

Abstract
Periodontitis is a chronic destructive infection of the tooth-supportive tissues, which is caused by pathogenic bacteria such as Actinobacillus actinomycetemcomitans. A severe form of periodontitis is found in Papillon-Lefèvre syndrome (PLS), an inheritable disease caused by loss-of-function mutations in the cathepsin C gene. Recently, we demonstrated that these patients lack the activity of the polymorphonuclear leukocyte (PMN)-derived serine proteinases elastase, cathepsin G, and proteinase 3. In the present study we identified possible pathways along which serine proteinases may be involved in the defense against A. actinomycetemcomitans. Serine proteinases are capable to convert the PMN-derived hCAP-18 into LL-37, an antimicrobial peptide with activity against A. actinomycetemcomitans. We found that the PMNs of PLS patients released lower levels of LL-37. Furthermore, because of their deficiency in serine proteases, the PMNs of PLS patients were incapable of neutralizing the leukotoxin produced by this pathogen, which resulted in increased cell damage. Finally, the capacity of PMNs from PLS patients to kill A. actinomycetemcomitans in an anaerobic environment, such as that found in the periodontal pocket, seemed to be reduced. Our report demonstrates a mechanism that suggests a direct link between an inheritable defect in PMN functioning and difficulty in coping with a periodontitis-associated pathogen.
Periodontitis is a chronic infection of the tooth-supportive tissues, which may lead to tooth loss. This disease is caused by pathogenic bacteria such as Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis that reside in the subgingival region (reviewed by Eley and Cox [21]). It is well known that acquired or inherited diseases afflicting the functioning of polymorphonuclear leukocytes (PMNs) cause an increased susceptibility to infections, including periodontitis (15, 32). However, the mechanism underlying the onset of periodontitis is largely unknown.
An extremely severe form of periodontitis is found in Papillon-Lefèvre syndrome (PLS; MIM# 245000) patients. Case studies on PLS patients in the last 25 years report contradictory findings. Several studies reported a depressed in vitro PMN chemotaxis (9, 10, 23, 24, 45, 68, 70, 74), whereas others reported normal values (10, 47, 68, 73). Similar conflicting results were found for phagocytosis (6, 7, 9, 10, 26, 27, 62, 73), the capacity to kill S. aureus (6, 10, 54, 62, 70), and the production of hydrogen peroxide (2, 5, 6, 10, 42, 55, 62, 68, 71). At first sight, these inconsistencies seem to support the notion that periodontitis in PLS patients is not caused by a general PMN defect.
As in other forms of periodontitis, PLS patients seem to have increased susceptibility to infections with periodontitis-associated pathogens such as A. actinomycetemcomitans (1, 5, 7, 13, 17, 18, 20, 22, 33, 41, 42, 46, 53, 59, 60, 67, 70, 74, 75, 76). This facultative anaerobic gram-negative pathogen produces virulence factors to promote its colonization and survival (61). Leukotoxin, the prime virulence factor of A. actinomycetemcomitans, is a 116-kDa pore-forming toxin belonging to the repeat-in-toxins (RTX) (43). Haubek et al. (30) demonstrated that A. actinomycetemcomitans strains producing high levels of leukotoxin were associated with early-onset periodontitis in children in Morocco. Subsequent studies on the occurrence of A. actinomycetemcomitans strains producing high levels of leukotoxin in periodontitis patients indicated that this association is not restricted to Moroccan patients (for a review, see reference 63). Studies on leukotoxin have indicated that it affects myeloid cells, such as PMNs and monocytes, and causes degranulation (35, 40). Cathepsin G and elastase from PMNs have been shown to be able to degrade this toxin extracellularly (34, 35). Whether serine proteinases solely are used by PMNs for degradation is unclear. It has been demonstrated that cysteine and other proteinases of bacteria are also capable of degrading leukotoxin (36).
