arginine residues. We were unable to detect MBD2 by Western blotting with this antibody (data not shown). This may be due to the structural difference between native MBD2 protein probed by immunoprecipitation and denatured MBD2 protein probed by Western blotting.
MBD2 coimmunopurifies with PRMT1 and PRMT5.
PRMT enzymes have been reported to associate with their substrates in a relatively stable manner (18, 26). The ability of endogenous MBD2 to coimmunopurify with the PRMT enzymes responsible for catalyzing its methylation was investigated by incubating immunopurified MBD2 with [3H]SAM. Following incubation, the reaction mixtures were analyzed by Western blotting or fluorography (Fig. 4A). The immunopurified MBD2 was labeled during the incubation, indicating that it had copurified with PMT. We analyzed these immunopurified MBD2 complexes by Western blotting with PRMT antibodies. PRMT5 and a protein that associates with PRMT5, called MEP50 (25), copurified with MBD2 (Fig. 4B). The abundant nuclear protein hnRNP A1, which is asymmetrically methylated in vivo (54), was immunoprecipitated under the same conditions and did not coprecipitate PRMT5 or MEP50 (Fig. 4B). FLAG-MBD2 complexes immunopurified from human cells were also analyzed by Western blotting with PRMT antibodies. This allowed us to probe both wild-type MBD2 and the MBD2(−RG41) mutant, which is not methylated in cells. FLAG-MBD2, when overexpressed, copurified with endogenous PRMT5 as expected and also with PRMT1 (Fig. 4C). The RG domain of MBD2 mediates the copurification with these PRMT enzymes, since no PRMT coprecipitated with the MBD2(−RG41) mutant. PRMT3 and PRMT4 were not detected as stable components of FLAG-MBD2 complexes. We conclude that MBD2 copurifies with PRMT1 and PRMT5 enzymes and that the MBD2 RG domain is essential for this copurification.
FIG. 4.

MBD2 coimmunopurifies with PMT and interacts with PRMT1 and PRMT5 in cells. IP, immunoprecipitation; IgG, immunoglobulin G. (A) Endogenous MBD2 complexes were immunoprecipitated from total human cell (Ramos) lysate by using MBD2 antibody, and IgG was used as a control. The immunoprecipitations were washed, resuspended in buffer containing [3H]SAM, and incubated at 37°C for 90 min. MBD2 was detached from the antibody beads by boiling in SDS sample buffer and analyzed by Western blotting with anti-MBD2 antibody or fluorography. Input represents cell lysate used for immunoprecipitations. The asterisk indicates the antibody heavy chain. (B) Endogenous MBD2 and hnRNP A1 complexes were immunoprecipitated from total human cell (Ramos) lysate. Corresponding IgGs were used as controls. The immunoprecipitations were boiled in SDS sample buffer and analyzed by Western blotting with the indicated antibodies. Input represents the total cell lysate used for the IP. (C) Human (293T) cells were transiently transfected with plasmids expressing FLAG-MBD2 or FLAG-MBD2(−RG41) proteins. FLAG proteins were prepared as described in the legend to Fig. 3B and analyzed by Western blotting with the indicated antibodies. Input represents pooled lysate from all transfections.
Depletion of PRMT1 and PRMT5 reduces the methylarginine content of MBD2 in vivo.
Since MBD2 copurifies with PRMT1 and PRMT5, we asked whether removing these enzymes from cells has any effect on the asymmetric and symmetric dimethylarginine contents of MBD2. Human cells (293T) transfected with vector (pRS) or vector containing sequences that generate siRNA against p53, PRMT1, or PRMT5 transcripts were selected with puromycin to obtain stable clones in which the targeted protein was knocked down by at least 50% (Fig. 5A). FLAG-MBD2 was transiently expressed in each of these stable cell lines. MBD2 expressed in PRMT1 knockdown cells had decreased levels of both asymmetric and symmetric dimethylarginine compared to the control cells (Fig. 5B). MBD2 expressed in PRMT5 knockdown cells had decreased levels of symmetric dimethylarginine. These results indicate that PRMT1 and PRMT5 methylate MBD2 in vivo. We also note that symmetric dimethylation of MBD2 depends on the presence of normal levels of endogenous PRMT1.
MBD2 with a reduced methylarginine content has a higher affinity for HDAC silencing complexes.
