

Abstract
T cell activation is characterized by a vast expansion of Ag-specific T cells followed by an equally extensive reduction in T cell numbers. This decline is due, in part, to activation-induced apoptosis of the responding T cells during repeated encounter with Ag. In the current study, we used solid-phase MHC class I/peptide monomers to cause activation-induced cell death (AICD) of previously activated CD8 T cells in an Ag-specific manner. AICD occurred rapidly and was mediated primarily by Fas–FasL interactions. Most interestingly, we observed that Th cells could provide survival signals to CTL significantly reducing the level of AICD. Both Th1 and Th2 subsets were capable of protecting CTL from AICD, and a major role for soluble factors in this protection was ruled out, as cell-to-cell contact was an essential component of this Th-mediated protection. Upon encounter with Ag-expressing tumor cells, CTL underwent significant apoptosis. However, in the presence of Th cells, the CTL not only were protected against death, but also had significantly greater lytic ability. In vivo tumor protection studies using peptide immunization showed that the activation of Ag-specific Th cells was crucial for optimal protection, but did not affect the magnitude of the CTL response in the lymphoid tissues. In this study, we examine the type of help that CD4 T cells may provide and propose a model of Th cell–CTL interaction that reduces CTL death. Our results show a novel role for Th cells in the maintenance of CTL responses.
In the past decade, it has become increasingly evident that CD4 Th cells play pivotal role in CTL immunity. Although this field has received considerable attention, the exact roles of Th cells remain controversial. CD4 T cell help can be classified into two separate temporal categories: During the initial priming phase of the CTL response, and later, during an ongoing CTL response. Although some groups have found that Th cells can enhance priming of CTL responses, particularly for weak Ags (1-3), Th cells are not absolutely required for primary CTL responses (4, 5). During priming Th cells can provide needed cytokines and growth factors (such as IL-2) to CTL to facilitate their expansion during an acute immune response. In addition, through cytokines and cellular interactions such as CD40L/CD40 Th cells can activate APC to up-regulate costimulatory molecules, surface MHC class I (MHC-I)3 and class II (MHC-II) molecules, and to produce cytokines such as IL-12, all of which serve to enhance the stimulatory capacity of the APC (6-8). It also has been proposed that a direct interaction between CTL and Th cells through CD40/CD40L could be important for the development of memory CTL, although other groups have shown that these molecules are not necessary (9). The presence of Th cells during CTL priming is believed to be essential for the formation of CD8 T cell memory responses (4, 5), although there seems to be some exceptions to this rule (10, 11).
Th cells also can provide help for the maintenance of CTL responses to both viruses and tumors (12, 13). In these systems, CTL responses were primed and then followed in the presence and absence of Th cells. The results showed that Th cells were required for full protection during tumor/viral challenge even after CTL priming. Although exact mechanisms for this phenomenon have yet to be defined, Th cells could be facilitating the reduction of the Ag load, altering CTL homing and migration, or providing growth/survival signals to CTL. Previous work by our group has shown that CTL expansion is enhanced in the presence of Th cells due to direct interactions between Th cells and CTL through costimulatory molecules such as MHC-II, 4-1BB, and CD27 (14).
In this study, we analyzed the effect that Th cells may have on activation-induced cell death (AICD) of CTL. After the initial expansion phase of the immune response, most T cells die and the host is left with a small, stable, memory T cell population (15, 16). This Ag-driven death is viewed as a regulatory mechanism to rein in immune responses and return lymphocyte numbers to homeo-static levels. However, AICD can be a double-edged sword. In chronic or high-dose viral challenge models, the immune system is depleted of responding T cells before eliminating the infectious agent (17). In tumor models, anti-tumor responses can be detected but are unable to eradicate the tumors. In both cases, AICD can bring an ongoing immune response to a premature conclusion. If CTL are unable to clear infected cells or tumor before they themselves succumb to apoptosis, then the infection will continue unabated or the tumor will grow unchecked. In this study, we established an in vitro model system of AICD in which we evaluated the ability of Th cells to prevent CTL apoptosis due to AICD. The results show that the presence of Th cells significantly reduced CTL susceptibility to AICD through a cell contact-dependent mechanism, indicating a novel method by which Th cells are able to help established CTL responses through increased CTL survival. The effect this protective function may have on both CTL maintenance and memory is discussed.
Materials and Methods
Mice
All mice were bred and maintained in a barrier facility at the Mayo Clinic (Rochester, MN). DO11.10, OT-I, C57BL/6, B6.lpr, and B6.gld mice were obtained from The Jackson Laboratory. OT-I.lpr mice were produced, bred, and maintained in our facility. 2C mice were provided by L. Pease (Mayo Clinic). TNFR1 and TNFR2 double-knockout (KO) mice were provided by P. Wettstein (Mayo Clinic). Animals were housed and used in accordance with Mayo Clinic Institutional Animal Care and Use Committee guidelines.
Cytokines, Abs, and other reagents
Recombinant human IL-2 (rhIL-2 (Aldesleukin); Chiron) was purchased from the Mayo Clinic Pharmacy. Murine IL-4, IL-10, IL-15, and TNF-α were purchased from PeproTech. Murine IL-12, human (h) LTα1β2, LTα2β1, and TGFβ1 were purchased from R&D Systems. Murine IFN-γ was purchased from Sigma-Aldrich. Human IL-7 was purchased from Endogen. 7-aminoactinomycin D (7-AAD), NG-methyl-l-arginine (l-NMMA), and CFSE were obtained from Molecular Probes. Biotinylated Abs to TNFR1 and TNFR2 were purchased from HBT. All purified and flourochrome-labeled Abs were purchased from BD Pharmingen. TRAIL-Fc, TNFR1,2-Fc, and Fas-Fc soluble fusion proteins for neutralization studies were purchased from Alexis Biochemicals. Butylated hydroxyanisole and N-acetyl-l-cysteine (NAC) were purchased from Sigma-Aldrich. MnTBAP, SB203580, Z-VAD-FMK, and Boc-D-FMK were purchased from Calbiochem. All peptides were purchased from A&A Labs, and stock solutions of 20 mg/ml were made in DMSO plus 0.1% trifluoroacetic acid and maintained in aliquots at −20°C. The following peptides were used in these studies: SIINFEKL (OVA257–264), SIYRYYGL (2C), TEWTSSNVM EERKIVK (OVA265–280), and ISQAVHAAHAINEAGR (OVA323–339). Kb/peptide monomers and tetramers were provided by L. Pease. We used Kb/monomers with the following peptides: SIINFEKL (Kb/OVA) and SIYRYYGL (Kb/SIYR). Lympholyte-M (Cedarlane Laboratories) was used to purify murine lymphocytes according to the manufacturer's protocol.
Generation of T cell cultures
Single-cell suspensions from spleen and lymph nodes of mice were washed once in RBC lysis buffer (0.15 M HCl, 0.1 mM Na2EDTA, 10 mM KHCO3(pH 7.3)), washed once in medium, and then resuspended in IMDM containing 5% FCS (HyClone), 5 × 10−5 M 2-ME, and 10 μg/ml gentamicin (all from Invitrogen Life Technologies). Cells from TCR transgenic mice were then cultured in 24-well tissue culture plates at 3–5 × 106 cells per well with 5 μg/ml their corresponding peptide Ag as well as 50 U/ml rhIL-2. Cells from non-TCR transgenic mice were placed in tissue culture plates coated with plate-bound anti-CD3 and anti-CD28 (1 μg/ml each for 3 h at 37°C) and cultured for 48 h. T cell cultures were then transferred to uncoated plates. CD4 and CD8 T cells were purified using magnetic beads (Miltenyi Biotec) or negative selection columns (R&D Systems) and put back in culture in medium containing 50 U/ml rhIL-2. All T cell populations were used for additional experiments 7–10 days after initial activation.
Cytotoxicity assays
For in vitro tumor restimulation, total splenocytes and lymph node cells were mixed, RBC lysed, and restimulated with irradiated EG.7 cells at a splenocytes-to-tumor cells ratio of 10:1 in the presence of 50 U rhIL-2 for 1 wk. These T cell cultures were then tested for their ability to kill 51Cr-labeled, OVA-expressing, and OVA-negative tumors. Target cells were pretreated with 100 U/ml IFN-γ to up-regulate MHC-I expression for 24 h. Target cells were then washed extensively and loaded with 51Cr for 1.5 h and washed again before use. Target cells were then incubated with various numbers of effector cells for 4 h. 51Cr released into the supernatant was analyzed on a Packard TopCount NXT (PerkinElmer).