PLS patients have, due to loss-of-function mutations, no cathepsin C activity (29, 72). Recently, we showed that cathepsin C is the activator of the PMN-derived serine proteinases elastase, cathepsin G, and proteinase 3 in human (16). The absence of cathepsin C not only leads to a loss of activity of the serine proteinases in PMNs of PLS patients but also to a reduction in the amount of the proteins themselves. Our data was confirmed by Pham et al. (54), who also demonstrated a severe reduction in serine proteinase activities in PLS patients. The importance of elastase, cathepsin G, and proteinase 3 in the immune system has been studied extensively. Studies of knockout mice reveal a crucial role in the defense against pathogens such as Staphylococcus aureus (57) and Escherichia coli (4, 48), Candida albicans (57), or Klebsiella pneumoniae (4). In addition, in vitro experiments showed that cathepsin G and elastase are able to kill A. actinomycetemcomitans and Capnocytophaga spp. (3). As mentioned above, elastase and cathepsin G are able to neutralize the leukotoxin of A. actinomycetemcomitans (34). In vitro, all three serine proteinases are able to convert the PMN-derived hCAP-18 (which is normally stored in the specific granules (44) into the antimicrobial peptide LL-37 (65). After exocytosis, however, only proteinase 3 seems to be capable of processing hCAP-18 (65). LL-37 has activity against a broad range of pathogens such as S. aureus, E. coli, Pseudomonas aeruginosa, and K. pneumoniae (25, 52, 64, 78) and also to A. actinomycetemcomitans (56, 69). In humans, the absence of LL-37 is associated with the Morbus Kostmann syndrome (56). These patients suffer from a congenital neutropenia and face severe periodontal disease during adulthood (56). No other deficiencies in LL-37 in human PMNs have been reported thus far.
Serine proteinases (e.g., elastase, cathepsin G, and proteinase 3), together with antimicrobial peptides (e.g., LL-37), form the basis of the oxygen-independent machinery PMNs can use to kill bacteria (12, 49). Since the periodontal pocket is characterized by a reduced oxygen tension (50), the defense against pathogens in this environment may depend predominantly on oxygen-independent means. Based on our previous finding that PLS patients lack elastase, cathepsin G, and proteinase 3 activity (16), we hypothesize that the etiology of periodontitis in these patients is due to a PMN defect, resulting in a compromised innate immune response to A. actinomycetemcomitans. It was the aim of the present study to identify the importance of elastase, cathepsin G, and proteinase 3 in the defense against A. actinomycetemcomitans.
MATERIALS AND METHODS
Reagents.
All chemicals were obtained from Sigma (Sigma Chemicals Co, St. Louis, MO) unless stated otherwise.
Patients and controls.
Three families with PLS patients participated in the present study. Samples of family A were available from two female PLS patients (AP1, 27 years old; AP2, 29 years old) and their parents (AC1 and AC2); samples of family B included one female PLS patient (BP1, 19 years old) and her parents (BC1 and BC2). (For further details on patient description, see reference 16.) Family C was a consanguineous family of Iranian origin with a male and a female PLS patient (CP1, 20 years old; CP2, 28 years old). Both patients from this family had hyperkeratotic plaques on their hand palms and foot soles, which extended to the knees and elbows. The female patient had suffered from severe periodontal disease and had been edentulous from the age of 14. The male patient had not lost any permanent teeth prematurely. Both patients, like the patients of family B, had suffered from severe liver abscess. Blood samples were available from both PLS patients and their mother (CC1). In most of the experiments the parents (having a similar genetic background) were used as controls. In some experiments, samples from healthy individuals were used as a reference (H) instead of one of the parents. The results of the experiments were compared to samples of five healthy individuals aged 22 to 45 years (controls were not tested for A. actinomycetemcomitans infection). During the course of the present study, family B, due to too-frequent sample collection, decided to stop participating. The Institutional Review Board on Human Studies approved this study, and all individuals involved gave their written consent, in accordance with the World Medical Association Declaration of Helsinki.
Sample isolation, sequencing, and mutation analysis.