Having generated MBD2 with a reduced methylarginine content, we used this system to analyze the consequences of impaired methylation on the molecular properties of MBD2. MBD2 is a component of HDAC silencing complexes that contain HDAC1, HDAC2, RbAp46, RbAp48, and MTA2 (8, 83). We compared HDAC silencing complexes copurifying with MBD2 from control cells and from PRMT1 or PRMT5 knockdown cells. We found significantly greater amounts of all of these HDAC complex proteins copurifying with MBD2 from both PRMT1 and PRMT5 knockdown cells (Fig. 5C). When methylation of MBD2 was impaired by another method, deletion of its RG domain, the population of MBD2 protein also copurified with larger amounts of the HDAC complex components, irrespective of the cellular environment (Fig. 5C). We also assessed the activity of HDAC enzyme copurifying with MBD2 from control and PRMT1 and PRMT5 knockdown cells. By use of standard HDAC assay conditions, 3H-acetylated histone H4 peptide was incubated with MBD2-HDAC complexes, and the enzymatic release of soluble [3H]acetate was measured. HDAC enzyme activity copurifying with MBD2 that is impaired in its methylarginine content was significantly higher than the HDAC activity copurifying with control MBD2 (Fig. 5D). These activity results corroborate the HDAC component protein level results. The simplest interpretation of these observations is that arginine methylation of MBD2 reduces its affinity for HDAC silencing complexes.
Arginine-methylated MBD2 has a lower affinity for methyl-CG DNA.
In vivo, MBD2 is recruited specifically to methyl-CG sites in chromatin (31, 59). The protein interacts in vitro with double-stranded DNA probes containing at least one methyl-CG (31, 44, 59). To determine whether arginine methylation of MBD2 has any consequences for the ability of the protein to interact with methyl-CG DNA, we purified FLAG-MBD2 in the presence of Empigen (to remove copurifying proteins) and subjected the protein to an in vitro methyl-CG DNA binding assay. Methyl-CG DNA (40 bp DNA containing 12 methyl-CGs) (40, 52) and the corresponding unmethylated-CG DNA were synthesized with biotin at the 5′ end and coupled to streptavidin-agarose. FLAG-MBD2 was incubated with each DNA probe, and bound and unbound fractions were analyzed (Fig. 6A). MBD2 bound to the methyl-CG DNA probe but not to the unmethylated DNA probe, demonstrating the specificity of the in vitro assay (Fig. 6A). The population of FLAG-MBD2 bound specifically to methyl-CG DNA was then subjected to step salt elutions with increasing NaCl concentrations. Protein in each elution was analyzed. The total population of FLAG-MBD2 was detected by Coomassie blue staining. The elution of total MBD2 peaks at 600 to 700 mM NaCl (Fig. 6B). When we analyzed the methylated population of MBD2 using ASYM24 and SYM10 antibodies, the peak elution of these methylated species was at 500 mM NaCl, with much less elution beyond 600 mM salt (Fig. 6B). This indicates that arginine-methylated MBD2 has a lower affinity for methyl-CG DNA than unmodified MBD2 and suggests that arginine methylation of MBD2 in cells may reduce its ability to bind to methyl-CG sites in chromatin.
FIG. 6.

MBD2 methylation controls the affinity of MBD2 for methyl-CG DNA. (A) FLAG-MBD2 was expressed in human (293T) cells and purified as described in the legend to Fig. 3B, except Empigen BB replaced Triton X-100 in the buffer. Biotinylated double-stranded DNA (40-mer containing 12 CGs, either unmethylated or 5-methylcytosine methylated on both strands) was immobilized on streptavidin-agarose. FLAG-MBD2 was incubated with the immobilized DNA in the presence of 0.25 mg/ml tRNA for 2.5 h. The unbound fraction was collected, the resin was washed in buffer containing 350 mM NaCl, and the bound fraction was detached from the resin by boiling in SDS sample buffer. Unbound and bound fractions were analyzed by SDS-PAGE, Coomassie blue staining, and Western blotting. Input represents FLAG-MBD2 added to the immobilized DNA. (B) FLAG-MBD2 was prepared and incubated with immobilized methyl-CG DNA as described for panel A. The resin was then step eluted with increasing concentrations of NaCl as shown. Unbound, bound, and eluted fractions were analyzed as described for panel A. The arginine-methylated population of MBD2 was detected by Western blotting with ASYM24 and SYM10 antibodies.
Highly methylated MBD2 has lower transcription repression activity in cells.
The molecular effects of methylating MBD2, reduced interaction with HDAC silencing complexes and reduced affinity for methyl-CG DNA, suggest that this posttranslational modification is inhibitory to MBD2 function as a transcription repressor. To test this prediction, we generated a series of RG domain mutants of MBD2. We expected that this would provide mutants with methylation content intermediate between the wild-type methylation of full-length MBD2 and the lack of methylation of the MBD2(−RG41) mutant. The mutants were expressed in and purified from human (293T) cells, and their methylarginine contents were analyzed by Western blotting as described previously. To our surprise, MBD2 mutants with partially deleted RG domains have a dramatically higher stoichiometry of asymmetric and symmetric dimethylarginine content than full-length MBD2 (Fig. 7A and B). To ensure that the observed increase in signal from the methylarginine antibodies was a consequence of increased arginine methylation rather than enhanced epitope recognition, we subjected cells expressing the MBD2 proteins to the in vivo methylation assay (Fig. 7C). Under normal conditions, there was no dramatic difference in the 3H labeling between the wild-type and mutant proteins. This might be attributed to the saturation of methyl groups on arginine prior to labeling (34, 69). When the cells are first treated with the methyltransferase inhibitor AdOx (to increase the unmethylated arginine content of cellular proteins), washed, and then subjected to labeling, the MBD2(−RG29) and MBD2(−RG23) mutants are indeed better acceptors of the methyl label than the wild-type protein. This indicates that these mutants, rather than having an enhanced affinity for the antibodies, are better PRMT substrates.
