

Abstract
The observation that leukocyte-endothelial cell (EC) interactions are localized to specific regions on the microvessel wall suggests that adhesion molecule distribution is not uniform. We investigated ICAM-1 distribution and leukocyte-EC interactions in blood perfused microvessels (< 80 μm) in cremaster muscle of anesthetized mice, using intravital confocal microscopy and immunofluorescent labeling. Variability of ICAM-1 expression directly determines leukocyte adhesion distribution within the venular microcirculation, and contributes to leukocyte rolling in arterioles during inflammation. The number of rolling interactions increased with ICAM-1 intensity (r2=0.69, p<0.05) and rolling velocity was lower in regions of higher ICAM-1 intensity. In controls, venular ICAM-1 expression was approximately two fold higher than in arterioles. Following TNF-α treatment, ICAM-1 expression was significantly increased, 2.8±0.2 fold in arterioles and 1.7±0.2 fold in venules (P<0.05). ICAM-1 expression on activated arteriolar ECs only reached the level of control venular ICAM-1. Arteriolar but not venular ECs underwent redistribution of ICAM-1 among cells; some cells increased and some decreased ICAM-1 expression, magnifying the variability of ICAM-1. TNF-α treatment increased the length of bright fluorescent regions per unit vessel length (42%, control; 70%, TNF-α) along the arteriolar wall, whereas no significant change was observed in venules (60%, control; 63%, TNF-α). The spatial distribution and expression levels of adhesion molecules in the microcirculation determine the timing and placement of leukocyte interactions, hence significantly impact the inflammatory response. That arteriolar ECs respond to TNF-α by upregulation of ICAM-1, although in a different way compared to venules, suggests an explicit role for arterioles in inflammatory responses.
Keywords: adhesion molecule expression, microvascular inflammation, in vivo, endothelial cell morphology
INTRODUCTION
Leukocyte transmigration into to the tissue is comprised of a complex series of events that are a function of timing and expression levels of various adhesion molecules.
Leukocyte rolling on and adhesion to the blood vessel wall are essential steps in the inflammatory cascade. These steps require multiple adhesion molecules, such as selectins, integrins or CAMs; a complicated mechanism integrates the functions of these molecules in the progression of the inflammatory cascade. A great deal is known about individual adhesion molecules and their interactions with leukocytes. For example, after initiation of inflammation, early rolling on P-selectin and later rolling on E-selectin is well established (1, 8, 9). Similarly, most evidence suggests that transition to firm adhesion requires selectin mediated rolling (5, 16); the principal molecules mediating firm adhesion are ICAM-1 and VCAM-1 (2, 25). ICAM-1 is a transmembrane glycoprotein with five extracellular IgG-like domains and a short cytoplasmic tail that associates with cytoskeletal proteins (16). ICAM-1 is constitutively expressed at low levels on the endothelial cell (EC) surface and is upregulated by inflammatory cytokines (16, 22). Known ligands for ICAM-1 are the β2-integrins LFA-1 and Mac-1. While ICAM-1 has been identified in isolated EC systems as the principal mediator of leukocyte adhesion, in situ its role appears to be more complex. Here, all the adhesion events overlap and interact in ways that are not fully understood, but which presumably must affect how and where leukocytes interact with the endothelium. What complicates the general picture is that in vivo there appears to be no clear separation of roles for the various families of adhesion molecules. Until recently, the major focus for the role of ICAM-1 has been its function as a mediator of leukocyte adhesion, but there is emerging evidence suggesting that ICAM-1, along with β2-integrins, can be essential for rolling (4, 27) and diapedesis (32). Importantly, ICAM-1 has also been identified as a critical signaling molecule connecting leukocyte adhesive interactions with downstream EC events (19, 30). To further complicate the in situ story, selectins have also been shown to contribute to neutrophil adhesion (17). The lack of a clear separation between the functions of these adhesion molecules emphasizes the importance of their expression levels and the placement of these molecules on endothelial cells (ECs). Both P and E-selectin mediated rolling can be highly variable (1, 7–10), and examination of neutrophil-EC interactions shows that rolling and transmigration occur in localized regions of the vessel (13, 31). Even though multiple factors may account for this, one main reason for this behavior is likely to be the variability in expression of adhesion molecules by the ECs (13).
Similar to the variability seen in rolling, there are localized microvessel wall regions that support neutrophil adhesion. Our preliminary observations (data not shown) indicate that leukocyte adhesion to venular wall is heterogeneous, and, of particular interest, support earlier studies (14, 29) indicating that in the presence of inflammatory cytokines, leukocytes also interact with arteriolar walls. It is established that mechanisms upregulating adhesion molecules in venules also upregulate their expression in arterioles (22). An increased number of interactions between leukocytes and arteriolar ECs in response to inflammatory stimuli implies that arterioles play a significant role in the inflammatory cascade. Because ICAM-1 is so important in leukocyte-EC interactions and signaling, understanding where it is and relatively how much (distribution) ICAM-1 is expressed in the microvasculature will help us to better understand the reasons for differences in leukocyte interactions with endothelium in different regions of the microvasculature, both locally, i.e. within different regions of individual microvessels, and broadly, across different microvascular vessel types. Therefore, the goals of this study were, first, to characterize the levels and patterns of ICAM-1 expression in arterioles and venules; second, to test the hypothesis that variability in expression of the primary adhesion molecule, ICAM-1, determines leukocyte adhesion distribution within the venular microcirculation; and third, to explore the distribution of ICAM-1 in arterioles, with the goal of relating the expression of this molecule to leukocyte-endothelial interactions in arterioles during inflammation.
MATHERIALS AND METHODS
Animal preparation
All procedures were approved by the Institutional Review Board of the University of Rochester.
C 57BL6J mice (Jackson laboratories) were initially anesthetized with sodium pentobarbital (65mg/kg I.P.) and maintained on supplemental anesthetic via a jugular catheter throughout the experiment. To insure a patent airway during the experiment, an endotracheal tube was inserted. Body temperature was maintained by placing the animal on a warmer during the experiment. The cremaster muscle was exposed using established methods (11, 15). Briefly, the right cremaster muscle was exteriorized and laid flat on a quartz pedestal. During preparation and observation the tissue was continuously superfused with warmed physiological solution with the following composition: (in mM) NaCl, 131.9; KCL, 4.7; CaCl, 2.0; MgSO4, 1.2, NaHCO3, 18; pH 7.4 at 36°C, and equilibrated with gas containing 0% O2, 5% CO2 and 95% N2 to maintain tissue PO2 <15 torr. Upon completion of the protocols, animals were euthanised by anesthetic overdose.
In selected animals, activated conditions were established in the tissue by intrascrotal injection of mouse recombinant TNF-α (Sigma-Aldrich): 0.5μg TNF-α in 0.25 ml saline was injected 4 hour prior to the start of the surgical preparation (12).
Micropipette cannulation and immunofluorescence labeling
Spatial distribution of endothelial ICAM-1 was determined by locally exposing the vessels to appropriately selected primary and secondary monoclonal antibodies. The approach to in vivo labeling and imaging of blood perfused microvessels has been described elsewhere (13, 31). Briefly, micropipettes were pulled from borosilicate capillary tubes (WPI) and beveled on an extra fine grinding wheel (Sutter) to create a needle like tip with a 12–15 μm diameter opening. After the surgery was completed the preparation was placed under the microscope for at least 15 minutes (stabilization period). Separate pipettes were filled with the appropriate primary and fluorescently tagged secondary antibodies and the primary feed arteriole to the cremaster was cannulated with each pipette sequentially. Cannulation took place in a main arteriole upstream of the microvascular region targeted for observation in order to avoid damage in the region to be observed. The main arteriole was occluded by another micropipette upstream of the cannulation site at the time of the antibody loading to allow a complete perfusion of the downstream regions with the antibody solution. Intravascular pressure during perfusion with the Ab solutions is typically of the order of 20 cm H2O in arterioles and 8 cm H2O in venules (24) which is in the physiological range. After Ab labeling and re-establishment of blood perfusion, microvessel diameters are typically not different from diameters before the labeling protocol.