Intracellular cytokine staining
T cell responses to Ag stimulation were measured by intracellular cytokine flow cytometry using the CytoFix/CytoPerm kit (BD Pharmingen) according to manufacturer's instructions. OT-I cells were placed in culture wells with or without plate-bound Kb/OVA for 6 h in the presence of monensin (GolgiStop; BD Pharmingen). Where indicated an equal number of CD4 T cells were mixed with CTL for 2 h before activation with Kb/OVA. Surface staining with anti-CD8 mAb (53.6-7) was performed for 20 min at 22°C. Cells were then fixed and permeabilized, and intracellular cytokine staining was performed with anti-IFN-γ mAb (clone XMG1.2) for 30 min at 4°C. Cells were then washed and fixed with 0.1% formaldehyde and analyzed by flow cytometry.
Apoptosis induction and flow cytometry analysis
Viable, activated T cells were isolated using Lympholyte-M and, as necessary, a subsequent passage through the Dead Cell Removal kit (Miltenyi Biotec) according to the manufacturer's instructions. Where necessary, T cells also were purified using magnetic beads as described above. T cells were then rested for 24 h in medium containing 50 U/ml IL-2. For mixed cell experiments, one cell type was then stained with 0.5–1.0 μM CFSE for 10 min at 37°C in medium without FCS and washed with complete medium. Twenty-four- or 48-well plates were coated for 2.5 h at 37°C with Kb/monomers at a concentration of 0.2 μg/ml unless otherwise noted. When used, inhibitors, cytokines, Abs, and/or Th cells were preincubated with the CD8 T cells for 2 h before transfer to Kb/monomer-coated plates. When plate-bound Abs were used, tissue culture plates were coated first with Kb/peptide monomers as described and then coated for an additional 3 h at 37°C with 1–10 μg/ml purified Ab. A total of 5–10 × 105 CTL cells per well was then cultured overnight in the Kb/monomer-coated plates in medium containing rhIL-2. Cells were then collected, stained with annexin V and 7-AAD, and analyzed on a FACScan (BD Biosciences). Twenty thousand CD8 T cells were routinely counted for each sample. To compare results between experiments, a normalized value (percentage specific death) was calculated as follows: ((percentage death in sample) − (percentage spontaneous death))/((percentage maximal death) − (percentage spontaneous death)) × 100.
Immunization and tumor challenge
Unless otherwise noted, C57BL/6 mice received nine daily injections of 100 μg of CpG 1826 (TCCATGACGTTCCTGACGTT) (CpG motifs in boldface) in PBS in the nape of the neck. On day 5, mice received peptide/protein along with the CpG in an IFA emulsion in the nape of the neck. Mice were routinely immunized with 50 μg of CTL peptide or OVA protein, 90 μg of appropriate Th peptides. For tumor measurements, mice were challenged at the indicated time points with 5 × 104 B16-OVA cells s.c. in the right flank. Tumor growth was measured every 3–4 days once tumors became palpable. Tumor size was measured by two perpendicular measurements of tumor diameter with spring-loaded calipers (The Dyer Company). In accordance with Mayo Clinic Institutional Animal Care and Use Committee policies, tumor-bearing animals were euthanized when tumors became ulcerated, mice became moribund, or when one of the two tumor diameter measurements was >20 mm. Both euthanasia and death due to tumor are considered equivalent end points.
ELISPOT assays
CD8 T cells were purified using Ab-coated magnetic beads (Miltenyi Biotec), and serial dilutions of the purified T cell cultures were mixed with peptide-pulsed/unpulsed EL-4 cells or with EG.7 cells (OVA-transfected EL4 cells) on plates coated overnight with anti-IFNγ Ab (Mabtech). ELISPOT plates were cultured for 48 h, washed gently, and incubated with a biotinylated anti-IFNγ Ab (Mabtech). Spots were developed using an avidin-peroxidase enzyme kit (ABC kit; Vector Laboratories) with 3-amino-9-ethylcarbazole as a substrate.
Statistical analyses
The p values for all comparisons between two groups were calculated using Student's t test. The lytic activity was compared using a two-way ANOVA. For survival experiments, Kaplan-Meier survival curves were plotted for each experiment and differences between groups were analyzed with the multiple comparison test using the Holm-Sidak method. In all cases, values of p < 0.05 were defined as significant.
Results
Kb/peptide monomers induce apoptosis of activated CD8 T cells
AICD is the result of repeated or continuous antigenic stimulation, which has been mimicked by using Abs to cross-link components of the TCR, such as the TCR-α or TCR-β chains or the CD3 signaling complex. Ongoing work in our laboratory examining the role for CD4 Th cells in CTL responses in a tumor model has indicated that the presence of Th cells during antigenic restimulation promotes CTL survival. To further explore this phenomenon, we developed an Ag-induced in vitro model of AICD. The initial goal was to establish a system of AICD what was devoid of APC but that would mimic MHC-peptide recognition by the TCR. To accomplish this, we tested the ability of plate-bound peptide-MHC-I monomers (Kb/OVA257–264) to induce apoptosis of previously activated CD8 T cells from OT-I TCR transgenic mice. As measured by Annexin-V/7-AAD staining, Kb/OVA257–264 monomers were found to be as effective as anti-CD3 mAb in inducing AICD of activated Ag-specific CTL from TCR transgenic OT-I mice (Fig. 1A). Similar results were obtained in numerous other apoptosis detection assays, including YO-PRO, a marker of cell membrane integrity; (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl-2H-tetrazolium) MTS cell viability assay, TUNEL, and by detection of activated caspase-3 using either mAbs or fluorescent caspase substrates (data not shown). In contrast with anti-CD3 Abs, which stimulate T cells in an Agnonspecific manner, Kb/peptide monomers should, in principle, induce AICD in an Ag-specific manner. Nevertheless, to confirm the Ag specificity of the Kb/peptide monomers in the induction of AICD, previously activated CTL from two different TCR transgenic mice, were restimulated with two types of plate-bound Kb/peptide monomers. In addition to OT-I cells, we also included in these experiments T cells from the 2C TCR transgenic mice, which recognize the SIYRYYGL (SIYR) peptide, which also is presented by the Kb molecule. The capacity of Kb/SIYR and Kb/OVA monomers to induce AICD in OT-I and 2C T cells was evaluated. As shown in Fig. 1B, 2C CD8 T cells were killed by Kb/SIYR, but not Kb/OVA, monomers, whereas the OT-I CD8 T cells died in the presence of Kb/OVA, but not with Kb/SIYR monomers. This type of AICD occurred only in previously activated CTL, because naive CTL from OT-I mice stimulated with Kb/OVA257–264 monomers failed to undergo apoptosis (Fig. 1C). The high background death observed in the naive T cell cultures was likely due to a lack of appropriate cytokines and growth factors. These cells were cultured in medium supplemented with IL-2 only, and it is likely that the lack of IL-7 contributed to the reduced viability (18). Nevertheless, we did not detect increased naive OT-I T cell death in the presence of Kb/OVA257–264 monomer. As shown in Fig. 1D, the use of tumor cells (B16-OVA) instead of plate-bound Ag also caused CD8 T cell death in an Ag-specific manner, indicating that peptide/MHC monomers correctly mimic cellular Ag presentation to effector CTL.
FIGURE 1.

Kb/peptide monomers induce AICD of CD8 T cells. A, Kb-OVA257–264 or anti-CD3 were coated onto 24-well plates at 1 μg/ml for 2.5 h at 37°C. Previously activated CD8 T cells from OT-I mice (see Materials and Methods) were cultured overnight alone or in the presence of either Kb/OVA or anti-CD3 Ab. T cells were then collected and stained with Annexin-V and 7-AAD and analyzed by flow cytometry. B,Kb/OVA257–264 or Kb/SIYR were coated as above and activated CD8 T cells from OT-I and 2C mice were cultured overnight in the presence or absence of the indicated monomer. The percentage of live OT-I (□) and live 2C (■) cells is shown. C, Previously activated CD8 OT-I T cells were purified by magnetic separation and naive OT-I CTL were purified using a CD3+CD62L+ T cell selection column. Both cell types were cultured overnight in the presence or absence of Kb/OVA and analyzed by flow cytometry as above. The percentage of live naive (■) and activated (□) CD8 T cells is shown. Results are representative of three separate experiments. D, A total of 1 × 106 OT-I cells were labeled with CFSE and were then cultured alone or in wells containing confluent monolayers of the indicated tumor cell lines or in wells precoated with Kb/OVA (0.2 μg/ml) overnight. (□), Conditions in the absence of Ag; (■), conditions with the relevant Ag (OVA). Cells were then collected and stained with Annexin-V and 7-AAD and analyzed by flow cytometry. E, Kb/OVA monomers (0.2 μg/ml) were coated onto 24-well plates at for 2.5 h at 37°C. A total of 2 × 106 OT-I cells was incubated for2hin culture medium in the presence or absence of the various inhibitors, and then half of the cells were transferred to wells containing Kb/OVA or anti-CD3. The remaining cells were incubated overnight without Kb/OVA. CI = BOC-FMK (60 μM), anti-FasL (5 μg/ml), and TNFR:Fc (5 ng/ml). After overnight incubation cells were collected and analyzed as described. F, Activated CD8+ T cells from wt and lpr mice were restimulated with a suboptimal dose of anti-CD3 (0.1 μg/ml) alone (■), with 5 μg/ml soluble FasL protein and 1 μg/ml enhancer protein (□), or with 50 ng/ml rTNFα ([unk]). After overnight culture, T cell viability was assessed as described. One representative experiment of three is shown. The percentage specific cell death calculated as follows: ((percentage death in sample − percentage spontaneous death)/(percentage maximal death − percentage spontaneous death)) × 100. Spontaneous death refers to the respective treatment wells without Kb-OVA257–264 or anti-CD3. Maximal death is determined by cells treated overnight with mitomycin C. Values of p are listed for treatments that differ significantly from untreated (medium alone) group. Results are representative of at six separate experiments.