To explore this unexpected observation further, we analyzed the HDAC complexes copurifying with the hypermethylated MBD2 mutants. These mutants were severely compromised in their ability to interact with HDAC silencing components (Fig. 7D). Therefore, all of the mutants consistently demonstrate that increasing levels of arginine methylation in MBD2 correlate with decreasing levels of copurifying HDAC silencing factors.
Armed with these hypermethylated mutants, we employed a transcription repression assay to assess the functional consequences of MBD2 arginine methylation. This cell-based assay was the original assay used to demonstrate the transcription repression function of the MBD proteins (8, 64). MBD2 was targeted to DNA by fusion to the Gal4 DNA binding domain (Gal4-MBD2). Transcription repression activity was detected by a plasmid carrying the firefly luciferase gene with a 5× Gal4 binding site upstream of the promoter. In this assay, Gal4-MBD2 repressed the transcription of the reporter gene in a dose-dependent manner (Fig. 7E, bars 1 to 3). Gal4-MBD2(−RG23), a hypermethylated mutant, was much less effective in repressing transcription, even when expressed at considerably higher protein levels than the Gal4-MBD2 wild type (Fig. 7E, bars 4 to 6). The TRD of MBD proteins is essential for recruiting HDAC silencing complexes that create a repressive chromatin structure (9, 58). When an MBD2 truncation mutant [MBD2(1-131)] that lacks the TRD was analyzed, it did not coimmunopurify with HDAC silencing complexes as anticipated (data not shown), and it did not repress transcription, even at a level of protein expression far higher than wild-type MBD2 (Fig. 7E, bar 7). These data show that the transcription repression activity of the Gal4-MBD2 wild type is ∼3-fold higher than that of Gal4-MBD2(−RG23). To test whether the difference in transcription repression efficiency between wild-type and mutant MBD2 proteins was a consequence of the hypermethylation of the mutant, we repeated the experiment with cells with PRMT1 and PRMT5 depleted. We found that the MBD2-RG23 mutant has enhanced repression activity in the cells that have lower levels of PRMT1 or PRMT5 (Fig. 7F). Wild-type MBD2 would also be expected to repress transcription more efficiently in these cells. We find that the difference in repression activity of wild-type MBD2 in control cells versus that in PRMT-deficient cells is slight. This is because the arginine methylation content of the wild-type protein in control cells is much lower than the arginine methylation content of the hypermethylated MBD2-RG23 mutant (Fig. 7B). Therefore, further lowering the methylation content of wild-type MBD2 in PRMT-depleted cells has a much less dramatic effect than the effect on the MBD2-RG23 mutant. We conclude that hypermethylated MBD2 is compromised in its function as a transcription repressor.
DISCUSSION
MBD2 is a new substrate for PRMT enzymes in vivo.
Arginine methylation of proteins, although documented for histones almost 40 years ago, has only recently come to be a focus of research efforts (5, 26, 57, 66). It is now known to be an essential modification in animals that controls a number of cellular functions, including chromatin activity and repair, RNA metabolism, and ribosome function (5, 48, 57). The mechanisms of control by arginine methylation of these processes have yet to be deciphered.
Here we report that MBD2 is methylated on arginines in cells. This is the first demonstration that an MBD protein family member is subject to methylation in vivo. We find that it is both asymmetrically and symmetrically dimethylated in vivo and that PRMT1 and PRMT5 can catalyze these methylations. When we remove over half of the PRMT1 or PRMT5 content of the cells by using RNA interference, MBD2 methylation is considerably reduced. The remaining MBD2 methylation may be attributable to the remaining PRMT1 and PRMT5 activity. Other PRMT family members may also contribute to this residual modification of MBD2.
Our experiments do not address whether an individual MBD2 molecule contains both asymmetric and symmetric dimethylated arginines or whether asymmetric and symmetric modifications are mutually exclusive. However, in cells with PRMT1 activity depleted, the total population of MBD2 molecules shows reduced asymmetric and symmetric dimethylarginine contents. This result suggests that symmetric modification of an individual MBD2 molecule by PRMT5 may depend upon prior asymmetric modification of MBD2 by PRMT1. To our knowledge, this is the first indication of communication between PRMT enzymes.