ICAM-1 expressed on the surface of the endothelium was labeled by localized perfusion using micropipette cannulation. Initially the vessel was perfused with rat anti-mouse monoclonal antibody in saline (MCA818, Serotec, 50 μg/ml) for 15 minutes; the first pipette was withdrawn and blood perfusion briefly restored before the second cannulation with the pipette containing goat anti-rat secondary fluorescent polyclonal antibody in saline (Alexa 488 anti-rat, Molecular Probes, 50 μg/ml), which was loaded for another 15 minutes. Following this perfusion, the micropipette was withdrawn and blood flow was reestablished in the target microvascular region. Finally, after all imaging protocols were completed, the vessel was perfused with fluorescent standard solution (0.05 mg/ml FITC-dextran in saline, 150 kDa M.W., Sigma-Aldrich) to normalize for differences in fluorescence produced by localized variability in the optical properties of the tissue. Intensity measurements for the standard solution were acquired for all vessels at a region of interest in the confocal plane at the center of the vessel and at least 5 μm away from the vessel wall. This step enabled comparison of the data among different vessels and animals. To confirm that there was no fluorescence contribution due to non-specific binding of the primary or secondary antibody, we performed separate control experiments using the same technical approach, where the cremaster muscle vasculature was perfused with (i) the relevant IgG binding antibody followed by the above secondary, or (ii) the secondary antibody alone. In both cases, mean fluorescence intensity was 24±0.9 and 26±1.8 intensity units respectively, with tissue background being 28±1.2 intensity units. In addition, we confirmed the specificity of the ICAM-1 labeling by performing the same procedure in ICAM-1 knockout mice (Jackson, Stock # 002867): mean fluorescence intensity in microvessels in these animals was 27±0.8 intensity units, which is not different from tissue background.
Intravital Microscopy
All images were acquired on an Olympus BX50WI microscope through an Olympus PlanF1 immersion objective (20×, 0.65 NA), giving a spatial resolution in our imaging system of 0.5μm. Images were recorded to half inch SVHS (SONY VO9500) for offline analysis. Fluorescence images were collected from tissue that was illuminated with a 20 mW Argon laser and were acquired through a Nipkow disk scanning confocal head (CSU 10, Yokogawa) connected to an intensified CCD camera (XR Mega10, Stanford Photonics). Laser power and camera gain settings were held constant throughout the whole set of experiments. Bright field images used to track leukocyte interactions were acquired through the confocal optical path.
Analyses
ICAM-1 intensity
Video images of intact blood perfused microvessels were captured and digitalized to 8 bit tiff images using a CG-7 frame grabber (Scion Corp.). Fluorescence intensity levels, as an index of ICAM-1 expression levels, were analyzed using NIH-Image software. Fluorescence was measured as gray scale units, where 0 = black and 255 = white, and was quantified as relative intensity using the intensity of the infused FITC-dextran solution sampled as described above. For all intensity measurements the laser power and camera gain settings were held constant, and the camera response was verified to be linear over the range used for these acquisitions. We also verified that the relationship between measured intensity and concentration of fluorochrome was linear using this acquisition system (r2 between 0.996 and 0.999 for the range of gain settings used in our acquisition system). This approach allows us to compare relative ICAM-1 expression distributions among microvascular regions. It is important to note that this approach does not allow specific quantification of the amounts of adhesion molecule protein expressed in situ. Fluorescent intensities on the vessel wall at the central plane of the vessel cross-section (Fig. 1) were collected by projecting a line 5-pixel wide along the wall and obtaining the intensity profile along that line. To make sure that the selected length of the vessel was all in the same focal plane we moved the focal plane in the vertical direction and confirmed that the entire measured length was contained within the same confocal “slice”. To compare ICAM-1 expression in different microvascular regions or between TNF-α activated and control microvessels we measured the fluorescent intensities along the vessel wall, and used predetermined criteria to categorize successive sampled regions as either “bright” or “dim”. Criteria for “bright” were determined by selecting 5 obviously very bright-looking 5 μm lengths along the vessel length and measuring their average intensity. “Bright” regions were defined as regions within ± 2 standard deviations of this mean intensity. An analogous procedure was used to define dim regions along the vessel. Thus, bright regions correspond to high ICAM-1 expression and dim regions are where ICAM-1 is low. In order to compare ICAM-1 expression among individual ECs, 5 obviously bright, in-focus ECs were selected along the vessel and their average intensity was determined. Individual ECs with average intensities within ± 2 standard deviations of that number were considered to have the “same” brightness, if not then their brightness was considered to be “different”
Figure 1. Some regions of the vessel wall express higher ICAM-1 levels than others.
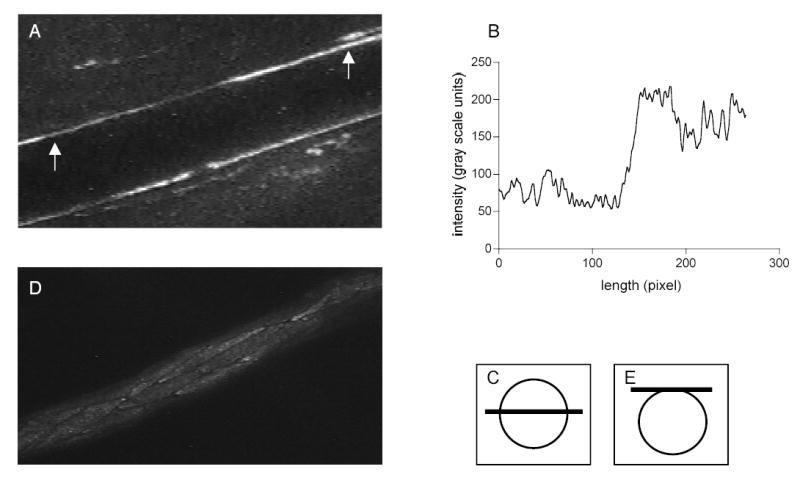
An arteriole was internally perfused with primary and fluorescently tagged secondary antibodies in sequence to label ICAM-1. ICAM-1 expression was quantified as a function of the fluorescence intensity. A: An example of variability of ICAM-1 expression in a blood perfused, confocally imaged arteriole at the central plane (shown in panel C). B: Corresponding fluorescence intensity plot of the region in between the arrows (shown in A). D: representative image of ICAM-1 labeled ECs lining the upper part of the vessel (focal plane on the top of the vessel as shown in panel E).
Leukocyte interactions
In addition to the quantification of fluorescence intensity, we also quantified leukocyte interactions with the vessel wall. Rolling leukocytes were defined as any leukocytes observed translating along the venular wall in contact with the endothelium that were moving slower than the hydrodynamic critical velocity as defined by Ley and Gaehtgens (18). The number of rolling leukocytes on the vessel wall was calculated by counting leukocytes rolling past a line perpendicular to the vessel axis per 40 sec time interval. Rolling leukocyte velocity was measured by tracking the distance rolled for a period of time greater than 0.15 sec (at least ten frames) within 50μm length defined for each observed vessel. Firmly adhered leukocytes were defined as cells that remained stationary for ≥ 30 seconds. The numbers and locations of firmly adhered leukocytes, and leukocyte rolling velocity, were quantified using trans-illuminated images, which were later superimposed onto the matched fluorescence images of ICAM-1 labeled ECs.
Statistics
Statistical tests were performed using Graphpad Prism (v 4.0) to undertake t-tests, ANOVA, linear regression or correlation analyses as appropriate. Statistical significance was set at P<0.05.
RESULTS
Using immunofluorescence labeling and confocal microscopy we undertook detailed characterization of the spatial distribution of ICAM-1 in the intact blood perfused microvasculature. Positioning the focal plane either in the center of the vessel or on the top, we were able to study ICAM-1 expression patterns on the vessel wall or on the surface of individual ECs lining the upper part of the vessel (Fig 1).