AICD in CD4 Th cells is considered to occur mostly via Fas–FasL interactions (19). On the other hand, there is considerable debate concerning which death receptors are responsible for AICD of CD8 T cells. FasL, TNFR1, TNFR2, and TRAIL have all been implicated to some extent in AICD of CD8 T cells (20-24). Additionally, AICD in CTL can occur in the absence of death receptor ligation, by the action of reactive oxygen intermediates (25) and through nonapoptotic pathways (26). We used two approaches to understand better the role that these pathways could play in the apoptotic cell death seen in our model system. The first approach was to use inhibitors to block specific pathways. In our experimental system, AICD induced by Kb/OVA257–264 monomers could be inhibited by either broad-spectrum caspase inhibitors (CI) or blockers of the Fas/FasL pathway, while an inhibitor of TNF-α (TNFR-Fc fusion protein) had no noticeable effect (Fig. 1E). Additional inhibitors of cell death pathways also were tested, and the results are summarized in Table I. Broad-spectrum CI, such as Z-VADFMK and Boc-D-FMK, blocked almost 40–50% of the AICD induced by Kb/OVA257–264 monomers. Similarly, other known inhibitors of Fas-mediated cell death such as anti-FasL Abs or soluble Fas fusion proteins were also able to block ∼35% of CTL AICD. In contrast, both NAC, an antioxidant known to decrease FasL expression on T cells, and MnTBAP, a superoxide dismutase mimetic capable of inhibiting AICD in other systems (27), showed only a modest protective effect, blocking ∼7% of CTL death. Similarly, other free radical inhibitors, such as butylated hydroxyanisole and l-NMMA, had no appreciable effect on CTL viability. Neither neutralizing Ab to TNF-α nor soluble rTNFR proteins were able to protect CTL AICD. We also observed reduced CTL death in the presence of a soluble TRAIL-Fc fusion protein; however, the protection observed was highly variable and did not meet the cutoff for statistical significance (Table I). We also tested whether the addition of exogenous TNF-α or a soluble death-inducing FasL construct could enhance AICD. Activated wild-type (wt) and lpr T cells were restimulated with a suboptimal dose of anti-CD3 mAb in the presence of soluble FasL or TNF-α. Although TNF-α had no effect, the addition of soluble FasL greatly enhanced death of wt, but not lpr, CD8 T cells (Fig. 1F). We also tested several of these inhibitors using tumor-cells as Ag instead of immobilized Kb/OVA monomers with similar results (data not shown). Namely, both neutralizing Abs to FasL and CI blocked CTL death, while the soluble TNFR-Fc protein had no effect. These results provide further evidence that our in vitro system is correctly modeling the death-inducing environment likely seen in vivo between CTL and tumor or infected cells.
Table 1.
Effect of death inhibitors on CTL AICD
| Inhibitor Type | Inhibitora | % AICD Inhibitionb | pc |
|---|---|---|---|
| Caspases | BOC-D-FMK | 43.6 ± 11.6 | 0.001* |
| ZVAD-FMK | 27.9 ± 10.1 | 0.002* | |
| Fas/FasL | Anti-FasL | 36.9 ± 13.2 | 0.001* |
| Fas:Fc | 35.9 ± 12.8 | 0.006* | |
| NAC | 7.5 ± 1.9 | 0.01* | |
| TNFα/TNFR | Anti-TNFα | 7.4 ± 8.3 | 0.3 |
| TNFR-Fc | −2.0 ± 1.1 | 0.15 | |
| TRAIL/TRAILR | 23.2 ± 16.5 | 0.07 | |
| O2/N2 Radicals | MnTBAP | 7.0 ± 4.1 | 0.04* |
| BHA | 1.9 ± 6.6 | 0.9 | |
| L-NMMA | −2.4 ± 1.3 | 0.4 |
ZVAD- and BOC-D-FMK are broad-spectrum CI. Anti-FasL (MFL3) and anti-TNFα (TN3 19.12) are neutralizing monoclonal Abs. Fas-Fc, TNFR-Fc and TRAILR-Fc are soluble fusion proteins with neutralizing activity towards FasL, TNF-α, and TRAIL respectively. NAC is an antioxidant known to reduce levels of FasL on T cells. MnTBAP is a superoxide dismutase mimetic and peroxynitrite scavenger. L-NMMA is a competitive inhibitor of nitric oxide synthase. Butylated hydroxyanisole is a phenolic antioxidant known to protect against TNF-α-mediated cytotoxicity. In each case, CTL were preincubated with the inhibitors for 2 h prior to overnight incubation in the presence of Kb-OVA257–264.
Percentage inhibition of AICD is determined as follows: (100 – (monomer-induced death with inhibitor/monomer-induced death without inhibitor) × 100. Numbers represent the average of triplicate samples ± SD of the mean.
Values of p were calculated using Student's t test. Reagents that significantly blocked CTL death (p < 0.05) are marked with an asterisk. The table lists combined results from at least four experiments per inhibitor.
Another approach to examine the role of Fas and TNF-α in AICD of CTL is to use T cells from mice lacking Fas or TNFR1 and TNFR2 and test their susceptibility to AICD. Because both of the TNFR have been shown to participate in AICD, we used double-KO mice lacking both the p55 and the p75 TNFR. Previously activated CD4 and CD8 T cells from TNFR double-KO mice showed similar sensitivity to anti-CD3 mAb-mediated AICD as cells derived from wt mice (data not shown). To test the role of Fas–FasL interactions, we also used the lpr and gld mice, which have mutations in Fas and FasL respectively (28, 29). When restimulated with anti-CD3, T cells from both lpr and gld mice show significant reduced apoptosis, compared with the wt controls (Fig. 2A). Similarly, CD8 T cells from OT-I mice bred onto the lpr background were less susceptible to Kb/OVA monomer-induced AICD than were normal OT-I cells (Fig. 2B).
FIGURE 2.

CTL AICD involves the Fas/FasL pathway and can be prevented by the addition of CD4+ Th cells without affecting CTL activation. A, Activated CD8 T cells were isolated from bulk lymphocyte cultures from B6 wt, lpr, or gld mice by magnetic bead separation and cultured in the presence or absence of anti-CD3 (1 μg/ml, precoated for 2.5 h at 37°C). Percentage of specific cell death was measured as explained in the legend to Fig. 1. B, Activated, CD8 T cells from OT-I and OT-I lpr mice were purified as above and cultured in the presence or absence of plate-bound Kb/OVA monomer (0.2 μg/ml precoated for 2.5 h at 37°C. After overnight culture, T cell viability was assessed with Annexin-V and 7-AAD and analyzed by flow cytometry. One representative experiment of three is shown. C, Kb/OVA257–264 monomers were coated onto 24-well plates at 1 μg/ml for 2.5 h at 37°C. Previously activated, CD8 T cells from OT-I mice were rested for 2 h with or without an equal number of previously activated CD4 T cells from DO11.10 mice. Half of each T cell culture was transferred to Kb/OVA257–264-coated plates and the other half was placed in wells without Kb/OVA257–264. All cells were incubated overnight, and cultures were then collected and analyzed as described in Materials and Methods. Th cells derived from OT-II and B6 mice gave similar results (Fig. 2F and data not shown). D, Coculture of CTL with Th cells does not block effector function of previously activated CTL. CTL were incubated with 51Cr-labeled B16 or B16-OVA targets in the presence or absence of activated Th cells. After 4 h of incubation, supernatant was analyzed for cytotoxicity using a 51Cr release assay. Tumor cell lysis in the presence of Th cells only was <5% (data not shown). Data are representative of two separate experiments. E, Activated OT-I T cells were incubated with (□) or without (■) solid-phase Kb/OVA257–264 monomers for 6 h, then stained for intracellular IFN-γ expression as described in Materials and Methods. F, CD4 Th cells were purified from bulk T cell cultures from B6 wt and lpr mice. CD8 OT-I cells were preincubated for 2 h with B6 (□), lpr ([unk]), or without Th cells (■), then cultured overnight with the indicated concentrations of Kb/OVA257–264. Percentage specific cell death was measured as described in Fig. 1 legend. Both B6 and lpr Th cells were equally efficient in reducing CTL AICD.