Peripheral heparin anticoagulated blood was collected by standard venipuncture from family C for mutation analysis of the cathepsin C gene. Genomic DNA was isolated from the blood samples by using the Puregene kit (Gentra Systems, Inc., Michigan) according to the manufacturer's protocol. Sequencing was performed as described previously (16). In family C, a previously reported mutation, c.815G>C/p.R272P, was found (72). Patients CP1 and CP2 were homozygous for this mutation, whereas the mother (CC1) was a heterozygous carrier. The patients showed no cathepsin C activity, and the mother had reduced (50%) levels. We confirmed that the patients were deficient for elastase, cathepsin G, and proteinase 3 activity (16).
Isolation of PMNs from peripheral blood.
Peripheral blood (heparin anticoagulated) was obtained from patients, parents, and healthy unrelated controls by standard venipuncture. The blood was diluted with phosphate-buffered saline (PBS) and layered on Percoll (density, 1.076 g/ml) or Ficoll (both from Pharmacia, Uppsala, Sweden). After centrifugation for 30 min at 1,000 × g, the supernatant was removed, and the red cell pellet was treated with lysis buffer (155 mM ammonium chloride, 0.1 mM EDTA, 10 mM KHCO3) to remove the erythrocytes. The PMNs were washed twice with PBS, collected by centrifugation for 5 min at 300 × g at 4°C, and resuspended in assay buffer (20 mM HEPES, 132 mM NaCl, 6 mM KCl, 1 mM MgSO4, 1.2 mM KH2PO4, 1 mM CaCl2, 5.5 mM glucose [pH 7.4]) (28). Cell viability was assessed by trypan blue exclusion and was generally more than 98%.
Enzyme activity.
Proteinase 3 activity was measured by the hydrolysis of N-t-but-Ala-Ala-Nva (Norvaline)-thiobenzylester (Elastin Products Co., Missouri) (39). The reaction buffer consisted of 100 mM HEPES (Gibco Laboratories, Grand Island, NY), 500 mM NaCl, 5 mM DTNB [5,5′-dithiobis(2-nitrobenzoic acid)], and 250 μM N-t-but-Ala-Ala-Nva-thiobenzylester (pH 7.5). The absorbance was measured at 405 nm for 15 min. All measurements were performed on Wallac 1420 VictorII (Perkin-Elmer Life Sciences, Turku, Finland).
Leukotoxin degradation by PMN lysates.
Leukotoxin degradation by PMN lysates was studied as follows. Leukotoxin, purified from the highly leukotoxic A. actinomycetemcomitans HK 1519 strain, was kindly provided by A. Johansson (University of Ume(angst)a, Ume(angst)a, Sweden) (36). PMN lysates of PLS patients and their parents from family A and family B and a healthy male (H1, 23 years old) control were equalized to protein content, which was determined by using a BCA assay kit (Pierce, Rockford, Illinois). An aliquot of 2 μg of protein was incubated with 150 ng of leukotoxin and incubated at 37°C for 3 h. Proteins in the samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. PMNs from five healthy individuals (22 to 45 years old) were treated similarly, with comparable results (data not shown).
Leukotoxic activity.
PMNs were resuspended in assay buffer at a concentration of 3.6 × 106 cells/ml. A 450-μl aliquot of cell suspension was incubated with 50 μl (150 ng) of leukotoxin. The samples were incubated at 37°C in air with 5% CO2 for 1 h. After the incubation, the samples were placed on ice to stop the reaction. PMNs were collected after centrifugation (300 × g, 5 min, 4°C). PMNs exposed to leukotoxin were fixed in 1% glutaraldehyde and 4% formaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4). The cells were washed and postfixed in 1% OsO4 in 0.1 M sodium cacodylate buffer. Next, the cells were dehydrated through a graded series of ethanol, pelleted, and embedded in epoxy resin (LX-112). Semithin sections were made with a diamond knife and stained according to the method of Richardson et al. (58). Sections were randomized and examined by using a ×60 objective lens on a Leica DM LB microscope (Leica, Wetzlar, Germany). According to their morphology, cells were classified as either viable or nonviable. Viable cells had intensely stained cytoplasm, normal-appearing nuclei, and discernible organelles; nonviable cells had weakly stained cytoplasm and were without clearly defined organelles (including the nucleus). At least 100 cells were counted per patient or control. The number of nonviable cells was expressed as a percentage of the total number of counted cells. Ultrathin sections were stained with uranyl and lead and then examined in a Philips EM 420 electron microscope (Philips Medical Systems, Eindhoven, The Netherlands). Images were digitized, and figures were prepared by using Adobe Photoshop (Adobe Systems, San Jose, CA).