The RG domain of MBD2 is essential for methylation. The whole domain is required for stable interaction with, and normal methyl transfer by, PRMT enzymes. A deletion mutant that lacks the whole domain [MBD2(−RG41)] does not stably interact with PRMT enzymes and is not methylated in cells. Interestingly, deletion mutants that lack part of the domain [MBD2(−RG29) and MBD2(−RG23)] also cannot stably interact with PRMT enzymes (data not shown), but they are hypermethylated compared to wild-type MBD2. We speculate that these deletions have created mutants with an increased substrate-enzyme turnover rate, resulting in their hypermethylation.
The RG domain spans about 40 aa and contains 14 RG repeats. We have attempted to identify the sites that are methylated, but so far, recovery of peptides from this large domain has not been successful. Outside the RG domain of MBD2, there are three more arginine residues in an RG/GR context. We cannot exclude the possibility that these arginines can also be methylated, although our in vivo protein methylation assay of the MBD2 RG domain deletion mutant did not show labeling even after long film exposures.
We note that a form of MBD2, termed MBD2b, which lacks the N-terminal RG domain, is also predicted to be expressed in cells (31). This form of MBD2 is immune to the arginine methylation we report here for full-length MBD2.
Methylation of MBD2 controls its molecular interactions.
Methylation preserves the positive charge of target arginine residues, increases hydrophobicity and bulk, and removes the amino hydrogens, thus reducing hydrogen bonding capacity. We show here that methylation of MBD2 alters both its protein-protein and protein-DNA interaction properties: methylation weakens both of these molecular contacts.
The effects of arginine methylation of other PRMT substrates on their protein-protein interaction capacities have been clearly documented: methylation in different contexts can promote or inhibit protein-protein contacts. For example, Sam68-SH3 protein interactions are inhibited by Sam68 methylation and Sm protein-SMN interactions are stimulated by Sm protein methylation (4, 24). The effects of protein arginine methylation, if any, on protein-nucleic acid contacts, have so far been elusive in experimental interrogation (5, 57). Our report of protein arginine methylation altering protein-DNA contacts is a clear demonstration that methylation can control a protein-nucleic acid complex.
Methylation of MBD2 inhibits its function as a transcription repressor.
We show several lines of molecular evidence that are in full accordance with, and led us to test, the hypothesis that methylation of MBD2 controls its function as a transcription repressor. First, arginine methylation-deficient MBD2 mutants have a higher affinity for HDAC silencing complexes than wild-type protein. Second, MBD2 protein in cells with reduced PRMT1 or PRMT5 activity has a reduced methylarginine content and displays a higher affinity for HDAC silencing complexes. Third, MBD2 mutants with partially disrupted RG domains are hypermethylated compared to wild-type protein and have a lower affinity for HDAC silencing complexes. Fourth, arginine-methylated MBD2 interacts with methyl-CG DNA with a lower affinity than the unmodified protein. Therefore, the molecular contacts involved in MBD2-mediated transcription repression that we analyzed are all inhibited by methylation.
Using a commonly employed cell-based transcription repression assay (8, 64), we found that the hypermethylated MBD2 mutants are functionally compromised: they are much less competent at repressing transcription than wild-type MBD2. The loss of function of the mutant can be rescued when the cellular levels of PRMT1 or PRMT5 are reduced. Together with the molecular consequences of MBD2 methylation, the data indicate that PRMT enzymes inhibit MBD2 activity in cells.
To address the possibility that the properties of the MBD2 mutants, defective HDAC complex formation and repression activity, are due to removal of the RG residues rather than aberrant methylation status, we analyzed their subcellular localization. Like the wild-type protein, the MBD2 mutants all localize to the nucleus (data not shown), indicating that they are at least structurally competent in terms of recognition and transport by nuclear import factors.
Our demonstration that a posttranslational modification of MBD2 inhibits activity at molecular and functional levels represents one of the first regulatory mechanisms for an MBD protein family member. A regulatory mechanism for one other MBD protein has been reported: MeCP2 is phosphorylated in cultured neuronal cells upon membrane depolarization. This event correlates with the release of MeCP2 from the brain-derived neurotrophic factor promoter (16, 56).
A recent study documenting and comparing the components of MBD2 and MBD3 complexes found that MBD2 is methylated in vitro by PRMT5 and this requires the RG domain of MBD2 (51). These data support our initial observation that led us into our molecular and functional study of MBD2 methylation in vivo.
Several questions are raised by our observations; most importantly, the physiological conditions under which MBD2 is methylated need to be explored (at the cellular level, cell type and developmental stage, and at the molecular level, where in the cell and when in the lifetime of an MBD2 molecule methylation is catalyzed and whether the methyl marks are reversible under any circumstance) (2, 19, 80).
Clues must come from considering these questions in the context of MBD2 biology. Inactivating MBD2 in the mouse causes maternal behavior deficiency, misexpression of the genes expressing cytokines interleukin 4 and gamma interferon during T-helper-cell differentiation, and defective responses to pathogen challenge (32, 38, 39). How these phenotypes relate to the molecular activity of MBD2 and the control mechanism we have uncovered needs to be addressed.