Effect of Th cells on CTL AICD
To determine whether Th cells would influence CTL susceptibility to AICD, activated CD4 T cells were added to previously activated CTL, and the mixed cultures were then restimulated with Kb/peptide monomers. In these circumstances, we observed that when Th cells were cocultured with CTL there was a marked reduction in CTL AICD (Fig. 2C). When these experiments were repeated (eight times), the amount of cell death prevented by the addition of Th cells ranged from 25 to 50% of the maximal (data not shown). One explanation for these results was that the presence of Th cells in these cultures reduced CTL death by physically blocking access to the Kb/OVA monomers and thus, preventing CTL activation. To test this possibility, CTL were mixed with or without Th cells (1:1 ratio) and then added to 51Cr-labeled adherent targets (monolayers of B16 or B16-OVA) for a 4-h cytotoxicity assay as described in Materials and Methods. As shown in Fig. 2D, the presence of Th cells in this experiment did not block CTL-mediated lysis, in fact, the lytic activity was significantly enhanced by the presence of Th cells ( p = 0.0006). Th cells do not have direct effects (lysis or up-regulation of MHC-I) on the tumor cells used for this assay (data not shown). Furthermore, IFN-γ production by OT-I CTL upon restimulation by Kb/OVA257–264 monomers was analyzed by intracellular cytokine staining and showed that the presence of Th cells did not block cytokine production by the CD8 T cells (Fig. 2E). OT-I CTL also were incubated with and without Kb/OVA257–264 monomers in the presence and absence of Th cells and then stained for T cell activation markers. As expected,CTL activation markers (CD44 and CD69) were up-regulated upon antigenic restimulation, and the naive cell marker CD62L was down-regulated. The presence of Th cells in these cultures did not significantly alter the pattern of expression of any of these activation markers (data not shown). Together, these results indicate that Th cells do not block CTL AICD merely by inhibiting CTL activation, cytokine secretion, or lytic effector function. Another possible explanation for the protective role of Th cells for CTL AICD is that activated Th cells express Fas on their surface and could serve as decoys for FasL on the CTL by simply absorbing FasL and thereby masking the death signal. To address this possibility, we generated CD4 T cells from BL/6 lpr mice (which lack functional Fas) as well as from wt mice and tested their ability to protect CTL. As shown in Fig. 2F, Th cells from lpr mice were as effective as those from wt animals in protecting CTL from AICD. These data indicate that Th cells are not simply adsorbing FasL to prevent Fas/FasL signaling on CTL.
The role of cytokines in the Th cell-derived survival signals to CTL
Having established that Th cells were able to provide a significant survival advantage to CTL by decreasing the level of AICD, we examined some of the potential mechanisms that could be involved. One hallmark of Th cell function is the production of cytokines. Some of these cytokines are known to affect T cell proliferation, differentiation, survival and effector function (30-34). Most of the members of the IL-2 family of cytokines (IL-2, IL-4, IL-7, and IL-15) have pro- and/or anti-apoptotic effects on T cells. Additionally, Th cells express a wide variety of costimulatory molecules on their cell surface, and the corresponding ligands for some of these receptors are found on CTL (35). Th cells in direct contact with CTL could provide costimulation to the CTL while being stimulated by Ag and could affect AICD.
We initially tested whether addition of IL-2 to the CTL cultures would alter their susceptibility to AICD and found that, even at concentrations 100× higher than normal (5000 U/ml), rIL-2 had no appreciable effect on CTL death (data not shown). We also tested numerous other recombinant cytokines (IL-4, IL-10, IL-12, IFN-γ, TNF-α, TGF-β, LTα, LTβ, and RANK) by adding them to CTL cultures shortly before Kb/OVA monomer restimulation (Table II). The addition of individual cytokines to T cell cultures did not appreciably affect CTL death. Because testing every combination of cytokines was not feasible, we selected another strategy to further study the role of soluble factors in rescuing CTL from AICD. Aside from regulatory T cells, CD4 T lymphocytes can be divided into two subsets based on cytokine secretion: Th1 cells (characterized by IFN-γ secretion) and Th2 cells (producing IL-4 and IL-5). The local cytokine microenvironment present at the time of Ag presentation to a naive CD4 T cell will determine which developmental pathway the responding cells proceed down; thus, in vitro generation of Th1 and/or Th2 populations can be produced by the addition of the appropriate cytokines to the T cell cultures (36, 37). As shown in Fig. 3A, the Th cells typically used in our experiments contained a mixture of IFN-γ producing Th1 cells and IL-4 secreting Th2 cells. To create populations of either Th1 or Th2 cells, we stimulated naive lymphocytes from DO11.10 mice with their appropriate antigenic peptide in the presence of IFN-γ and IL-4 neutralizing Ab to produce Th1 cells, or in the presence of IL-4 and anti-IFN-γ Ab to produce Th2 cells. As shown by intracellular cytokine staining, these priming conditions allowed us to develop relatively pure cultures of the individual T cell subsets (Fig. 3A). Mixed, Th1, and Th2 CD4+ T cells were then cocultured with OT-I CTL in the presence of Kb/OVA257–264 monomers (Fig. 3B). Both Th1 and Th2 Th cells were equally capable of providing survival signals to CTL. In additional experiments, medium from Th cell cultures was centrifuged and filtered to remove Th cells. CTL were then resuspended in the CD4 T cell-conditioned medium before restimulation by Kb/OVA257–264 monomers and found to be as susceptible to AICD as those CTL placed in normal medium (Fig. 3C). All of these cultures had similar levels of CTL death, indicating that soluble factors produced by Th cells were unable to protect CTL. To further test the potential role of Th cell-derived soluble factors in decreasing CTL AICD, we used culture vessels containing semipermeable membranes to physically separate the Th cells from CTL while still allowing diffusion of soluble factors. The CTL were then activated by Kb/OVA257–264 monomers, and viability was assessed 18 h later. As shown in Fig. 3D, Th cells required cell-to-cell contact to protect the CTL against AICD, because the effect was absent in the cultures where the Th cells and CTL were separated by the semi-permeable membranes.
Table 2.
Effect of various recombinant cytokines on CTL AICDa
| Cytokine | Concentration | % AICD Inhibitionb | pc |
|---|---|---|---|
| IL-4 | 15 ng/ml | 5.0 ± 7.7 | 0.32 |
| IL-10 | 10 ng/ml | 11.8 ± 17.2 | 0.31 |
| IL-12 | 0.1 ng/ml | 13.4 ± 13.7 | 0.43 |
| IL-15 | 10 ng/ml | 2.5 ± 19.1 | 0.46 |
| IFNγ | 100 IU/ml | 7.3 ± 13.5 | 0.39 |
| TGFβ | 10 ng/ml | 11.2 ± 15.1 | 0.16 |
| RANK | 1 ng/ml | 1.9 ± 7.1 | 0.54 |
| LTα1β2 | 25 ng/ml | −6.0 ± 11.3 | 0.22 |
| LTα2β1 | 1 ng/ml | 7.7 ± 15.5 | 0.27 |
Kb-OVA257–264 monomers were coated onto 24-well plates at 1 μg/ml for 2.5 hours at 37°C. Previously activated CD8 T cells from OT-1 mice were preincubated with medium alone or with the indicated cytokines for 2 h before overnight incubation on the Kb-OVA257–264-coated plate. Cells were then stained with Annexin-V and 7-AAD and analyzed as described. Each cytokine was tested in at least four separate experiments. This table summarizes data from multiple experiments. Numbers represent the average of two separate experiments ± SD of the mean.
Percentage inhibition of CTL AICD has been calculated to allow comparison between different experiments.
Values of p were calculated using Student's t test
FIGURE 3.