Degranulation of PMNs.
For degranulation experiments, PMNs were resuspended in assay-buffer to a concentration of 107 cells/ml. Cells were preincubated at 37°C for 5 min with 10 μg of cytochalasin B/ml and subsequently stimulated with 2.5 μM formylmethionyleucylphenylalanine (fMLP) or 4 μg of phorbol 12-myristate 13-acetate (PMA)/ml for 15 min. Similarly, cells were preincubated at 37°C in assay buffer and then stimulated with 1 μM ionomycin for 15 min. Cells were placed on ice water to stop the stimulation and pelleted by centrifugation (300 × g, 10 min, 4°C). The supernatants were collected and stored at −20°C.
SDS-PAGE and immunoblotting.
SDS-PAGE under reducing conditions and immunoblotting were performed by using Mini-Protean cells and mini Trans-Blot Electrophoretic Transfer cells according to the manufacturer's instructions (Bio-Rad, Hercules, CA). For the LL-37 blots, polyvinylidene difluoride membranes were blocked, incubated with primary antibodies to LL-37 (65), and subsequently incubated with horseradish peroxidase-conjugated secondary antibodies or with alkaline phosphatase-conjugated secondary antibodies for the PMN cell lysates. For the leukotoxin blot, the membrane was incubated with a primary polyclonal rabbit antibody against leukotoxin (provided by A. Johansson, University of Umeå, Umeå, Sweden) and afterward incubated with a horseradish peroxidase-conjugated secondary antibody (all secondary antibodies were from Dako, Glostrup, Denmark). Immunoreactive bands were visualized by fluorescence using ECL-Plus (Amersham, Buckinghamshire, United Kingdom) or with nitroblue tetrazolium salt-phosphate in 0.1 M Tris-HCl buffer (pH 9.5).
Bacterial killing CFU assay.
A. actinomycetemcomitans HK921, a highly leukotoxic strain of A. actinomycetemcomitans from the JP2 clone, was obtained from M. Kilian, University of Aarhus, Aarhus, Denmark (31). The high leukotoxic activity (up to 20 times) of this strain is caused by a mutation in the promoter region (8). A. actinomycetemcomitans HK921 was grown aerobically in BHI medium for 1 to 2 days in the presence of 5% CO2. Bacteria were washed twice with PBS and resuspended in assay buffer. In preliminary experiments (data not shown) with the PMNs of healthy controls we found that incubation of the PMNs at a ratio of 10:1 (PMN to bacteria) resulted in 80 to 90% killing after 120 min. PMNs (107) from PLS patients (AP1 and AP2) and their parents (AC1 and AC2) and bacteria (108) were incubated in the presence of 10% serum in an anaerobic hood (80% N2, 10% H2, 10% CO2) at 37°C for 120 min with shaking. At set time points, samples were taken, diluted, and plated on horse blood agar plates. Plates were cultured at 37°C for 1 to 2 days. CFU were counted as a measure for survival of the bacteria. The number of bacteria at the start of the experiment was set at 100%. The number of bacteria at subsequent time points was expressed relative to the initial 100%. To exclude the influence of leukotoxin-neutralizing antibodies, we performed the experiments with rabbit serum that showed no immunoreactivity with the leukotoxin of A. actinomycetemcomitans (37).
Statistics.
Data are presented as means and were analyzed by using GraphPad Software (San Diego, CA). Differences were tested by using the Mann-Whitney test and considered significant when the P value was <0.05 (two-tailed analysis).
RESULTS
Serine proteinases are essential in the neutralization of leukotoxin.