ICAM-1 expression by ECs under control conditions
ICAM-1 expression levels were indicated by the normalized intensity of fluorescently labeled surface ICAM-1 in blood-perfused vessels. As expected from previously published data, we found that under control conditions, venular ECs expressed ICAM-1. The expression levels were sufficient to allow fluorescence intensity to be quantified. Importantly, under the same control conditions, ICAM-1 expression was clearly detected as quantifiable fluorescence in arteriolar ECs. Normalization of the fluorescence intensity in each vessel using the FITC-dextran standard solution enabled comparison between different vessels and tissues. Figure 2 shows that the average ICAM-1 expression by venular ECs was 2-fold greater than that in arteriolar ECs. Inspections of the fluorescence intensity distribution on the vessel walls showed that there were repeated patterns having regions with higher and lower ICAM-1 expression (bright and dim regions). This was true for both arterioles and venules under control conditions. The lengths of these bright regions ranged from 60 to 110 μm in arterioles and from 80 to 130 μm in venules, with average values of 83±4.1 μm (n=12 vessels) and 111±5.1 μm (n=12) respectively. Likewise the lengths of the dim regions ranged from 74 to 140 μm in arterioles and from 80 to 170 μm in venules, with average values of 100±7.2 μm (n=10) and 105 ±7.7 μm (n=10) respectively (Fig 3).
Figure 2. Expression levels of ICAM-1 are different between control and TNF-α treated arterioles and venules.
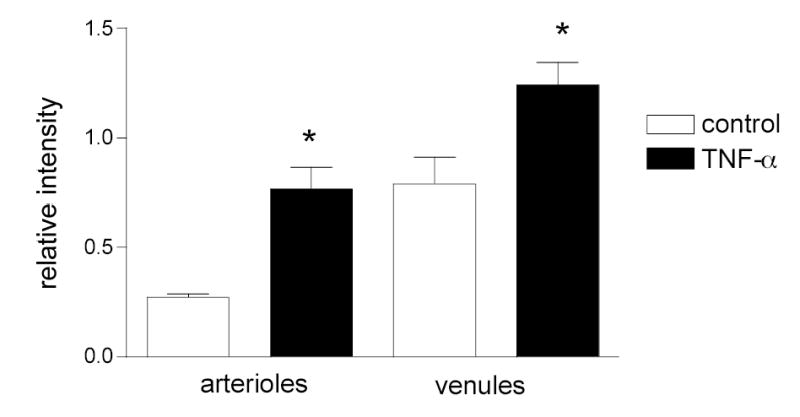
Both arteriolar and venular ECs express ICAM-1 under control conditions and its levels significantly increase following TNF-α treatment. ECs were labeled for ICAM-1. Average fluorescence intensity of ECs was obtained for both control and TNF-α treated mice. The intensity was normalized to FITC-dextran and the background noise was subtracted. N= 40 cells for each group.
* significantly different from the appropriate control group (Anova, p<0.01). There is no significant difference between columns 2 and 3.
Figure 3. ICAM-1 distribution on the vessel wall is not uniform.
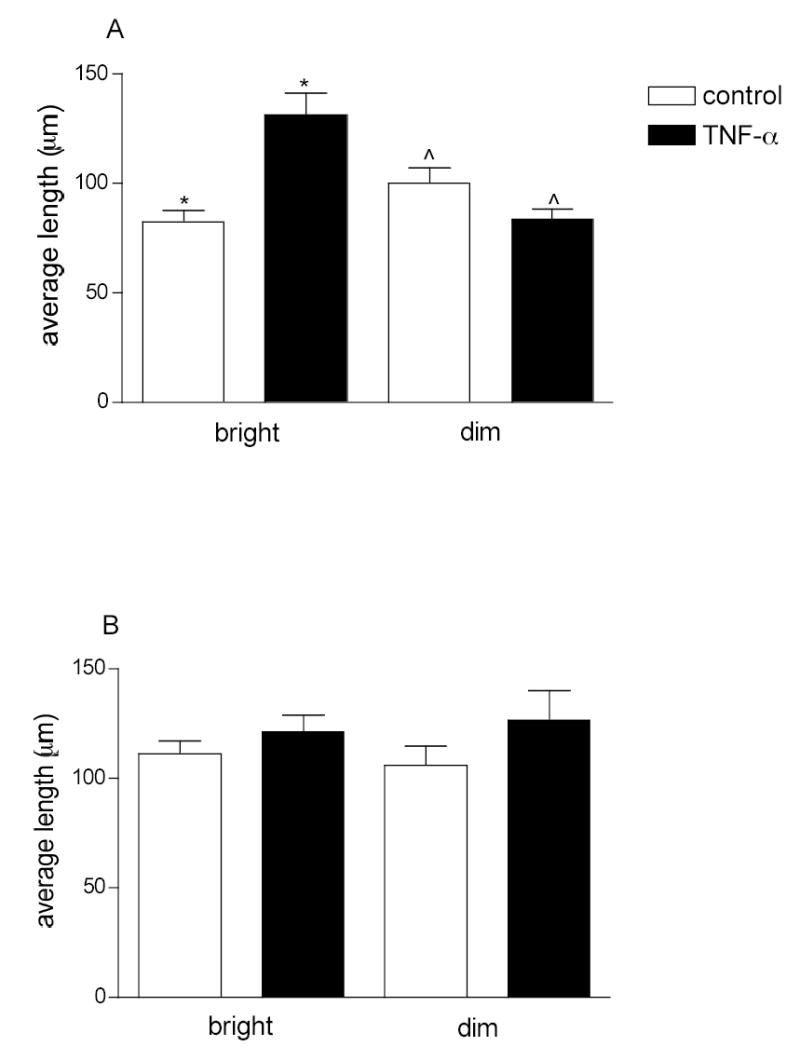
Arterioles (A) and venules (B) were labeled for ICAM-1. Fluorescence intensity along the vessel wall was quantified and pooled into bright and dim regions using predetermined criteria (see Methods). The data are presented as average lengths of sampled bright and dim regions (n=10–12 vessels). Length distribution was normalized to the total sampled length of the vessel. TNF-α treatment significantly increased the average length of the bright regions in arterioles but it had no effect on the average lengths of either bright or dim regions in venules.
*, ^ significantly different from each other (Anova, p<0.01).
To determine the overall expression of ICAM-1 on the vessel wall we quantified the percentage of bright pixels per 1 mm length of vessel wall. We found that under normal conditions more than half (63%) of the venular wall length was bright, whereas only 42% was classified as bright in arterioles (Fig 4). Moreover, in addition to the length difference, the intensity of these bright regions in venules was substantially higher than in arterioles (0.65 vs 0.30 intensity units normalized to FITC-dextran in venules vs arterioles respectively). The finding that more of the ECs lining the venular wall expressed higher levels of ICAM-1 and that these expression levels were on average 2-fold higher that those in arterioles likely explains the occurrence of leukocyte adhesion in venules and not in arterioles under control conditions (Fig 5).
Figure 4. TNF-α treatment increased the length of the bright regions in arterioles but not in venules.
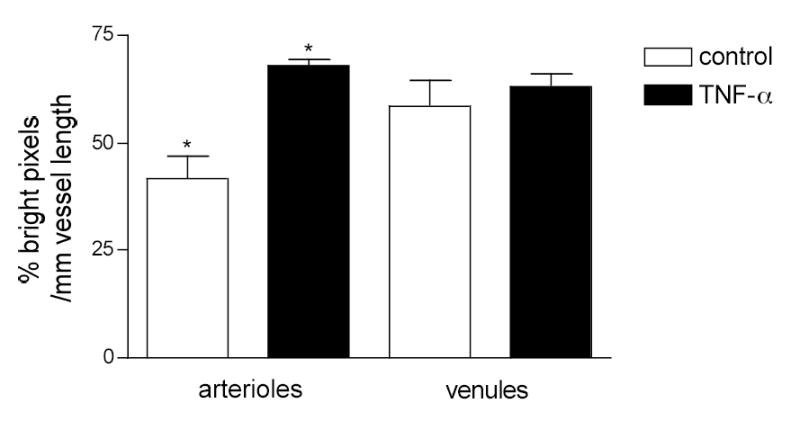
Arterioles and venules were labeled for ICAM-1. The percentage of bright pixels per mm length of vessel wall was quantified in control and TNF-α treated arterioles and venules.