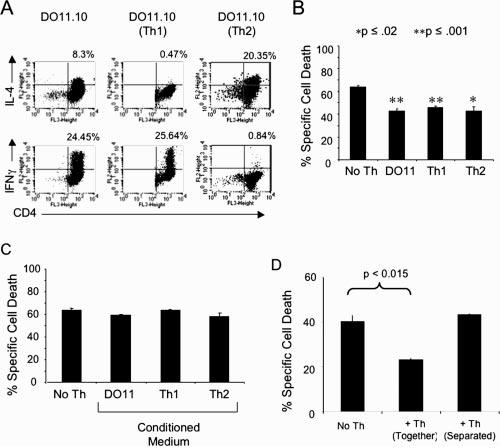
Both Th1 and Th2 cells are able to protect CTL from AICD in a cell contact-dependent manner. A, Lymphocytes and splenocytes from D011.10 mice were cultured under normal conditions or under conditions promoting Th1 or Th2 development (as described in Materials and Methods) for 6 days. An aliquot of Th cells from each condition was then washed and put in fresh medium containing 3 μg/ml Con A (Sigma-Aldrich). Two hours later, GolgiStop was added, and the cultures were incubated for another 4 h. The T cell cultures were collected and stained for intracellular INFγ and IL-4 using BD Pharmingen's Cytofix/Cytoperm kit according to manufacturer's protocol. B, Purified Th cells from the Th1, Th2, and mixed cultures, characterized as described in A, were preincubated for 2 h with CFSE-labeled CTL and then transferred to Kb/OVA257–264-coated wells. After overnight culture, CTL viability was assessed with Annexin-V and 7-AAD and analyzed by flow cytometry. C, OT-I cells were preincubated for 2 h in normal or Th-conditioned medium (medium collected after centrifugation/filtration of Th cell cultures), and transferred to Kb-OVA257–264-coated wells. Percentage specific cell death was measured as described in the legend to Fig. 1. D, Th cells were either placed in culture together with OT-I CTL or were separated by a semipermeable membrane chamber. T cell cultures were then transferred to Kb/OVA-coated plates and incubated overnight. After overnight culture CTL viability was assessed as described above. Data are representative of three separate experiments.
Although costimulation is thought to be an essential component of naive CD8 T cell priming, it is not required for subsequent recognition of Ag-expressing target cells by effector CTL. However, the presence of costimulation upon subsequent Ag encounter could have an impact on the ultimate fate (survival vs death) of the CTL (14, 38, 39). To assess the role of costimulation by Th cells in providing protection against AICD in CTL, we used plate-bound or soluble Abs to cross-link various costimulatory molecules together with Kb/peptide monomers. As shown in Figs. 4, A and B, certain types of costimulation selectively altered CTL viability. Costimulation through CD2, CD48, and, to some extent, CTLA-4, showed a level of protection similar to what had been observed using Th cells. The protective effect of anti-CTLA-4 Abs may be due to dampening of the activating stimulus by the Kb/monomers. These results also showed that costimulation via 4-1BB, CD30, CD27, and OX40 did not have an appreciable effect on CTL death in this model system. Costimulation via CD28 increased the level of CTL AICD, as has been observed previously (40). The various Abs used in the above experiments had similar protective effects when used in either bound or soluble form (data not shown). Although several studies have that direct T cell–T cell interactions through CD40/CD40L are possible, when we tested both plate-bound Abs to CD40 and soluble rCD40L proteins, we found that they had no effect on CTL AICD (data not shown). We conducted several experiments using Abs to block cellular interactions by preincubating the Th cells with various Abs to cell surface molecules on the Th cells and found that they were still able to protect CTL (data not shown). These results are not unexpected given the ability of these Abs to protect CTL from AICD in both plate-bound and soluble form.
FIGURE 4.

Costimulatory signals affect both AICD and TCR tetramer staining. Kb/OVA monomer (0.2 μg/ml) was coated onto 24-well plates at for 2.5 h at 37°C, and the plates were then washed 2× with PBS. A, The plates were then coated with 5 μg/ml of the indicated Abs for an additional 2.5 h and extensively washed before the addition of OT-I cells. Isotype-matched rat IgG (■) is the negative control for this experiment. The plates were then cultured overnight before determining the percentage specific cell death (measured as described in Fig. 1 legend). B, Identical experiment as described in A, but these Abs were of hamster origin, so the results are compared with hamster (Ham) IgG (■). Five micrograms per milliliter of the indicated Abs was added to each well concurrently with OT-I cells, and percentage specific cell death was measured as described above. C–E, A total of 1 × 106 OT-I cells was treated with 5 μg/ml anti-CD2 Ab, control Ab (isotype-matched Ig), or Th cells for 2 h before transfer to Kb-OVA-coated plate. After overnight culture cells were stained with PE-labeled Kb/OVA257–264 tetramer (C and E) or with Abs to Vβ5.2 (D).
Although both anti-TCR (and anti-CD3) Abs and tetramers bind to the TCR, there are substantial differences in how they bind. Abs bind to various antigenic locations on the TCR, but more importantly, they bind with very high affinity and do not require any other protein interactions. Tetramers, in contrast, consist of MHCI/peptide complexes and bind to the CDR1, CDR2, and CDR3 regions with much lower affinity than do Ab–Ag interactions. Furthermore, like all other TCR–MHC-I interactions, optimal tetramer binding requires the participation of the CD8 coreceptor. Disruption of lipid rafts reduces tetramer staining but does not alter surface TCR levels, indicating that tetramer binding to TCR requires lipid raft integrity (41).
To determine whether the presence of Th cells alter expression of CTL surface markers, these molecules were analyzed on CTL restimulated in the presence and absence of Th cells. Restimulation of OT-I cells with plate-bound Kb/OVA monomer altered the expression levels of some of surface molecule on CTL. As expected, CD25, CD44, and CD69 were markedly up-regulated, whereas CD62L levels decreased after restimulation. Notably, the levels of Fas, FasL, 4-1BB, TCR-α, TCR-β, and OX40 on CTL did not change in the presence or absence of Th cells (data not shown). Most interestingly, we observed consistent changes in tetramer staining of CTL in the presence of certain costimulatory signals. When CTL were pretreated with plate-bound Abs that block AICD (anti-CD2), tetramer staining of CTL was greatly increased (Fig. 4C), whereas tetramer staining was not enhanced in the presence of isotype controls or Abs to costimulatory molecules that do not affect AICD (4-1BB). In all cases, total TCR levels, assessed by Abs to TCRVβ, remained constant (Fig. 4D). Importantly, the increase in tetramer staining observed with anti-CD2-treated CTL also was observed when the CTL were cultured in the presence of Th cells (Fig. 4E). Because total TCR levels on the CTL did not change under any of these conditions, these results indicate that the TCR molecules already present on the cell surface become more accessible to tetramer (MHC/peptide) binding.
In vivo effects of Th cells in CTL function
Having identified a novel mechanism of CD4 T cell help in ongoing CTL responses, we sought to determine a physiological role for this effect. We used the well-characterized B16-OVA tumor model to examine the role of Th cells in maintaining CTL responses to tumors. We first tested the ability of OVA-specific Th cells to protect mice from B16-OVA. Mice were immunized as described in Materials and Methods, and 12 days after immunization, mice were challenged with B16-OVA subcutaneously on the right flank (Fig. 5A) (42). Tumors in the mice treated with either pan HLA DR-binding epitope (PADRE) or OVA323–339 Th peptides grew with kinetics similar to tumors in the untreated mice. In the OVA265–280 Th (another helper epitope that lies proximal to the OVA CTL epitope)-immunized group, one of five mice never developed tumor and survived for >4 mo after challenge, whereas the remaining mice developed tumors with growth kinetics similar to the other groups. Tumors in this group (OVA265–280) were visible 2–3 days later than in the other groups, and in all groups, the tumor-bearing mice died within 2 wk of developing palpable tumors. Similar results were seen in a repeat experiment, except that all mice in the OVA265–280 group also succumbed to tumor. The differences between the immunized groups were not statistically significant in either experiment.
FIGURE 5.

A, Mice received nine daily injections of CpG and the indicated peptide immunization in IFA on day 5 (see Materials and Methods). One week after immunization, mice were challenged with 5 × 104 B16-OVA s.c., and tumor growth and survival was monitored daily. B, Mice were immunized with peptides as described above. Mice were challenged with tumor for 21 days later. Four weeks after immunization, lymph nodes were isolated, and CD8+ T cells were purified by negative selection and titrated against EG.7 (OVA-transfected EL4) tumor cells in a 48-h ELISPOT assay. Background responses to the parental EL-4 tumor line have been subtracted.