The steady-state subcellular localization of PRMT1 has been variously reported as predominantly nuclear (23, 76) and predominantly cytoplasmic (18, 35). PRMT5 is predominantly cytoplasmic (23, 70), although it was reported to be on chromatin in chromatin immunoprecipitation experiments (21, 47, 67). Newly synthesized MBD2 may be methylated as it emerges from ribosomes, but it is also possible that the modifications take place in the nucleus. It will be important to determine the preferred substrate for each PRMT (monomeric MBD2, MBD2 assembled in HDAC complexes, and MBD2 assembled in methyl CG-chromatin complexes).
Finally, our findings provide the first link between DNA methylation and protein arginine methylation. Since the molecular functions and biology of MBD proteins have yet to be fully reconciled (43), there remains the possibility that MBD proteins have functions in addition to their roles in the DNA methylation system of chromatin control. If this is the case, methylation of MBD2 may also be relevant to other as yet uncovered aspects of MBD2 protein function.
Acknowledgments
We thank the researchers mentioned in Materials and Methods for the plasmids and antibodies used in this study. We thank Frank Uhlmann, Anne McBride, and Janet Cronshaw for their comments on the manuscript.
This work was supported by Cancer Research UK.
REFERENCES
- 1.Amir, R. E., I. B. Van den Veyver, M. Wan, C. Q. Tran, U. Francke, and H. Y. Zoghbi. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23:185-188. [DOI] [PubMed] [Google Scholar]
- 2.Bannister, A. J., and T. Kouzarides. 2005. Reversing histone methylation. Nature 436:1103-1106. [DOI] [PubMed] [Google Scholar]
- 3.Beard, C., E. Li, and R. Jaenisch. 1995. Loss of methylation activates Xist in somatic but not in embryonic cells. Genes Dev. 9:2325-2334. [DOI] [PubMed] [Google Scholar]
- 4.Bedford, M. T., A. Frankel, M. B. Yaffe, S. Clarke, P. Leder, and S. Richard. 2000. Arginine methylation inhibits the binding of proline-rich ligands to Src homology 3, but not WW, domains. J. Biol. Chem. 275:16030-16036. [DOI] [PubMed] [Google Scholar]
- 5.Bedford, M. T., and S. Richard. 2005. Arginine methylation an emerging regulator of protein function. Mol. Cell 18:263-272. [DOI] [PubMed] [Google Scholar]
- 6.Berger, J., and A. Bird. 2005. Role of MBD2 in gene regulation and tumorigenesis. Biochem. Soc. Trans. 33:1537-1540. [DOI] [PubMed] [Google Scholar]
- 7.Bird, A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16:6-21. [DOI] [PubMed] [Google Scholar]
- 8.Bird, A. P., and A. P. Wolffe. 1999. Methylation-induced repression—belts, braces, and chromatin. Cell 99:451-454. [DOI] [PubMed] [Google Scholar]
- 9.Boeke, J., O. Ammerpohl, S. Kegel, U. Moehren, and R. Renkawitz. 2000. The minimal repression domain of MBD2b overlaps with the methyl-CpG-binding domain and binds directly to Sin3A. J. Biol. Chem. 275:34963-34967. [DOI] [PubMed] [Google Scholar]
- 10.Boisvert, F. M., J. Cote, M. C. Boulanger, P. Cleroux, F. Bachand, C. Autexier, and S. Richard. 2002. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J. Cell Biol. 159:957-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boisvert, F. M., J. Cote, M. C. Boulanger, and S. Richard. 2003. A proteomic analysis of arginine-methylated protein complexes. Mol. Cell Proteomics 2:1319-1330. [DOI] [PubMed] [Google Scholar]
- 12.Bourc'his, D., and T. H. Bestor. 2004. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431:96-99. [DOI] [PubMed] [Google Scholar]
- 13.Bourc'his, D., G. L. Xu, C. S. Lin, B. Bollman, and T. H. Bestor. 2001. Dnmt3L and the establishment of maternal genomic imprints. Science 294:2536-2539. [DOI] [PubMed] [Google Scholar]
- 14.Chen, R. Z., S. Akbarian, M. Tudor, and R. Jaenisch. 2001. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27:327-331. [DOI] [PubMed] [Google Scholar]
- 15.Chen, T., and E. Li. 2004. Structure and function of eukaryotic DNA methyltransferases. Curr. Top. Dev. Biol. 60:55-89. [DOI] [PubMed] [Google Scholar]
- 16.Chen, W. G., Q. Chang, Y. Lin, A. Meissner, A. E. West, E. C. Griffith, R. Jaenisch, and M. E. Greenberg. 2003. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302:885-889. [DOI] [PubMed] [Google Scholar]
- 17.Cook, J. R., J. H. Lee, Z. H. Yang, C. D. Krause, N. Herth, R. Hoffmann, and S. Pestka. 2006. FBXO11/PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues. Biochem. Biophys. Res. Commun. 342:472-481. [DOI] [PubMed] [Google Scholar]