* TNF-α significantly increased (p<0.05) the length of the bright regions in arterioles but not in venules. (n=20 vessels per group)
Figure 5. Delivery of leukocytes to the vessel wall following TNF-α treatment is the same in arterioles and venules but adhesion is different.
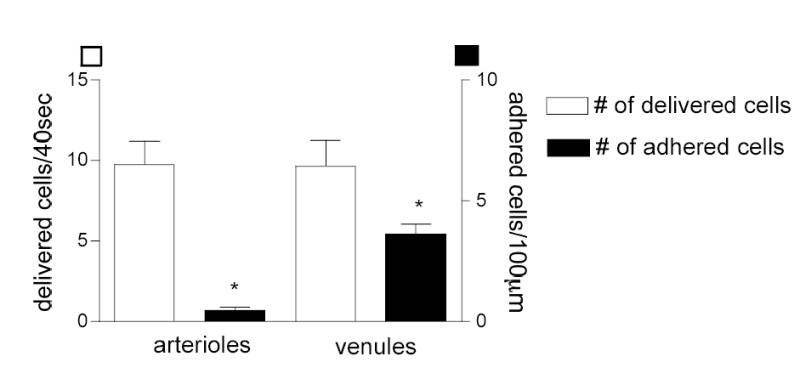
Mouse cremaster muscle was treated locally with TNF-α four hours prior to data collection. Delivered flux (visible fraction of leukocytes near the wall/40 sec) and the number of adhered leukocytes (leukocytes/100 μm vessel wall) were quantified in both arterioles and venules. The number of adhered leukocytes in venules is significantly higher than in arterioles (* significant different from each other, p<0.05) but the potential number of interactions (i.e. the delivered cells) is not different between the two vessel types (n=10 vessels per group).
ICAM-1 expression after activation with TNF-α
Acute inflammation results in upregulation of selectin molecules expressed by the ECs (10) and increased leukocyte-ECs interactions (12, 14, 16, 29). The overall expression of other adhesion molecules such as ICAM-1 also increases following proinflammatory stimuli (19) but the expression patterns and location of that increase is not known and is addressed in these experiments. In mice pretreated with TNF-α four hours prior to data collection (see Methods), ICAM-1 expression was quantified as described earlier. As expected, following TNF-α treatment ICAM-1 expression in venules significantly increased by about two fold (1.7±0.2 fold, P < 0.05), whereas, interestingly, in arterioles it was increased even more (2.8±0.2 fold) (Fig 2). In arterioles the increase in ICAM-1 expression was a result of an overall increase in the bright regions per unit vessel length (42% in control vs. 70% in TNF-α) which corresponded to an increase in the average length of the individual bright regions (from 83±5.4 to 130±9.7 μm, n=10). Thus these data imply that in arterioles, new ECs that in control conditions did not express much ICAM-1, were activated by TNF-α treatment, resulting in there being a greater number of cells expressing high levels of ICAM-1. In contrast to arterioles, the total length that was bright per unit vessel length in venules remained unchanged (~ 63%) and the average length of bright regions in venules was not significantly altered (111±5.9 vs 122±6.1 μm), suggesting that the cells that expressed ICAM-1 in control conditions were the ones that upregulated their expression levels with TNFα stimulation.
To further explore the ICAM-1 expression patterns among individual ECs, we asked how likely it was that adjacent ECs would have similar ICAM-1 expression. To do this, we quantified fluorescence in adjacent pairs of ECs by positioning the focal plane on the top of the vessel where individual ECs could be observed (Fig 1D,E). We found (Fig 6) that in venules under control and TNF-α activated conditions ICAM-1 expression levels were similar (i.e. within 2 standard deviations, as described in Methods) in most of the sampled ECs (85% similar in controls and 80% with TNF-α). For comparison, we also sampled individual ECs from throughout the tissue and compared randomly assigned pairs (Fig 6). The same experiment in arterioles showed contrasting results. In control mice, most (72%) of the adjacent ECs expressed similar levels of ICAM-1, whereas TNF-α treatment resulted in greater variability of ICAM-1 expression among ECs, with 46% of sampled adjacent pairs of ECs expressing different levels of ICAM-1 from each other (Fig 6).
Figure 6. ICAM-1 expression between adjacent ECs in arterioles but not in venules is significantly different following TNF-α.
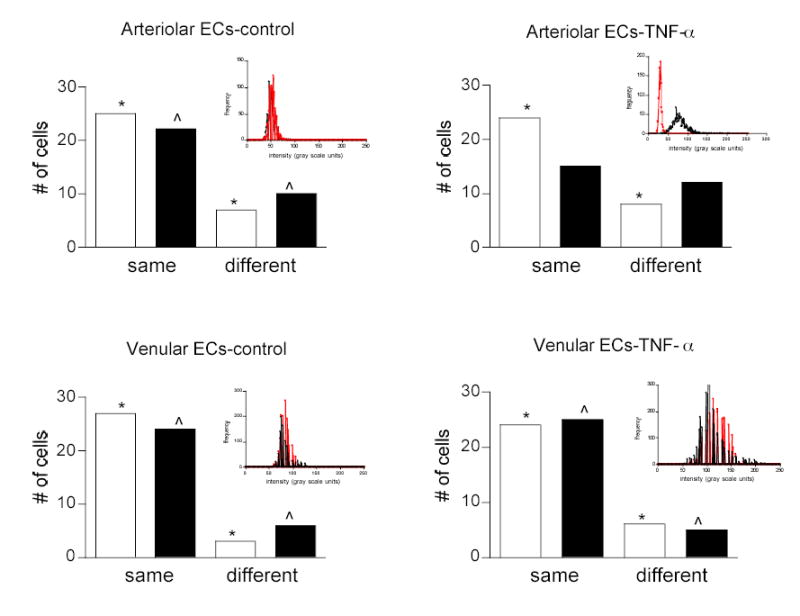
Arterioles and venules were labeled for ICAM-1. Adjacent pairs of ECs (black bars) were sampled and their average fluorescent intensity was quantified and compared. To establish the extent to which adjacent cells could have the same intensity due to random chance, individual ECs were sampled from throughout the tissue, randomly paired (white bars) and their average fluorescence was measured. Cells with intensities within 2 standard deviations were considered to be the same, otherwise they were labeled as different. Following TNF-α treatment, in arterioles but not in venules a different population of ECs was activated resulting in a greater number of adjacent ECs expressing different levels of ICAM-1 *, ^ - mean fluorescence intensity of the cells in each indicated group is significantly different (P < 0.05). Inset are histograms of intensity distribution for a typical pair (one red, one black) of adjacent ECs for each group. Note that in controls, the histograms for the cell pair overlap in both arterioles and venules, whereas following TNF-α treatment in arterioles one cell decreased and one cell increased its ICAM-1 expression compared to venules where both cells increased their ICAM-1 expression.
Thus overall, we conclude that even though most ECs lining the venular wall initially express ICAM-1 these same ECs in response to TNF-α significantly increased their ICAM-1 expression. Arterioles on the other hand, initially expressing much less ICAM-1, underwent redistribution of ICAM-1 among the cells in response to TNF-α, where some cells increased and some cells decreased their ICAM-1 expression, magnifying the variability of ICAM-1.