Because we were interested in examining the effect of Th cells on maintaining CTL responses, we next immunized mice with CTL peptide in the presence or absence of various Th epitopes. We allowed 3 wk for the primary immune responses to subside before tumor challenge. Several mice from each group were sacrificed on the day of tumor challenge to determine immune function in each group. CTL responses were quantified using both ELISPOT and chromium release assays. All immunized groups had similar numbers of Ag-specific CTL (Fig. 5B) and had similar lytic activity against Ag-expressing tumor lines (data not shown). Despite the similar levels of cell-mediated immune responses in the primary lymphoid organs, when survival was monitored following tumor challenge, we saw significant differences between groups. As shown in Fig. 6A, nonimmunized animals rapidly succumbed to tumor within 3–4 wk, whereas mice immunized with both CTL and OVA Th peptides showed significantly greater protection than that seen with CTL peptide alone or with CTL peptide and the unrelated PADRE Th peptide. As we did not see differences in CTL responses in the lymphoid organs that would account for the anti-tumor immunity observed, we hypothesize that both tumor-specific CTL and Th cells are acting in a coordinated fashion at the tumor site to mediate rejection, possibly by enhancing CTL expansion or survival. In protected mice, tumors were too small for analysis, and for those tumors that did grow, once they were palpable they consisted mostly of necrotic tissue, which further hampered direct analysis of in situ immune responses. To circumvent this obstacle, we reimmunized mice as before and then pooled the lymph nodes from all animals within each group. We then purified both CD4 and CD8 T cells from the lymph nodes by magnetic beads using negative selection. Instead of in vivo tumor challenge, the CD8 T cells were cultured with EG.7 tumor cells in the presence or absence of CD4 T cells isolated from the same group. Fig. 6B shows the net expansion of these CTL cultures 8 and 17 days after restimulation. Although all of the CD8 T cell cultures initially expanded, only those cultures containing activated CD4 cells (groups immunized with PADRE, OVA323–339, or OVA265–280 peptides) continued to expand beyond day 8. In each culture containing activated Th cells, we also saw fewer dead cells. Additionally, these experiments showed no difference between tumor-specific and irrelevant CD4 T cell help. These results indicate the in vitro presence of Th cells during restimulation with tumor allows for more robust expansion and decreased death of these CTL.
FIGURE 6.

Tumor protection and ex vivo restimulation of CTL. A, Mice received nine daily injection of CpG and peptide immunization on day 5 as described in Materials and Methods. Three weeks after immunization, mice were challenged with 5 × 104 B16-OVA s.c. Mice were regularly monitored for tumor growth and survival. The combined data from two separate experiments with similar results are shown here. B, Mice were immunized with CpG and peptides as described in Materials and Methods. One week later, CD4 and CD8 cells were purified from the pooled lymph nodes of all five mice in each group by magnetic bead separation. Purified CD8 T cells were incubated with (solid lines) or without (dotted lines) an equal number of CD4 T cells from the same immunization group and with irradiated EG.7 cells. All cultures were maintained in complete medium with 50 U/ml rhIL-2. Flow cytometric analysis was done to count live CD8 T cells at the indicated time points. Results are representative of two separate experiments.
Discussion
In this study, we have developed a model system in which to study CTL AICD in an Ag-specific manner. Two lines of evidence overwhelmingly indicate that Fas–FasL interactions are required for CTL death in our system: 1) lpr and gld mice exhibit greatly decreased susceptibility to AICD, and 2) Fas/FasL signaling inhibitors or CI also block AICD. We did not see any decrease in AICD with inhibitors of TNF-α signaling or with TNFR KO mice. Other groups have shown a role for TRAIL in AICD of CTL (23, 24). In our study, we observed inhibition of cell death by blocking the TRAIL pathway but the protective effect of inhibiting TRAIL was variable and did not quite meet the cutoff for statistical significance.
Although our findings are in line with numerous other reports showing that murine CTL are sensitive to Fas/FasL-mediated AICD (43-47), as mentioned in the Introduction, there is considerable controversy over the apoptotic pathways operating on CD8 T cells undergoing AICD. The different results seen by various groups may be related to numerous factors including the type of T cells used (cloned CTL lines vs freshly isolated T cells), the type of stimulation (in vitro vs in vivo, with peptide vs mitogen), and the length of time between priming and restimulation. There also are additional mechanisms that down-regulate T cell responses. Several reports have shown that activated CTL can enter an anergic state termed Ag-induced nonresponsiveness (AINR). These CTL are unable to produce IL-2 yet retain both lytic function and the ability to secrete IFNγ and can be rescued by the addition of exogenous IL-2 (48, 49). These findings are different from our results, which show that CTL are actively dying and that this death cannot be reversed by the provision of exogenous cytokines. These differences highlight the divergent mechanisms used to curb T cell responses. In fact, all CTL cultures in our study were maintained in IL-2, which may have bypassed AINR and allowed the cells to continue to the stage where AICD played a role. In addition, we used CTL cultures 7–10 days after initial stimulation, whereas typical AINR is thought to occur 3–4 days after stimulation. Cytokine deprivation is another circumstance by which T cell responses are shutdown. As Ag becomes limiting, cytokine levels decrease and T cells begin to undergo apoptosis. This mechanism of T cell death has been shown to be Fas independent and to be controlled by Bim, a pro-apoptotic member of the Bcl-2 family (50, 51). Again, the present study may have avoided this type of cell death by the addition of IL-2 to the CTL cultures. This additional program of T cell death further illustrates the redundant mechanisms by which the immune response is shut down. The common theme to each of these pathways is that CTL responses are tightly controlled by the availability of CD4 T cell help. In the case of cytokine deprivation and AINR, Th cells provide the critical cytokines to CTL, whereas in the case of AICD, direct costimulatory signals from Th cells can block CTL death. These different regulatory mechanisms are likely to be involved at different time points along the developmental pathway of an effector CTL, with AINR coming into effect shortly after stimulation to ensure that primary CTL responses to do not undergo additional unnecessary expansion, whereas cytokine deprivation and AICD act later to ensure that immune responses are curtailed at the appropriate time.
A key finding in our study is that the presence of CD4 Th cells at the site of Ag recognition by activated CTL may be able to significantly decrease CTL AICD. This novel mechanism of T cell help may explain, in part, the dependence of both long-term CTL responses and CD8 T cell memory on Th cells. Our results indicated that this protective signal did not affect CTL activation, cytokine secretion or cytolytic activity. One published report indicates that Fas+ B cells are able to protect T cells from AICD by redirecting FasL expression on T cells away from those T cells (52). It was a distinct possibility that the Th cells were acting through a similar mechanism to protect CTL. However, given that both wt and lpr Th cells were equally capable of protecting CTL, the protective effect cannot be explained by Th cells sequestering FasL. Our results indicate that cytokines, either individually or in combination (such as Th-conditioned medium), were unable to alter CTL susceptibility to death, nevertheless we cannot rule out the possibility that they modulate the survival signal provided by direct contact with Th cells. Our data show that cell-to-cell contact was essential for this protective effect, and that mimicking costimulation by the use of plate-bound Abs to certain molecules (CD2 and CD48) also was able to protect CTL from AICD. Blocking experiments to abrogate this cell–cell signaling were inconclusive, and we are not able to pinpoint Th-mediated survival signals to any single cell surface molecule. The Abs used may not block receptor-ligand binding or may bind to CTL once the two cell types are mixed. A more straightforward approach to study the role of these costimulatory molecules would be to use Th cells from receptor or ligand KO mice. However, these experiments would be quite complex because multiple costimulatory molecule pairs can work in concert or synergistically to promote CTL survival.
We should point out that the CD8 and CD4 T cells used in our studies express both CD2 and CD48, so if CD2 or CD48 were solely responsible for the protective effect, it is unclear why CTL could not provide this signal to one another. One explanation for this may come from the fact that we noticed a considerable decrease in CD2 expression on CTL undergoing apoptosis and it may be that the Th cells maintained the high levels of CD2 necessary to provide the protective signal (data not shown). As costimulation is known to aggregate TCR into lipid rafts, we hypothesized that both Th cells and plate-bound Abs to certain costimulatory molecules redistribute the TCR on the CTL surface in a manner where it becomes more accessible for MHC-I peptide binding. We were able to detect significant changes in tetramer binding, and consequently, lipid raft aggregation on CTL in the presence of both Th cells and Abs known to reduce AICD. Such redistribution of TCR on the cell surface would alter TCR signaling events and could be responsible for the increased survival of CTL in the presence of Th cells. The overall results illustrate a novel mechanism of CD4 T cell help for CTL immune responses by providing close-range survival signals, which may promote viral or tumor clearance by prolonging CTL responses at the Ag site. The requirement for close contact between Th cell and CTL is reminiscent of the contacts between Th cell and APC with respect to CD40–CD40L interactions, and more recently, directed cytokine secretion targeted toward the APC (53).