- 18.Cote, J., F. M. Boisvert, M. C. Boulanger, M. T. Bedford, and S. Richard. 2003. Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol. Biol. Cell 14:274-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuthbert, G. L., S. Daujat, A. W. Snowden, H. Erdjument-Bromage, T. Hagiwara, M. Yamada, R. Schneider, P. D. Gregory, P. Tempst, A. J. Bannister, and T. Kouzarides. 2004. Histone deimination antagonizes arginine methylation. Cell 118:545-553. [DOI] [PubMed] [Google Scholar]
- 20.Eden, A., F. Gaudet, A. Waghmare, and R. Jaenisch. 2003. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300:455. [DOI] [PubMed] [Google Scholar]
- 21.Fabbrizio, E., S. El Messaoudi, J. Polanowska, C. Paul, J. R. Cook, J. H. Lee, V. Negre, M. Rousset, S. Pestka, A. Le Cam, and C. Sardet. 2002. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 3:641-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng, Q., and Y. Zhang. 2001. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. 15:827-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frankel, A., N. Yadav, J. Lee, T. L. Branscombe, S. Clarke, and M. T. Bedford. 2002. The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J. Biol. Chem. 277:3537-3543. [DOI] [PubMed] [Google Scholar]
- 24.Friesen, W. J., S. Massenet, S. Paushkin, A. Wyce, and G. Dreyfuss. 2001. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 7:1111-1117. [DOI] [PubMed] [Google Scholar]
- 25.Friesen, W. J., A. Wyce, S. Paushkin, L. Abel, J. Rappsilber, M. Mann, and G. Dreyfuss. 2002. A novel WD repeat protein component of the methylosome binds Sm proteins. J. Biol. Chem. 277:8243-8247. [DOI] [PubMed] [Google Scholar]
- 26.Gary, J. D., and S. Clarke. 1998. RNA and protein interactions modulated by protein arginine methylation. Prog. Nucleic Acid Res. Mol. Biol. 61:65-131. [DOI] [PubMed] [Google Scholar]
- 27.Goll, M. G., and T. H. Bestor. 2005. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74:481-514. [DOI] [PubMed] [Google Scholar]
- 28.Goll, M. G., F. Kirpekar, K. A. Maggert, J. A. Yoder, C. L. Hsieh, X. Zhang, K. G. Golic, S. E. Jacobsen, and T. H. Bestor. 2006. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311:395-398. [DOI] [PubMed] [Google Scholar]
- 29.Guy, J., B. Hendrich, M. Holmes, J. E. Martin, and A. Bird. 2001. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27:322-326. [DOI] [PubMed] [Google Scholar]
- 30.Hata, K., M. Okano, H. Lei, and E. Li. 2002. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129:1983-1993. [DOI] [PubMed] [Google Scholar]
- 31.Hendrich, B., and A. Bird. 1998. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 18:6538-6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hendrich, B., J. Guy, B. Ramsahoye, V. A. Wilson, and A. Bird. 2001. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 15:710-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hendrich, B., and S. Tweedie. 2003. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 19:269-277. [DOI] [PubMed] [Google Scholar]
- 34.Herrmann, F., M. Bossert, A. Schwander, E. Akgun, and F. O. Fackelmayer. 2004. Arginine methylation of scaffold attachment factor A by heterogeneous nuclear ribonucleoprotein particle-associated PRMT1. J. Biol. Chem. 279:48774-48779. [DOI] [PubMed] [Google Scholar]
- 35.Herrmann, F., J. Lee, M. T. Bedford, and F. O. Fackelmayer. 2005. Dynamics of human protein arginine methyltransferase 1 (PRMT1) in vivo. J. Biol. Chem. 280:38005-38010. [DOI] [PubMed] [Google Scholar]
- 36.Horike, S., S. Cai, M. Miyano, J. F. Cheng, and T. Kohwi-Shigematsu. 2005. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37:31-40. [DOI] [PubMed] [Google Scholar]
- 37.Howell, C. Y., T. H. Bestor, F. Ding, K. E. Latham, C. Mertineit, J. M. Trasler, and J. R. Chaillet. 2001. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 104:829-838. [DOI] [PubMed] [Google Scholar]
- 38.Hutchins, A. S., D. Artis, B. D. Hendrich, A. P. Bird, P. Scott, and S. L. Reiner. 2005. Cutting edge: a critical role for gene silencing in preventing excessive type 1 immunity. J. Immunol. 175:5606-5610. [DOI] [PubMed] [Google Scholar]
- 39.Hutchins, A. S., A. C. Mullen, H. W. Lee, K. J. Sykes, F. A. High, B. D. Hendrich, A. P. Bird, and S. L. Reiner. 2002. Gene silencing quantitatively controls the function of a developmental trans-activator. Mol. Cell 10:81-91. [DOI] [PubMed] [Google Scholar]