Differences in shape and size of venular and arteriolar ECs
So far we have learned that in response to TNF-α ICAM-1 expression changes are different in arterioles and in venules. We speculate that one of the reasons for this different response might be different morphological properties of ECs. For example, cells with higher surface area may need much higher expression levels of ICAM-1 to provide the same ICAM-1 density to mediate leukocyte interactions compared to their smaller neighbors: alternatively, expression density might affect redistribution or clustering (time and placing) of ICAM-1 on the cell surface. Thus, we conducted a study to determine the size and the shape of both arteriolar and venular ECs. Since ICAM-1 labels the surface of the ECs and leaves the junctional regions unstained, ICAM-1 labeled ECs have a very clear outline of their borders (Figs 1D and 7). Thus we used ICAM-1 labeled regions as a marker of EC surface area. To confirm that the cell outline was indeed a cell border, in separate experiments we labeled the ECs for the junctional protein VE-cadherin using rat anti-mouse CD144 antibody (#555289, BD Pharmigen, 50 μg/ml) followed by the Alexa488 (goat anti-rat, Molecular Probes, 50 μg/ml) secondary antibody. Length and width of the ECs was measured by projecting a line across the longest major and minor axis (Fig 7): the results are summarized in Table 1. The values of arteriolar EC dimensions measured following ICAM-1 labeling (length 96.9±1.7 μm; width 13.0±0.2 μm n=104) were not significantly different from those measured using VE-cadherin (length 97±2.9 μm; width 12.4±0.5 μm n=42). The agreement in the measured values for EC dimensions by both markers also confirms that ICAM-1 is indeed expressed on the entire surface of the EC. The analysis shows that arteriolar ECs are much longer than venular ECs, with an overall length to width ratio of 7.45 compared to 2.66 in venules; ECs in venules appeared more rounded due to their overall shorter major axis. Also, an approximation of the average EC surface area (average length x average width) showed that arteriolar ECs were almost twice as big as venular ECs (1200 μm2 vs 600 μm2). Thus one explanation for the rarity of adhesion events in arterioles compared to venules may be that because the luminal surface area of arteriolar ECs is roughly twice that of venular ECs, they simply cannot reach sufficient surface density of ICAM-1. The surface area of the EC, and the density of ICAM-1 expressed on that cell, undoubtedly will affect the amount of fluorescent signal we detect and the way that leukocytes interact with that cell.
Figure 7. Representative arteriolar ECs labeled for ICAM-1 (top), VE-cadherin (middle) and ICAM-1 redistributed to the tips (bottom).
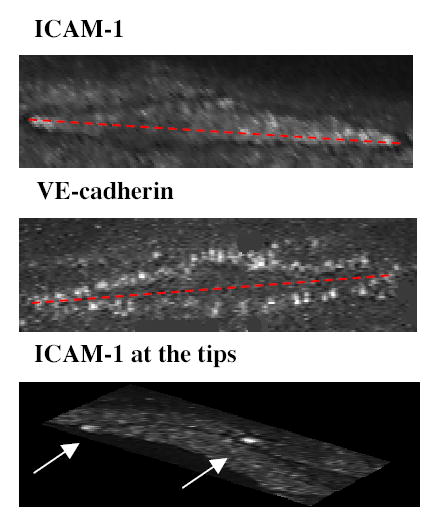
Arterioles were internally perfused with the appropriate primary and fluorescently tagged secondary antibody to separately label for ICAM-1 or VE-cadherin as described in methods. The length and width of the cells were measured by projecting a line across the longest major and minor axis of the EC as illustrated by the dotted red line. Following TNF-α treatment some ECs in arterioles were found to redistribute ICAM-1 to their upstream tips (indicated by arrows).
TABLE 1.
Dimensions of ECs in arterioles and venules.
ECs were immunofluorescently labeled for ICAM-1 (see Methods) enabling borders of ECs to be visualized (see Fig 1, panel D). Length and width of the ECs were measured by projecting a line across the longest major and minor axes (see Fig, 7). Sampled vessels were grouped according to vessel diameter (overall mean diameter, 44.6±3.7 μm).
| Vessel type | Vessel diameter (μm) | EC length (μm) | EC width (μm) | n |
|---|---|---|---|---|
| arterioles | < 30 | 100.3±4.6 | 10.9.3±0.4 | 20 |
| 30 – 50 | 99.7±2.8 | 13.0±0.4 | 50 | |
| > 50 | 90.7±2.9 | 15.1±0.4 | 34 | |
| All EC | 96.9±1.7 | 13.0±0.2 | 104 | |
| venules | < 30 | 40.2±1.9 | 11.3±0.5 | 20 |
| 30 – 50 | 40.5±0.7 | 16.0±0.4 | 47 | |
| > 50 | 40.9±0.9 | 18.2±0.5 | 43 | |
| All EC | 40.5±1.2 | 15.2±0.5 | 110 |
TNF-α induces rolling but not adhesion in arterioles
We next asked how these differences in ICAM-1 expression and EC morphology were reflected in leukocyte function in arterioles vs venules during inflammation. We were able to confirm earlier findings (12, 26, 32) that treatment with the pro-inflammatory cytokine TNF-α increased the number of firmly adhered leukocytes in venules. This treatment also induced leukocyte rolling in arterioles, which again is in agreement with previously published data(14, 29). Surprisingly, we found that despite the dramatic increase in ICAM-1 expression in both arterioles and venules, leukocytes were rarely found to adhere in arterioles. We saw on average 0.4±0.2, (n=10 vessels) adhered leukocytes per 100 μm in arterioles versus 5.5±0.6 (n=10) in venules (Fig 5). The rarity of leukocyte adhesion events in arterioles together with the fact that the delivered fluxes of leukocytes (visible leukocyte fraction near the vessel wall/40 sec) were found to be the same in arterioles and venules (9.66±1.59 and 9.77±1.41 respectively) suggests that the main reason for the low number of adhered leukocytes in arterioles is the adhesion molecule levels and expression patterns rather than the shear environment. This argument is supported by previous work from our laboratory (12) that showed that leukocyte velocity, but not the number of rolling leukocytes (the leukocyte delivery) is shear dependent.
Variability in ICAM-1 expression correlates with leukocyte-EC interactions
Our next goal was to determine whether the observed leukocyte-EC interactions could be accounted for by the expression patterns of ICAM-1 that we observed in venules and arterioles respectively. In order to determine the relation between leukocyte adhesion and ICAM-1 expression in venules, we measured leukocyte interactions systematically in specified regions of the vessel wall where we also quantified the fluorescence intensity. To do this, the vessel wall was broken into a grid of rectangles of 40×15 μm2: this dimension was selected based on the average length and width of an actual venular EC (Table 1). The number of adhered leukocytes and the average fluorescence within each rectangle were correlated and are presented in Fig. 8A. We found that in venules the number of firmly adhered leukocytes increased with increased ICAM-1 expression (r2=0.69, P<0.05). This finding supports the idea that the TNF-α induced increase in the number of firmly adhered leukocytes in venules is mediated via ICAM-1. Interestingly however, adherent leukocytes are still observed in venules in ICAM-1 knockout mice despite that adhesion in each condition is overall significantly reduced (Fig 8B). This indicates that ICAM-1 is not the sole mediator of adhesion in this model, and, importantly, raising the question of what is the specific role of ICAM-1 in mechanisms underlying the inflammatory response. In contrast to venules, in arterioles the pro-inflammatory stimulus resulted in an increased number of rolling but not firmly adhered leukocytes. Since leukocyte rolling on the EC surface is known to be important in the cascade of events leading to leukocyte adhesion and transmigration (17, 25, 27) we quantified the number of rolling leukocytes in areas of different ICAM-1 expression. The number of leukocytes rolling on the vessel wall was calculated by counting leukocytes rolling past a line perpendicular to the vessel axis. ICAM-1 intensity was quantified over a length of 50 μm centered on the same region of the vessel wall. The number of rolling leukocytes in arterioles was significantly higher at the regions of higher ICAM-1 intensity (r2=0.56, P<0.05, Fig. 9A). The same analysis in venules yielded no correlation between the number of rolling leukocytes and ICAM-1 expression (r2=0.11, P>0.05, data not shown). Interestingly, the leukocytes that rolled over the higher ICAM-1 intensity regions in arterioles had characteristically slower rolling velocities than those that rolled over lower intensity regions (Figure 9B), suggesting that some aspect of the adhesion molecule expression that mediates leukocyte rolling differs in a way that is a function of ICAM-1 expression. Thus, although increased ICAM-1 expression failed to support leukocyte adhesion in arterioles as it did in venules, it appears likely that it may be involved in leukocyte rolling in arterioles. In turn this suggests that arterioles can play an important role in the initial responses of the inflammatory cascade.