It is clear that more work needs to be done to elucidate the intracellular changes that occur in protected CTL that confer resistance to AICD. Preliminary results have not shown significant changes in key components of the Fas/FasL pathway, and more work needs to be done to shed light on the mechanism(s) of protection. Although our in vitro experiments did not directly address the need for Ag-specific CD4 T cell help, the requirement for close physical proximity between Th cells and CTL would indicate that Ag specificity is essential. This hypothesis is supported by our in vivo tumor protection studies and in published results from other groups showing the necessity of Ag specific Th cells for optimal tumor protection (54, 55). Our in vitro studies clearly showed that CD4 Th cells in close proximity to CTL re-encountering Ag are able to protect the CD8 T cells from AICD (Figs. 2, C and F, and 3, B and D). There are several lines of evidence to indicate that this effect may play a role during in vivo immune responses. First, optimal tumor protection in our in vivo studies was dependent on Ag-specific Th activity (Fig. 6A). Tumor challenge occurred after the peak of the primary immune response, and we did not detect significant differences in either CTL numbers or effector function that could account for the differences in protection. This, as well as the fact that the nonspecific Th epitope, PADRE, did not provide protective immunity, indicates that the improved protection was likely due to a local effect at the tumor site. Although we were unable to examine immune responses directly in the tumor, we do show that in vivo primed CTL, once cultured with tumor cells in the presence of Th cells, exhibit sustained expansion, whereas CTL cultured in the absence of Th cells exhibit a brief expansion followed by significant decrease in cell numbers. The results from our in vitro model indicate that substantial numbers of CTL are undergoing AICD upon encounter with Ag-expressing tumor cells, and that in the presence of Th cells, this AICD is significantly reduced. One outcome of this protective effect is illustrated by the noticeable increase in CTL cytotoxicity in the presence Th cells (Fig. 2D). This enhanced killing effect also is one possible explanation for the improved tumor protection seen with both CTL and Ag-specific Th cells.
Although the exact mechanism of specific protection of CTL by Th cells is still unknown, our data support the following model of protection: Th cells present at the tumor site are able to physically interact with CTL via adhesion and costimulatory molecules. This interaction provides survival signals through costimulatory molecules (such as CD2) to the CTL, leading to lipid raft aggregation and TCR recruitment into the lipid rafts. This, in turn, alters the signals received by the CTL upon interaction with an Ag-expressing target cell. Under normal circumstances, CTL–target cell interactions lead to two signals in the CTL: 1) the release of perforin/granzymes onto the target cell, and 2) a death-inducing stimulus for the CTL. These CTL are able to kill a limited number of target cells before succumbing to AICD. Protected CTL receive additional antiapoptotic stimuli that increase resistance to AICD allowing them to persist at the Ag site. We postulate that these helped CTL would be better able to clear infection/tumor and also may be more likely to generate a CD8 T cell memory pool. These close interactions between Th cells and CTL are most likely to occur in either the lymphatics or at the site of Ag deposition where enclosed space or chemokine gradients have drawn sufficient numbers of both CD4 and CD8 T lymphocytes together for the cell-to-cell contact to occur. The implications of this are far reaching and can have significant impact on the immune responses. This antiapoptotic effect may explain, in part, the need for CD4 T cell help to maintain CTL responses to viruses such as lymphocytic choriomeningitis virus, CMV, HIV, and hepatitis viruses (56-58) as well as the ability of Th cells to contribute to autoimmune disease (59, 60). Immune regulation of CD4 T cell help can, therefore, control the outcome of CTL responses, and the data presented here show that the presence of Th cells can promote CTL longevity, possibly facilitating clearance of the infection and generation of CD8 T cell memory.
Although the importance of CD4 cells in CTL responses is recognized, only a few mechanisms have been put forth to explain their dramatic effects on effector function of CTL. In this study, we have shown a novel mechanism of T cell help. By providing direct survival signals to CTL, Th cells block AICD and increase the functional life span of the CTL response. By allowing more CTL to escape AICD, Th cells also may promote greater CTL memory generation. This mechanism has direct implications for the outcome of immune responses to tumors and pathogens and may be a factor in autoimmune disease as well. Further work is needed to elucidate the exact mechanism(s) by which Th cells protect CTL and to further define the physiological role of this protective effect. Future studies also should examine the duration and timing of the protection provided by Th cells. On one hand, this signal may function similarly to CD40L conditioning of APC. Just as conditioned APC are then able to fully stimulate CTL responses without subsequent need for Th cells, protected CTL may exhibit sustained protection from AICD, enabling them to clear Ag. Alternatively, provision of this protective signal may need to closely coincide with Ag restimulation, thereby providing regulatory control to limit the ability of CTL to escape apoptosis.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported in part by National Institutes of Health Grants P50CA91956, T32AI07425, R01CA103921, and R01CA80782.
Abbreviations used in this paper: MHC-I, MHC class I; AICD, activation-induced cell death; KO, knockout; rhIL-2, recombinant human IL-2; 7-AAD, 7-aminoactinomycin D; l-NMMA, NG-methyl-l-arginine; NAC, N-acetyl-l-cysteine; CI, caspase inhibitor; wt, wild type; AINR, Ag-induced nonresponsiveness; PADRE, pan HLA DR-binding epitope.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Keene JA, Forman J. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J. Exp. Med. 1982;155:768–782. doi: 10.1084/jem.155.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franco A, Tilly DA, Gramaglia I, Croft M, Cipolla L, Meldal M, Grey HM. Epitope affinity for MHC class I determines helper requirement for CTL priming. Nat. Immunol. 2000;1:145–150. doi: 10.1038/77827. [DOI] [PubMed] [Google Scholar]
- 3.Wang JC, Livingstone AM. Cutting edge: CD4+ T cell help can be essential for primary CD8+ T cell responses in vivo. J. Immunol. 2003;171:6339–6343. doi: 10.4049/jimmunol.171.12.6339. [DOI] [PubMed] [Google Scholar]
- 4.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 6.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 7.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 8.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 9.Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297:2060–2063. doi: 10.1126/science.1072615. [DOI] [PubMed] [Google Scholar]
- 10.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat. Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marzo AL, Vezys V, Klonowski KD, Lee SJ, Muralimohan G, Moore M, Tough DF, Lefrancois L. Fully functional memory CD8 T cells in the absence of CD4 T cells. J. Immunol. 2004;173:969–975. doi: 10.4049/jimmunol.173.2.969. [DOI] [PubMed] [Google Scholar]
- 12.Hunziker L, Klenerman P, Zinkernagel RM, Ehl S. Exhaustion of cytotoxic T cells during adoptive immunotherapy of virus carrier mice can be prevented by B cells or CD4+ T cells. Eur. J. Immunol. 2002;32:374–382. doi: 10.1002/1521-4141(200202)32:2<374::AID-IMMU374>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 13.Zajac AJ, Murali-Krishna K, Blattman JN, Ahmed R. Therapeutic vaccination against chronic viral infection: the importance of cooperation between CD4+ and CD8+ T cells. Curr. Opin. Immunol. 1998;10:444–449. doi: 10.1016/s0952-7915(98)80119-2. [DOI] [PubMed] [Google Scholar]
- 14.Giuntoli RL, II, Lu J, Kobayashi H, Kennedy R, Celis E. Direct costimulation of tumor-reactive CTL by helper T cells potentiate their proliferation, survival, and effector function. Clin. Cancer Res. 2002;8:922–931. [PubMed] [Google Scholar]
- 15.Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis: immune regulation in a dynamic and unpredictable antigenic environment. Annu. Rev. Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- 16.Budd RC. Activation-induced cell death. Curr. Opin. Immunol. 2001;13:356–362. doi: 10.1016/s0952-7915(00)00227-2. [DOI] [PubMed] [Google Scholar]