Serine proteinases have been demonstrated to be able to neutralize the leukotoxin produced by A. actinomycetemcomitans (34). We studied the lack of serine proteinase activity in the PMNs of PLS patients on the degradation of leukotoxin. PMN lysates were obtained from two unrelated families, family A (parents AC1 and AC2, patients AP1 and AP2) and family B (parents BC1 and BC2, patient BP1), and a healthy control (H1) and incubated with 150 ng of leukotoxin for 3 h (Fig. 1A). In both families the PMN lysates were equally capable of cleaving the leukotoxin compared to the control. After 3 h of incubation the intact leukotoxin was no longer detectable, and fragments of the leukotoxin were detectable. In contrast, the samples of the PMN lysates of the PLS patients of both families showed fragments of the leukotoxin, but the greater part of the intact leukotoxin was still detected.
FIG. 1.

Degradation of leukotoxin by PMNs and cytotoxic action of leukotoxin. (A) Western blot detection of cleavage of leukotoxin by PMN lysates of family A and family B. Aliquots of 150 ng of leukotoxin were incubated with equal protein amounts (2 μg) of PMN cell lysates for 3 h at 37°C. As a control, an equal quantity of leukotoxin was incubated in assay buffer (left lane, L) or incubated with PMN lysates isolated from a healthy donor (H1). (B) Western blot detection of leukotoxin in supernatants after a 1-h incubation of leukotoxin at 150 ng with PMNs (1.8 × 106 cells) isolated from family A and family C at 37°C. Proteins in equal volumes of supernatants were separated by SDS-PAGE and blotted onto a polyvinylidene difluoride membrane. Leukotoxin and fragments were detected with polyclonal antibodies raised against the leukotoxin. As a control, 150 ng of leukotoxin was incubated in assay buffer (right lane, L). A sample of a healthy individual (H2) was also included. Family A: parents AC1 and AC2 and patients AP1 and AP2. Family B: parents BC1 and BC2 and patient BP1. Family C: parents CC1 and CC2 and patient CP1. (C) On the left side, two micrographs are shown of PMNs isolated from a parent of family A (AC1); on the right, two micrographs are shown of PMNs isolated from a PLS patient of family A (AP1). The top micrographs show PMNs incubated in assay buffer for 1 h; the bottom micrographs show PMNs incubated in the presence of 150 ng of leukotoxin. Note the absence of discernible organelles in the patient's PMNs upon exposure to leukotoxin. (D) Histomorphometric analysis of the percentage of “nonviable” cells compared to the total number of cells. Controls, n = 4; patients, n = 4. The mean ± the standard deviation is given. *, P < 0.05 (Mann-Whitney test).
Next, we studied the importance of serine proteinases in the protection of PMNs to exposure to the leukotoxin (Fig. 1B). For this purpose, PMNs were isolated from PLS patients from two unrelated families (AP1, AP2, CP1, and CP2) and their parents (AC1, AC2, and CC1) and incubated with leukotoxin at 150 ng. Since there was no sample available from the father of family C, a sample of a female healthy (H2, aged 45) donor was used instead. The supernatants were analyzed for the presence of leukotoxin (Fig. 1B). In the samples of the parents (AC1, AC2, and CC1) and the healthy donor (H2) there was almost no intact leukotoxin at 116 kDa or fragments of the leukotoxin present, indicating that the leukotoxin had been degraded. The samples of the patients (AP1, AP2, CP1, and CP2) clearly showed the presence of the intact leukotoxin. Based on the intensity of the staining, the amount of leukotoxin was comparable to the initial quantity of leukotoxin (Fig. 1B, right lane). By using microscopy we studied in detail the condition of the PMNs after exposure to the leukotoxin (Fig. 1C). Electron microscopy revealed that the PMNs of the parent (AC1) had not been heavily affected by the leukotoxin compared to cells that were not exposed to the leukotoxin (Fig. 1C, left graphs). The majority of the PMNs had a “viable” appearance, as signified by an intense staining of cytoplasm and nuclei. The nuclei had a normal multilobed appearance. Furthermore, granules and other intracellular compartments were clearly visible. A similar result was found for the other parents and the healthy donor. In contrast, the PMNs of the patient (AP1) were severely damaged (Fig. 1C, right graphs). The majority of the PMNs of the patients stained vaguely both in the cytoplasm and in the nucleus. No organelles or granules were observed in the cytoplasm. PMNs of other patients (AP2, CP1, and CP2) showed similar phenomena. We quantified the percentage of “nonviable” cells in sections of PMNs of controls and of PLS patients by using light microscopy (Fig. 1D). It showed that a significantly higher percentage of PLS patient's cells was affected (P < 0.05).