Figure 8. Effect of ICAM-1 expression on leukocyte adhesion in venules.
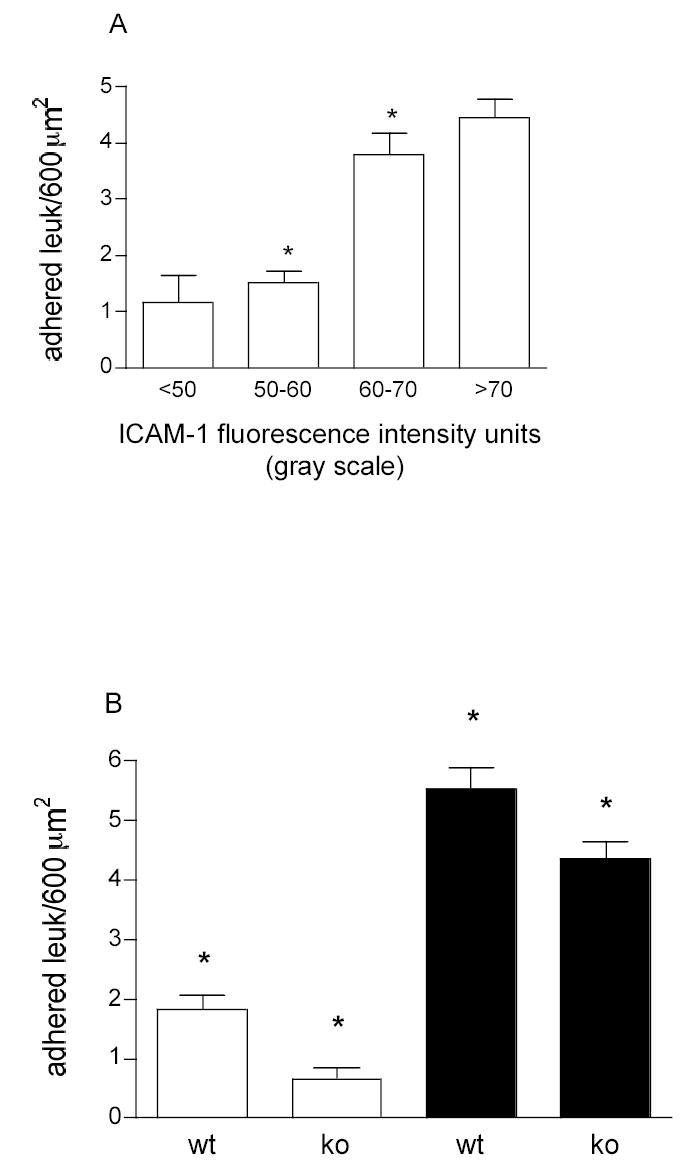
To quantify the number of leukocytes adhered to the upper part of the venules the vessel wall was divided into a grid of rectangles of 40×15 μm2. Adhered leukocytes were counted in these rectangles along the vessel using transilluminated images with the focal plane positioned at the top of the vessel (see panel E, Fig1). Later, ICAM-1 was fluorescently labeled. Fluorescent intensity (ICAM-1 expression) was measured within the rectangles positioned on the same areas where adhesion was quantified. N=23 regions. A. in wild type (WT) animals the leukocyte adhesion increases with the increase in ICAM-1 intensity (r2 = 0.69, p<0.05). B. Under control (white bars) and TNF-α (black bars) the number of adhered leukocytes was quantified in WT and ICAM-1 knockout (KO) mice using a similar approach. While the number of adhesion events significantly decreased in KO mice (P<0.001, Anova), adhered leukocytes were still observed. * Significantly different from each other P < 0.05.
Figure 9. Leukocyte-EC interactions in arterioles are affected by ICAM-1 distribution.
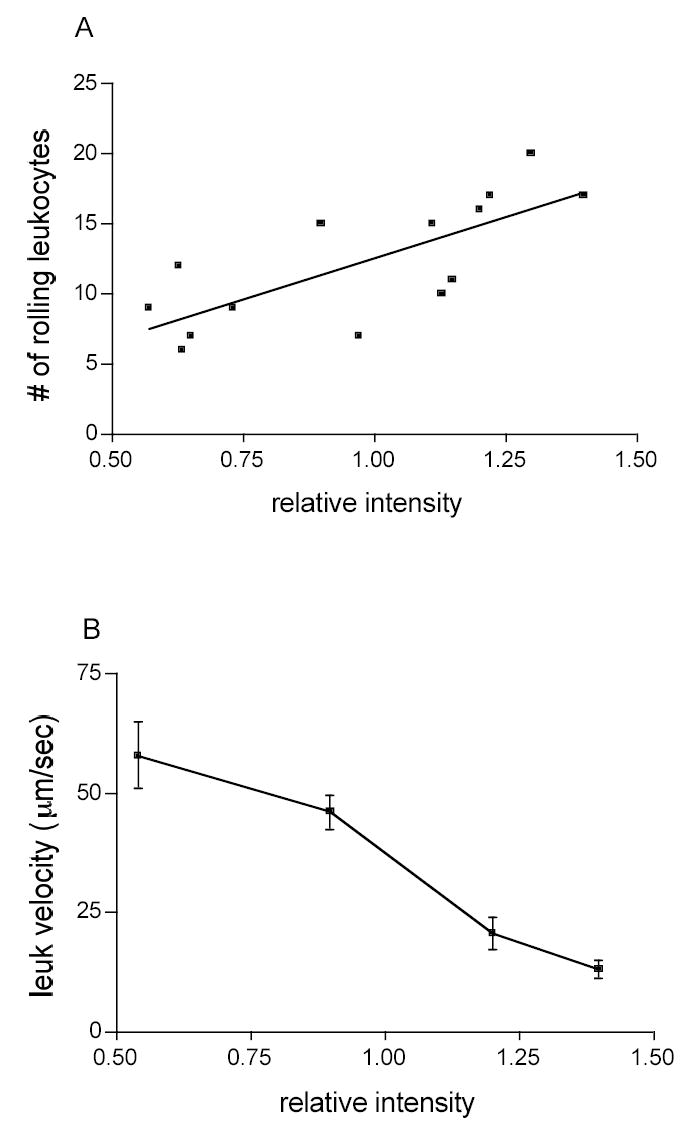
The number and velocity of rolling leukocytes were quantified using transilluminated images and later superimposed onto matching images of fluorescently labeled arterioles. The X-axes represent the values of ICAM-1 fluorescent intensity normalized to the values of fluorescent intensity of FITC-dextran (see Methods). A. the number of rolling leukocytes increased (r2=0.56, P<0.05) with an increase in fluorescent intensity (relative ICAM-1 expression). B. leukocytes that rolled over higher ICAM-1 intensity regions had characteristically slower rolling velocities than those that rolled on lower intensity regions (n = 3 vessels per group).
DISCUSSION
We observed that rolling in both venules and arterioles, and adhesion in venules, were localized to specific regions of the vessel. Our data suggest that the main reason for this is not the shear environment but the distribution of the adhesion molecules. Similarly to previous findings for P-selectin (13), we show that the distribution of ICAM-1 on the EC surface is not uniform. Moreover, ICAM-1 spatial distribution varied within arterioles and venules as well as between these two types of vessels. During inflammation leukocytes are recruited from the blood into the tissue in a series of events that involves leukocyte rolling, leukocyte adhesion and finally leukocyte transmigration. Each of these steps requires multiple regulatory mechanisms and is a necessity for subsequent steps. We speculate that the right timing and placement of leukocyte-EC interactions is a key for an efficient inflammatory response. Leukocyte-EC interactions increase under inflammatory conditions in both venules and arterioles, but, importantly, transmigrating leukocytes were observed in venules but not in arterioles and only at specific regions of the vessel wall (data not shown). Thus our data support the idea that leukocyte-EC interactions are important in both arterioles and venules, but that their contribution to the integrated inflammatory response differs in the two microvascular regions.