- 40.Jeffery, L., and S. Nakielny. 2004. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 279:49479-49487. [DOI] [PubMed] [Google Scholar]
- 41.Jones, P. L., G. J. Veenstra, P. A. Wade, D. Vermaak, S. U. Kass, N. Landsberger, J. Strouboulis, and A. P. Wolffe. 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19:187-191. [DOI] [PubMed] [Google Scholar]
- 42.Kaneda, M., M. Okano, K. Hata, T. Sado, N. Tsujimoto, E. Li, and H. Sasaki. 2004. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429:900-903. [DOI] [PubMed] [Google Scholar]
- 43.Klose, R. J., and A. P. Bird. 2006. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31:89-97. [DOI] [PubMed] [Google Scholar]
- 44.Klose, R. J., S. A. Sarraf, L. Schmiedeberg, S. M. McDermott, I. Stancheva, and A. P. Bird. 2005. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 19:667-678. [DOI] [PubMed] [Google Scholar]
- 45.Kondo, E., Z. Gu, A. Horii, and S. Fukushige. 2005. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol. Cell. Biol. 25:4388-4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kriaucionis, S., and A. Bird. 2003. DNA methylation and Rett syndrome. Hum. Mol. Genet. 12(Spec. no. 2):R221-R227. [DOI] [PubMed] [Google Scholar]
- 47.Kwak, Y. T., J. Guo, S. Prajapati, K. J. Park, R. M. Surabhi, B. Miller, P. Gehrig, and R. B. Gaynor. 2003. Methylation of SPT5 regulates its interaction with RNA polymerase II and transcriptional elongation properties. Mol. Cell 11:1055-1066. [DOI] [PubMed] [Google Scholar]
- 48.Lee, D. Y., C. Teyssier, B. D. Strahl, and M. R. Stallcup. 2005. Role of protein methylation in regulation of transcription. Endocr. Rev. 26:147-170. [DOI] [PubMed] [Google Scholar]
- 49.Lee, J., and M. T. Bedford. 2002. PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep. 3:268-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee, J., J. Sayegh, J. Daniel, S. Clarke, and M. T. Bedford. 2005. PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J. Biol. Chem. 280:32890-32896. [DOI] [PubMed] [Google Scholar]
- 51.Le Guezennec, X., M. Vermeulen, A. B. Brinkman, W. A. Hoeijmakers, A. Cohen, E. Lasonder, and H. G. Stunnenberg. 2006. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol. Cell. Biol. 26:843-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lewis, J. D., R. R. Meehan, W. J. Henzel, I. Maurer-Fogy, P. Jeppesen, F. Klein, and A. Bird. 1992. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69:905-914. [DOI] [PubMed] [Google Scholar]
- 53.Li, E., C. Beard, and R. Jaenisch. 1993. Role for DNA methylation in genomic imprinting. Nature 366:362-365. [DOI] [PubMed] [Google Scholar]
- 54.Liu, Q., and G. Dreyfuss. 1995. In vivo and in vitro arginine methylation of RNA-binding proteins. Mol. Cell. Biol. 15:2800-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luikenhuis, S., E. Giacometti, C. F. Beard, and R. Jaenisch. 2004. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc. Natl. Acad. Sci. USA 101:6033-6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinowich, K., D. Hattori, H. Wu, S. Fouse, F. He, Y. Hu, G. Fan, and Y. E. Sun. 2003. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302:890-893. [DOI] [PubMed] [Google Scholar]
- 57.McBride, A. E., and P. A. Silver. 2001. State of the arg: protein methylation at arginine comes of age. Cell 106:5-8. [DOI] [PubMed] [Google Scholar]
- 58.Meehan, R. R. 2003. DNA methylation in animal development. Semin. Cell Dev. Biol. 14:53-65. [DOI] [PubMed] [Google Scholar]
- 59.Meehan, R. R., J. D. Lewis, S. McKay, E. L. Kleiner, and A. P. Bird. 1989. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 58:499-507. [DOI] [PubMed] [Google Scholar]
- 60.Morgan, H. D., F. Santos, K. Green, W. Dean, and W. Reik. 2005. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 14(Spec. no. 1):R47-R58. [DOI] [PubMed] [Google Scholar]
- 61.Najbauer, J., and D. W. Aswad. 1990. Diversity of methyl acceptor proteins in rat pheochromocytoma (PC12) cells revealed after treatment with adenosine dialdehyde. J. Biol. Chem. 265:12717-12721. [PubMed] [Google Scholar]
- 62.Nakielny, S., S. Shaikh, B. Burke, and G. Dreyfuss. 1999. Nup153 is an M9-containing mobile nucleoporin with a novel Ran-binding domain. EMBO J. 18:1982-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nan, X., H. H. Ng, C. A. Johnson, C. D. Laherty, B. M. Turner, R. N. Eisenman, and A. Bird. 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386-389. [DOI] [PubMed] [Google Scholar]