Processing of hCAP-18.
By using enzymes and antimicrobial peptides, PMNs are very well equipped to handle bacteria in an anaerobic environment such as the periodontal pocket. LL-37 is an important antimicrobial peptide in PMNs, and its precursor hCAP-18 is stored spatially separated from the PMN-derived serine proteinases. Upon degranulation and exocytosis of the contents of the secretory granules in the extracellular environment, proteinase 3 has been demonstrated to process hCAP-18 into LL-37 (65). Although PLS patients have normal levels of serum hCAP-18 (data not shown), the lack of proteinase 3 activity in PLS patients may affect its processing to LL-37. We studied the extracellular processing of endogenous hCAP-18 in the PMNs of PLS patients of family A and compared this with their parents. We stimulated PMNs to degranulate by the use of various secretagogues (65). After 15 min of incubation, supernatants were collected and analyzed by Western blotting for the presence of hCAP-18, intermediate fragments (not fully processed hCAP-18 fragments), and LL-37.
The Western blot analysis clearly showed that hCAP-18 (at 18 kDa) was present in the exocytosed material of PMNs of the PLS patients and their parents (Fig. 2A). Since ionomycin is known to be the more potent secretagogue (65), the net yield of hCAP-18 was the highest. The striking result on the Western blot, however, was the yield in LL-37. Whereas the parents (AC1 and AC2) demonstrated demarcated bands at 4 kDa, most likely being LL-37, the PLS patients (AP1 and AP2) showed lower levels of LL-37. In the case of the weaker secretagogues PMA and fMLP, increased exposure times were necessary to visualize them. Another notable observation on the Western blot was the more pronounced presence of intermediate, not fully processed hCAP-18 fragments in the samples of the patients. Densitometric analysis of these bands confirmed that the PMNs of PLS patients had produced less LL-37 than the PMNs of their parents and that their intermediate fragments were more pronounced (Table 1) . Previously, we have shown that PLS patients have no proteinase 3 activity (16). To confirm our previously reported findings, we measured proteinase 3 activity in exocytosed material of the PMNs stimulated with ionomycin (Fig. 2B) The parents (AC1 and AC2) demonstrated proteinase 3 activity, whereas the patients (AP1 and AP2) were negative.
FIG. 2.

Immunoblot of hCAP-18/LL-37 of exocytosed material of PMNs isolated from PLS patients and their parents from family A. (A) Equal volumes (10 μl) of exocytosed material were analyzed. PMNs were stimulated with PMA (4 μg/ml), fMLP (2.5 μM), or ionomycin (1 μM) for 15 min at 37°C. Supernatants were analyzed by Western blotting with monoclonal antibodies to LL-37. hCAP-18 and LL-37 are indicated by arrows. Intermediate fragments indicate partially processed hCAP-18. The lower panel is the lower half of the immunoblot shown in the upper panel with a longer exposure time. Parents are indicates by AC1 and AC2; patients are indicated by AP1 and AP2. All samples were run on one gel. (B) Proteinase 3 activity in ionomycin exocytosed material of PMNs of family A. Samples (10 μl) were analyzed for proteinase 3 activity. Activity is expressed as the increase in absorption at 405 nm over time. Blanc, sample consisting of the reaction mixture was included in this assay to correct for spontaneous decay of the substrate. Note that the lines of the patients and the blanc coincide. The family A parents are indicated by AC1 and AC2; the family A patients are indicated by AP1 and AP2.
TABLE 1.