Previous studies have shown that ICAM-1 is expressed in a wide variety of tissues, and that in most of these tissues, its expression levels increase during inflammation (21, 30). There is also evidence that in ECs of different origins (such as lung microvascular ECs vs arteriolar ECs) there are differences in the signaling pathways activated by ICAM-1 ligation (30), again suggesting that the differences in expression levels among ECs of different origins are likely reflected in different signaling roles and ultimately in leukocyte-endothelial cell interactions.
When sampling the ICAM-1 distribution we identified two populations of ECs; cells expressing higher and cells expressing lower levels of ICAM-1. This was true for both control and TNF-α activated microvessels. Importantly, the expression patterns, levels, and the response to the proinflammatory stimulus were very different in arterioles compared to venules. Most ECs comprising the venular wall were found to express high levels of ICAM-1 creating long bright regions which are on the order of 2–4 venular ECs in length. Following TNF-α treatment, adjacent venular ECs did not show a significant difference in either ICAM-1 expression or spatial distribution, but the total ICAM-1 expression levels were elevated nearly 2 fold. This indicates that the venular wall can be viewed as regions containing multiple ECs where the majority of these cells exhibit high ICAM-1 expression levels. Following TNF-α stimulation the same ECs further upregulated their ICAM-1 expression. A potential consequence of this extensive surface area of high ICAM-1 expression is a high number of leukocyte-EC interactions in venules in inflammation. On the other hand, arterioles, which do not exhibit significant leukocyte-EC interactions under control conditions, were found to have fewer and shorter regions of high ICAM-1 expression. Furthermore, in contrast to venules, which responded to TNF-α by increasing the ICAM-1 expression levels in those ECs that were already expressing significant levels of ICAM-1, the increased ICAM-1 expression in arteriolar ECs occurred in individual cells that had not been identified during control as significant expressors of ICAM-1. We infer this because we found that in arterioles the increase in ICAM-1 expression was due to an increased percentage of bright length per unit vessel and because the ICAM-1 expression between adjacent ECs in arterioles had significantly changed following TNF-α treatment, indicating that cells that were initially not expressing significant ICAM-1 levels were turned on. Surprisingly we discovered that this variability was a result of increased ICAM-1 expression by some of the ECs and an actual decrease in ICAM-1 expression by others. The cells that apparently decreased their expression were found to be darker than the background intensity; they were confirmed to be viable because they had an intact nucleus by live DNA stain (data not shown). One possible explanation might be that after activation, ICAM-1 internalization rate was increased and that this is one of the self-regulatory mechanisms for leukocyte-EC interactions. Additionally, in arterioles but not venules, some ECs were found to redistribute the ICAM-1 molecules to their upstream tips, again suggesting that regulation of surface expression of ICAM-1 is different in arterioles compared to venules (see Fig. 7). Whether this redistribution to upstream extremities of the ECs is flow dependent is beyond the scope of this study: it is known that ICAM-1 expression can be upregulated in a shear dependent manner (20, 28)
Even though leukocyte-EC interactions were not observed in arterioles under control conditions in this study we emphasize the potential relevance of arterioles in the inflammatory response. In particular, we identified TNF-α induced leukocyte rolling in arterioles, along with the ~3 fold dramatic increase in ICAM-1 expression. Moreover, we found that the number of rolling leukocytes was significantly higher at the regions of higher ICAM-1 intensity and their rolling velocities were characteristically slower than those rolling over the regions of lower ICAM-1 intensity (Fig. 9), suggesting either that ICAM-1 may be directly involved in leukocyte rolling in arterioles, or, alternatively, that it acts in synergy or simply co-localizes with other, more relevant molecules. Further support for the conclusion that ICAM-1 is necessary but not sufficient for leukocyte-EC interactions is that compared to WT mice, mice deficient in ICAM-1 have reduced numbers of adhered and transmigrating leukocyte and significantly increased neutrophil-rolling velocities during inflammation (Fig. 8B, (27)). Fig. 8B shows that, in the absence of ICAM-1 expression, other adhesion molecules mediate a greater fraction of the total leukocyte adhesive interactions during TNF-a activation compared to control conditions, where a greater fraction is mediated by ICAM-1. This further suppports the idea that ICAM-1 acts in concert with other molecules in mediating leukocyte adhesion events, and suggests that the relative importance of the various adhesion molecules might itself be regulated. ICAM-1 is also involved in downstream inflammatory steps, specifically EC Ca2+ signaling as well as rearrangement of inter-endothelial junctions (19). These steps are a part of an essential change in vessel barrier properties to allow transport of both leukocytes and macromolecules into the tissue during the inflammatory cascade. Even though we did not observe leukocyte transmigration from arterioles, the dramatic increase in ICAM-1 expression could be associated with other aspects of barrier function, for example in preliminary studies (23) we have shown that arteriolar permeability increases with TNF-α.
Despite the increase in ICAM-1 expression in arterioles, leukocyte adhesion events were very rare compared to venules (fig 5). There are several possible explanations for this observation. First, an important difference between arterioles and venules is the shear environment. However, we measured shear rates in typical arterioles and found that it was of the order of 400 s−1, which is similar to the venular shear rate measured previously in the same tissue in our laboratory (12). This argues that the lack of adhesion in arterioles is not due to the shear environment. Second, as shown in Fig. 2, the relative intensity of ICAM-1 fluorescence in TNF-α activated arterioles was of the same order as venules under control conditions, thus it is possible that the amount of ICAM-1 expressed by arteriolar ECs is not sufficient to support leukocyte adhesion but is enough to activate ICAM-1 mediated signaling. Third, our estimate of EC surface area in both arterioles and venules suggests that arteriolar ECs are much larger than venular ECs (Table 1). Larger ECs will need higher total ICAM-1 expression to support the same adhesion molecule density (and hence level of interactions) as the smaller, venular ECs. Fourth, there is evidence that under many conditions, leukocyte-EC adhesion may depend less on ICAM-1 and instead involve additional adhesion molecules, such as E-selectin or VCAM-1 (3, 6, 17). As the levels and expression patterns of these molecules in arterioles remain to be determined, it is possible that their levels remained insufficient to provide the necessary environment for leukcocyte adhesion. Finally, given the well established differences in cell and molecular phenotype between arterioles and venules, it appears likely that the regulation and expression of various chemokines will be different, with consequent differences in probability of leukocyte-EC interactions in the two vessel types. Despite the differences in incidence of leukocyte-EC interactions in the two vessel types that we observed, the increased ICAM-1 expression and the induced leukocyte rolling during inflammation argues that arterioles play a role in the inflammatory response.
In summary, this study shows that spatial variations in leukocyte-EC adhesion interactions in venules and leukocyte-EC rolling interactions in arterioles correlates with the expression patterns of ICAM-1 on the endothelial surface. These expression patterns are different in arterioles versus venules. Our findings suggest that arterioles have an important role in the inflammatory cascade. Out study lends further support to the thesis that spatial distribution as well as expression levels of adhesion molecules in the microcirculation will determine the timing and the placement of leukocyte interactions and hence significantly impact the inflammatory response.
Acknowledgments
We thank Ms J.M. Kuebel for her experience and contributions. This work was supported by NIH grants HL 18208 and HL 75186.