- 64.Ng, H. H., Y. Zhang, B. Hendrich, C. A. Johnson, B. M. Turner, H. Erdjument-Bromage, P. Tempst, D. Reinberg, and A. Bird. 1999. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 23:58-61. [DOI] [PubMed] [Google Scholar]
- 65.Nuber, U. A., S. Kriaucionis, T. C. Roloff, J. Guy, J. Selfridge, C. Steinhoff, R. Schulz, B. Lipkowitz, H. H. Ropers, M. C. Holmes, and A. Bird. 2005. Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum. Mol. Genet. 14:2247-2256. [DOI] [PubMed] [Google Scholar]
- 66.Paik, W. K., and S. Kim. 1967. Enzymatic methylation of protein fractions from calf thymus nuclei. Biochem. Biophys. Res. Commun. 29:14-20. [DOI] [PubMed] [Google Scholar]
- 67.Pal, S., S. N. Vishwanath, H. Erdjument-Bromage, P. Tempst, and S. Sif. 2004. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 24:9630-9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Panning, B., and R. Jaenisch. 1996. DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev. 10:1991-2002. [DOI] [PubMed] [Google Scholar]
- 69.Pawlak, M. R., S. Banik-Maiti, J. A. Pietenpol, and H. E. Ruley. 2002. Protein arginine methyltransferase I: substrate specificity and role in hnRNP assembly. J. Cell. Biochem. 87:394-407. [DOI] [PubMed] [Google Scholar]
- 70.Rho, J., S. Choi, Y. R. Seong, W.-K. Cho, S. H. Kim, and D.-S. Im. 2001. PRMT5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J. Biol. Chem. 276:11393-11401. [DOI] [PubMed] [Google Scholar]
- 71.Rollins, R. A., F. Haghighi, J. R. Edwards, R. Das, M. Q. Zhang, J. Ju, and T. H. Bestor. 2006. Large-scale structure of genomic methylation patterns. Genome Res. 16:157-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Samaco, R. C., A. Hogart, and J. M. LaSalle. 2005. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 14:483-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sansom, O. J., J. Berger, S. M. Bishop, B. Hendrich, A. Bird, and A. R. Clarke. 2003. Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat. Genet. 34:145-147. [DOI] [PubMed] [Google Scholar]
- 74.Santos, F., A. H. Peters, A. P. Otte, W. Reik, and W. Dean. 2005. Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev. Biol. 280:225-236. [DOI] [PubMed] [Google Scholar]
- 75.Sarraf, S. A., and I. Stancheva. 2004. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 15:595-605. [DOI] [PubMed] [Google Scholar]
- 76.Tang, J., J. D. Gary, S. Clarke, and H. R. Herschman. 1998. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem. 273:16935-16945. [DOI] [PubMed] [Google Scholar]
- 77.Tudor, M., S. Akbarian, R. Z. Chen, and R. Jaenisch. 2002. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc. Natl. Acad. Sci. USA 99:15536-15541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wade, P. A., A. Gegonne, P. L. Jones, E. Ballestar, F. Aubry, and A. P. Wolffe. 1999. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat. Genet. 23:62-66. [DOI] [PubMed] [Google Scholar]
- 79.Walsh, C. P., J. R. Chaillet, and T. H. Bestor. 1998. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20:116-117. [DOI] [PubMed] [Google Scholar]
- 80.Wang, Y., J. Wysocka, J. Sayegh, Y. H. Lee, J. R. Perlin, L. Leonelli, L. S. Sonbuchner, C. H. McDonald, R. G. Cook, Y. Dou, R. G. Roeder, S. Clarke, M. R. Stallcup, C. D. Allis, and S. A. Coonrod. 2004. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 306:279-283. [DOI] [PubMed] [Google Scholar]
- 81.Xu, G. L., T. H. Bestor, D. Bourc'his, C. L. Hsieh, N. Tommerup, M. Bugge, M. Hulten, X. Qu, J. J. Russo, and E. Viegas-Pequignot. 1999. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402:187-191. [DOI] [PubMed] [Google Scholar]
- 82.Young, J. I., E. P. Hong, J. C. Castle, J. Crespo-Barreto, A. B. Bowman, M. F. Rose, D. Kang, R. Richman, J. M. Johnson, S. Berget, and H. Y. Zoghbi. 2005. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 102:17551-17558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang, Y., H. H. Ng, H. Erdjument-Bromage, P. Tempst, A. Bird, and D. Reinberg. 1999. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13:1924-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao, X., T. Ueba, B. R. Christie, B. Barkho, M. J. McConnell, K. Nakashima, E. S. Lein, B. D. Eadie, A. R. Willhoite, A. R. Muotri, R. G. Summers, J. Chun, K. F. Lee, and F. H. Gage. 2003. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl. Acad. Sci. USA 100:6777-6782. [DOI] [PMC free article] [PubMed] [Google Scholar]