Densitometric analysis of the immunoreactive bands of a Western blot of hCAP-18/LL-37 of exocytosed material of PMNs isolated from PLS patients and their parents of family Aa
| Secretagogue | Mean amt (%) ± SDb
|
|||||
|---|---|---|---|---|---|---|
| hCAP-18
|
Intermediate-size fragments
|
LL-37
|
||||
| Parents | Patients | Parents | Patients | Parents | Patients | |
| fMLP | 46.0 ± 0.1 | 65.9 ± 5.4 | 15.4 ± 3.7 | 27.2 ± 9.6 | 38.6 ± 6.4 | 6.9 ± 4.2 |
| PMA | 48.1 ± 4.7 | 61.0 ± 0.3 | 13.6 ± 0.1 | 28.6 ± 3.4 | 38.3 ± 4.7 | 10.4 ± 3.8 |
| Ionomycin | 40.5 ± 0.1 | 46.7 ± 1.1 | 10.3 ± 0.7 | 20.6 ± 6.11 | 49.2 ± 0.8 | 32.7 ± 5.1 |
See Fig. 2A.
The total amounts of immune reactive protein were determined by densitometry. Individual bands for hCAP-18, intermediate fragments, and LL-37 are expressed as percent values relative to the total immunoreactive protein (100%). The total immunoreactive protein was expressed per secretagogue (fMLP, PMA, and ionomycin). n = 2 for both parents and patients. Due to the small sample numbers in the groups, the differences were not compared statistically.
Serine proteinases are crucial in the anaerobic killing of A. actinomycetemcomitans.
PLS patients are often infected with periodontitis-associated pathogens, A. actinomycetemcomitans in particular. We demonstrated that PLS patients had, next to the absence of serine proteinase activity and the reduced amounts of proteins themselves, also reduced levels of LL-37 and that PMNs were incapable of effectively neutralizing the leukotoxin produced by A. actinomycetemcomitans. Since the high leukotoxic activity of A. actinomycetemcomitans results in a reduced killing efficiency by PMNs (37), it is possible that PLS patients are even less efficient in killing this pathogen. In the following experiments we determined whether a high leukotoxic activity of A. actinomycetemcomitans, together with reduced levels of serine proteinase activity and reduced levels of LL-37 in PMNs of PLS patients, might cause difficulties in dealing with this particular pathogen in an anaerobic environment. The incubation of A. actinomycetemcomitans with the PMNs of PLS patients (AP1 and AP2) and their parents (AC1 and AC2) showed that there is a delay in killing for the PLS patients (Fig. 3). After 2 h of incubation, this delay seemed more pronounced in that the PMNs of the parents had killed 82% of the A. actinomycetemcomitans, whereas the PMNs of PLS patients had killed only 61% of the bacteria. Unfortunately, there was no material for other PLS patients available to verify these data.
FIG. 3.
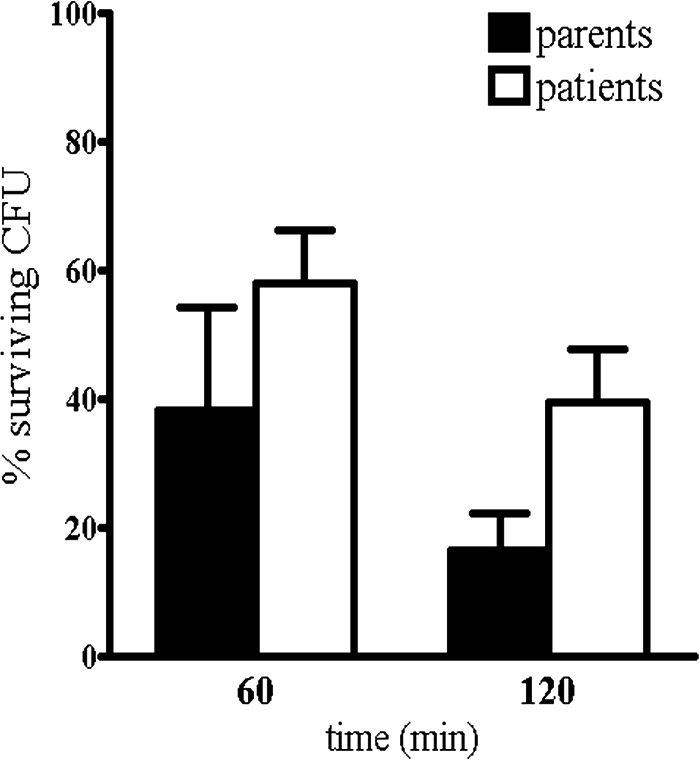
Anaerobic killing of A. actinomycetemcomitans by PMNs. PMNs isolated from parents and patients of family A were incubated with A. actinomycetemcomitans (ratio 1:10) at 37°C for 60 or 120 min under anaerobic conditions. Serial dilutions of cell lysate samples were plated on blood agar plates and cultured for 48 h, and the number of CFU was counted. Columns indicate the mean percentage ± the SD of surviving CFU as a measure for the number of bacteria killed. Parents, n = 2; patients, n = 2. Due to the small sample number in the groups, the differences were not compared statistically.