- 17.Wherry EJ, McElhaugh MJ, Eisenlohr LC. Generation of CD8+ T cell memory in response to low, high, and excessive levels of epitope. J. Immunol. 2002;168:4455–4461. doi: 10.4049/jimmunol.168.9.4455. [DOI] [PubMed] [Google Scholar]
- 18.Okamoto Y, Douek DC, McFarland RD, Koup RA. IL-7, the thymus, and naive T cells. Adv. Exp. Med. Biol. 2002;512:81–90. doi: 10.1007/978-1-4615-0757-4_11. [DOI] [PubMed] [Google Scholar]
- 19.Van Parijs L, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity. 1996;4:321–328. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 20.Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 21.Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 22.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumor necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 23.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Lorenzo MJ, Alava MA, Gamen S, Kim KJ, Chuntharapai A, Pineiro A, Naval J, Anel A. Involvement of APO2 ligand/TRAIL in activation-induced death of Jurkat and human peripheral blood T cells. Eur. J. Immunol. 1998;28:2714–2725. doi: 10.1002/(SICI)1521-4141(199809)28:09<2714::AID-IMMU2714>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 25.Williams MS, Henkart PA. Role of reactive oxygen intermediates in TCR-induced death of T cell blasts and hybridomas. J. Immunol. 1996;157:2395–2402. [PubMed] [Google Scholar]
- 26.Davidson WF, Haudenschild C, Kwon J, Williams MS. T cell receptor ligation triggers novel nonapoptotic cell death pathways that are Fas-independent or Fas-dependent. J. Immunol. 2002;169:6218–6230. doi: 10.4049/jimmunol.169.11.6218. [DOI] [PubMed] [Google Scholar]
- 27.Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, Marrack PC. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–744. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 30.Van Parijs L, Refaeli Y, Lord JD, Nelson BH, Abbas AK, Baltimore D. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity. 1999;11:281–288. doi: 10.1016/s1074-7613(00)80103-x. [DOI] [PubMed] [Google Scholar]
- 31.Ohta N, Hiroi T, Kweon MN, Kinoshita N, Jang MH, Mashimo T, Miyazaki J, Kiyono H. IL-15-dependent activation-induced cell death-resistant Th1 type CD8 αβ+NK1.1+ T cells for the development of small intestinal inflammation. J. Immunol. 2002;169:460–468. doi: 10.4049/jimmunol.169.1.460. [DOI] [PubMed] [Google Scholar]
- 32.Pawelec G, Hambrecht A, Rehbein A, Adibzadeh M. Interleukin 10 protects activated human T lymphocytes against growth factor withdrawal-induced cell death but only anti-fas antibody can prevent activation-induced cell death. Cytokine. 1996;8:877–881. doi: 10.1006/cyto.1996.0117. [DOI] [PubMed] [Google Scholar]
- 33.Ayroldi E, Zollo O, Cannarile L, D'Adamiom F, Grohmann U, Delfino DV, Riccardi C. Interleukin-6 (IL-6) prevents activation-induced cell death: IL-2-independent inhibition of Fas/fasL expression and cell death. Blood. 1998;92:4212–4219. [PubMed] [Google Scholar]
- 34.Refaeli Y, Van Parijs L, Alexander SI, Abbas AK. Interferon γ is required for activation-induced death of T lymphocytes. J. Exp. Med. 2002;196:999–1005. doi: 10.1084/jem.20020666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh CS, Heimberger AB, Gold JS, O'Garra A, Murphy KM. Differential regulation of T helper phenotype development by interleukins 4 and 10 in an αβ T-cell-receptor transgenic system. Proc. Natl. Acad. Sci. USA. 1992;89:6065–6069. doi: 10.1073/pnas.89.13.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, Romagnani S. Natural killer cell stimulatory factor (interleukin 12 (IL-12)) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J. Exp. Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ayroldi E, Migliorati G, Cannarile L, Moraca R, Delfino DV, Riccardi C. CD2 rescues T cells from T-cell receptor/CD3 apoptosis: a role for the Fas/Fas-L system. Blood. 1997;89:3717–3726. [PubMed] [Google Scholar]
- 39.Collette Y, Benziane A, Razanajaona D, Olive D. Distinct regulation of T-cell death by CD28 depending on both its aggregation and T-cell receptor triggering: a role for Fas-FasL. Blood. 1998;92:1350–1363. [PubMed] [Google Scholar]
- 40.Yu XZ, Martin PJ, Anasetti C. CD28 signal enhances apoptosis of CD8 T cells after strong TCR ligation. J. Immunol. 2003;170:3002–3006. doi: 10.4049/jimmunol.170.6.3002. [DOI] [PubMed] [Google Scholar]
- 41.Drake DR, III, Braciale TJ. Cutting edge: lipid raft integrity affects the efficiency of MHC class I tetramer binding and cell surface TCR arrangement on CD8+ T cells. J. Immunol. 2001;166:7009–7013. doi: 10.4049/jimmunol.166.12.7009. [DOI] [PubMed] [Google Scholar]
- 42.Davila E, Celis E. Repeated administration of cytosine-phosphorothiolated guanine-containing oligonucleotides together with peptide/protein immunization results in enhanced CTL responses with anti-tumor activity. J. Immunol. 2000;165:539–547. doi: 10.4049/jimmunol.165.1.539. [DOI] [PubMed] [Google Scholar]
- 43.Wasem C, Arnold D, Saurer L, Corazza N, Jakob S, Herren S, Vallan C, Mueller C, Brunner T. Sensitizing antigen-specific CD8+ T cells for accelerated suicide causes immune incompetence. J. Clin. Invest. 2003;111:1191–1199. doi: 10.1172/JCI16344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raftery MJ, Behrens CK, Muller A, Krammer PH, Walczak H, Schonrich G. Herpes simplex virus type 1 infection of activated cytotoxic T cells: Induction of fratricide as a mechanism of viral immune evasion. J. Exp. Med. 1999;190:1103–1114. doi: 10.1084/jem.190.8.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei CH, Yagita H, Masucci MG, Levitsky V. Different programs of activation-induced cell death are triggered in mature activated CTL by immunogenic and partially agonistic peptide ligands. J. Immunol. 2001;166:989–995. doi: 10.4049/jimmunol.166.2.989. [DOI] [PubMed] [Google Scholar]
- 46.Liu ZX, Govindarajan S, Okamoto S, Dennert G. Fas-mediated apoptosis causes elimination of virus-specific cytotoxic T cells in the virus-infected liver. J. Immunol. 2001;166:3035–3041. doi: 10.4049/jimmunol.166.5.3035. [DOI] [PubMed] [Google Scholar]
- 47.Sobek V, Balkow S, Korner H, Simon MM. Antigen-induced cell death of T effector cells in vitro proceeds via the Fas pathway, requires endogenous interferon-γ and is independent of perforin and granzymes. Eur. J. Immunol. 2002;32:2490–2499. doi: 10.1002/1521-4141(200209)32:9<2490::AID-IMMU2490>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 48.Tham EL, Mescher MF. The poststimulation program of CD4 versus CD8 T cells (death versus activation-induced nonresponsiveness) J. Immunol. 2002;169:1822–1828. doi: 10.4049/jimmunol.169.4.1822. [DOI] [PubMed] [Google Scholar]
- 49.Tham EL, Mescher MF. Signaling alterations in activation-induced nonresponsive CD8 T cells. J. Immunol. 2001;167:2040–2048. doi: 10.4049/jimmunol.167.4.2040. [DOI] [PubMed] [Google Scholar]
- 50.Davey GM, Kurts C, Miller JF, Bouillet P, Strasser A, Brooks AG, Carbone FR, Heath WR. Peripheral deletion of autoreactive CD8 T cells by cross presentation of self-antigen occurs by a Bcl-2-inhibitable pathway mediated by Bim. J. Exp. Med. 2002;196:947–955. doi: 10.1084/jem.20020827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc. Natl. Acad. Sci. USA. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang JK, Ju ST, Marshak-Rothstein A. Protection of T cells from activation-induced cell death by Fas+ B cells. Eur. J. Immunol. 2000;30:931–937. doi: 10.1002/1521-4141(200003)30:3<931::AID-IMMU931>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 53.Huse M, Lillemeier BF, Kuhns MS, Chen DS, Davis MM. T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 2006;7:247–255. doi: 10.1038/ni1304. [DOI] [PubMed] [Google Scholar]
- 54.Tsutsui S, Sonoda K, Sumiyoshi K, Kitamura K, Toh Y, Kitamura M, Kuwano H, Sugimachi K, Okamura S. Prognostic significance of immunological parameters in patients with esophageal cancer. Hepatogastroenterology. 1996;43:501–509. [PubMed] [Google Scholar]
- 55.Cho Y, Miyamoto M, Kato K, Fukunaga A, Shichinohe T, Kawarada Y, Hida Y, Oshikiri T, Kurokawa T, Suzuoki M, et al. CD4+ and CD8+ T cells cooperate to improve prognosis of patients with esophageal squamous cell carcinoma. Cancer Res. 2003;63:1555–1559. [PubMed] [Google Scholar]
- 56.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cardin RD, Brooks JW, Sarawar SR, Doherty PC. Progressive loss of CD8+ T cell-mediated control of a γ-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 1996;184:863–871. doi: 10.1084/jem.184.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 59.Kirberg J, Bruno L, von Boehmer H. CD4+8-help prevents rapid deletion of CD8+ cells after a transient response to antigen. Eur. J. Immunol. 1993;23:1963–1967. doi: 10.1002/eji.1830230835. [DOI] [PubMed] [Google Scholar]
- 60.Kurts C, Carbone FR, Barnden M, Blanas E, Allison J, Heath WR, Miller JF. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J. Exp. Med. 1997;186:2057–2062. doi: 10.1084/jem.186.12.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]