References
- 1.Abbassi O, Kishimoto TK, McIntire LV, Anderson DC, Smith CW. E-selectin supports neutrophil rolling in vitro under conditions of flow. J Clin Invest. 1993;92:2719–2730. doi: 10.1172/JCI116889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barringhaus KG, Phillips JW, Thatte JS, Sanders JM, Czarnik AC, Bennett DK, Ley KF, Sarembock IJ. Alpha4beta1 integrin (VLA-4) blockade attenuates both early and late leukocyte recruitment and neointimal growth following carotid injury in apolipoprotein E (−/−) mice. J Vasc Res. 2004;41:252–260. doi: 10.1159/000078646. [DOI] [PubMed] [Google Scholar]
- 3.Chesnutt BC, Smith DF, Raffler NAMLS, White EJ, Ley K. Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation. 2006;13:99–109. doi: 10.1080/10739680500466376. [DOI] [PubMed] [Google Scholar]
- 4.Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99:336–341. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]
- 5.Forlow SB, Ley K. Selectin-independent leukocyte rolling and adhesion in mice deficient in E-, P-, and L-selectin and ICAM-1. Am J Physiol Heart Circ Physiol. 2001;280:H634–641. doi: 10.1152/ajpheart.2001.280.2.H634. [DOI] [PubMed] [Google Scholar]
- 6.Foy DS, Ley K. Intercellular adhesion molecule-1 is required for chemoattractant-induced leukocyte adhesion in resting, but not inflamed, venules in vivo. Microvasc Res. 2000;60:249–260. doi: 10.1006/mvre.2000.2272. [DOI] [PubMed] [Google Scholar]
- 7.Goetz DJ, el-Sabban ME, Pauli BU, Hammer DA. Dynamics of neutrophil rolling over stimulated endothelium in vitro. Biophys J. 1994;66:2202–2209. doi: 10.1016/S0006-3495(94)81016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones DA, Abbassi O, McIntire LV, McEver RP, Smith CW. P-selectin mediates neutrophil rolling on histamine-stimulated endothelial cells. Biophys J. 1993;65:1560–1569. doi: 10.1016/S0006-3495(93)81195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung U, Bullard DC, Tedder TF, Ley K. Velocity differences between Land P-selectin-dependent neutrophil rolling in venules of mouse cremaster muscle in vivo. Am J Physiol. 1996;271:H2740–2747. doi: 10.1152/ajpheart.1996.271.6.H2740. [DOI] [PubMed] [Google Scholar]
- 10.Jung U, Ley K. Regulation of E-selectin, P-selectin, and intercellular adhesion molecule 1 expression in mouse cremaster muscle vasculature. Microcirculation. 1997;4:311–319. doi: 10.3109/10739689709146794. [DOI] [PubMed] [Google Scholar]
- 11.Kim MB, Sarelius IH. Distributions of wall shear stress in venular convergences of mouse cremaster muscle. Microcirculation. 2003;10:167–178. doi: 10.1038/sj.mn.7800182. [DOI] [PubMed] [Google Scholar]
- 12.Kim MB, Sarelius IH. Regulation of leukocyte recruitment by local wall shear rate and leukocyte delivery. Microcirculation. 2004;11:55–67. doi: 10.1080/10739680490266199. [DOI] [PubMed] [Google Scholar]
- 13.Kim MB, Sarelius IH. Role of shear forces and adhesion molecule distribution on P-selectin-mediated leukocyte rolling in postcapillary venules. Am J Physiol Heart Circ Physiol. 2004;287:H2705–2711. doi: 10.1152/ajpheart.00448.2004. [DOI] [PubMed] [Google Scholar]
- 14.Kunkel EJ, Jung U, Ley K. TNF-alpha induces selectin-mediated leukocyte rolling in mouse cremaster muscle arterioles. Am J Physiol. 1997;272:H1391–1400. doi: 10.1152/ajpheart.1997.272.3.H1391. [DOI] [PubMed] [Google Scholar]
- 15.Lau KS, Grange RW, Isotani E, Sarelius IH, Kamm KE, Huang PL, Stull JT. nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiol Genomics. 2000;2:21–27. doi: 10.1152/physiolgenomics.2000.2.1.21. [DOI] [PubMed] [Google Scholar]
- 16.Lawrence MB, Springer TA. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell. 1991;65:859–873. doi: 10.1016/0092-8674(91)90393-d. [DOI] [PubMed] [Google Scholar]
- 17.Ley K, Allietta M, Bullard DC, Morgan S. Importance of E-selectin for firm leukocyte adhesion in vivo. Circ Res. 1998;83:287–294. doi: 10.1161/01.res.83.3.287. [DOI] [PubMed] [Google Scholar]
- 18.Ley K, Gaehtgens P. Endothelial, not hemodynamic, differences are responsible for preferential leukocyte rolling in rat mesenteric venules. Circ Res. 1991;69:1034–1041. doi: 10.1161/01.res.69.4.1034. [DOI] [PubMed] [Google Scholar]
- 19.Lorenzon P, Vecile E, Nardon E, Ferrero E, Harlan JM, Tedesco F, Dobrina A. Endothelial cell E- and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J Cell Biol. 1998;142:1381–1391. doi: 10.1083/jcb.142.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKinney VZ, Rinker KD, Truskey GA. Normal and shear stresses influence the spatial distribution of intracellular adhesion molecule-1 expression in human umbilical vein endothelial cells exposed to sudden expansion flow. J Biomech. 2006;39:806–817. doi: 10.1016/j.jbiomech.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Meyer K, Brown MF, Zibari G, Panes J, McMillan RW, McDonald JC, Granger DN. ICAM-1 upregulation in distant tissues after hepatic ischemia/reperfusion: a clue to the mechanism of multiple organ failure. J Pediatr Surg. 1998;33:350–353. doi: 10.1016/s0022-3468(98)90460-2. [DOI] [PubMed] [Google Scholar]
- 22.Nabah YN, Mateo T, Cerda-Nicolas M, Alvarez A, Martinez M, Issekutz AC, Sanz MJ. L-NAME induces direct arteriolar leukocyte adhesion, which is mainly mediated by angiotensin-II. Microcirculation. 2005;12:443–453. doi: 10.1080/10739680590960962. [DOI] [PubMed] [Google Scholar]
- 23.Sarelius IH, Kuebel JM, Huxley VH. Solute permeability of in situ skeletal muscle microvessels (Abstracct) Faseb Journal. 2005;19:A179. [Google Scholar]
- 24.Sarelius IH, Kuebel JM, Wang J, Huxley VH. Macromolecule permeability of in situ and excised rodent skeletal muscle arterioles and venules. Am J Physiol Heart Circ Physiol. 2006;290:H474–480. doi: 10.1152/ajpheart.00655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon SI, Hu Y, Vestweber D, Smith CW. Neutrophil tethering on E-selectin activates beta 2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immunol. 2000;164:4348–4358. doi: 10.4049/jimmunol.164.8.4348. [DOI] [PubMed] [Google Scholar]
- 26.Smith ML, Olson TS, Ley K. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. 2004;200:935–939. doi: 10.1084/jem.20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steeber DA, Tang ML, Green NE, Zhang XQ, Sloane JE, Tedder TF. Leukocyte entry into sites of inflammation requires overlapping interactions between the L-selectin and ICAM-1 pathways. J Immunol. 1999;163:2176–2186. [PubMed] [Google Scholar]
- 28.Sultan S, Gosling M, Abu-Hayyeh S, Carey N, Powell JT. Flow-dependent increase of ICAM-1 on saphenous vein endothelium is sensitive to apamin. Am J Physiol Heart Circ Physiol. 2004;287:H22–28. doi: 10.1152/ajpheart.00880.2003. [DOI] [PubMed] [Google Scholar]
- 29.Thorlacius H, Lindbom L, Raud J. Cytokine-induced leukocyte rolling in mouse cremaster muscle arterioles in P-selectin dependent. Am J Physiol. 1997;272:H1725–1729. doi: 10.1152/ajpheart.1997.272.4.H1725. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Pfeiffer GR, 2nd, Stevens T, Doerschuk CM. Lung microvascular and arterial endothelial cells differ in their responses to intercellular adhesion molecule-1 ligation. Am J Respir Crit Care Med. 2002;166:872–877. doi: 10.1164/rccm.2201007. [DOI] [PubMed] [Google Scholar]
- 31.Wojciechowski JC, Sarelius IH. Preferential binding of leukocytes to the endothelial junction region in venules in situ. Microcirculation. 2005;12:349–359. doi: 10.1080/10739680590934763. [DOI] [PubMed] [Google Scholar]
- 32.Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–592. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]