DISCUSSION
The results presented here provide new information about the biological mechanisms underlying the pathogenesis of a severe form of early-onset periodontitis as evidenced in patients suffering from Papillon-Lefèvre syndrome (PLS). Although periodontitis in PLS is often considered to be related to defective PMN functioning, no conclusive evidence has come forward in support of this assumption. The present results suggest that PMNs from PLS patients have difficulty in coping with A. actinomycetemcomitans in an anaerobic environment. Our results thus provide evidence for a mechanism to link a monogenetic defect in PMN functioning with one of the well-known periodontitis associated pathogens in humans. The microbiological status of PLS patients suggests that a majority of them are infected with A. actinomycetemcomitans (1, 5, 7, 13, 17, 18, 20, 22, 33, 41, 42, 46, 53, 60, 67, 70, 71, 74, 75, 76). This is in line with our finding that their resistance to this pathogen may be compromised.
The periodontal pocket is unique: it forms an anaerobic environment with a more or less specific bacterial flora (50). The anaerobic environment forces PMNs to use compounds other than those generated via oxygen, for which PMNs are well adapted. They release, for instance, increased levels of serine proteinases (12). Extensive research has revealed multiple roles of serine proteinases in inflammation and host defense, including a role in the neutralization of toxins. Our present data demonstrate that the PMNs of PLS patients are unable to degrade the leukotoxin of A. actinomycetemcomitans, which is the prime virulence factor for this pathogen (19, 77). Leukotoxin selectively kills human leukocytes (77) and increases the release of proinflammatory interleukin-1β from human macrophages (40). Our observation that the PMNs from PLS patients are more susceptible to lysis by exogenously added leukotoxin supports earlier studies showing that serine protease-mediated degradation of leukotoxin is essential for the neutralization (34). The importance of this leukotoxin in the pathogenesis of periodontitis is illustrated by the high prevalence of early-onset periodontitis in a group of children in Morocco (30) who all carried a strain of A. actinomycetemcomitans producing an extremely high level of leukotoxin. Early-onset periodontitis is a relatively rare phenomenon; it suggests that dealing with the leukotoxin seems to be crucial, particularly in children to adequately cope with an A. actinomycetemcomitans infection.
Serine proteinases, and more specifically proteinase 3, have also been suggested to play an essential role in the processing of hCAP-18 into the antimicrobial peptide LL-37 (65). We demonstrated that the PMNs of PLS patients release reduced levels of LL-37. Since LL-37 possesses antimicrobial activity against A. actinomycetemcomitans (69), inhibition of processing and release of this antimicrobial peptide may result in reduced bacterial clearance (14). This is supported by the observation that the absence of LL-37 in Morbus Kostmann is associated with a high prevalence of A. actinomycetemcomitans and severe periodontal disease (56). Sorensen et al. (65) have demonstrated that depletion of proteinase 3 from exocytosed material of PMNs results in a marked decreased processing of hCAP-18 to LL-37. By using a natural human model deficient in serine proteinase activity (PLS) (16, 54), we have clearly shown the presence of processed hCAP-18 in exocytosed material of PMNs, despite the absence of serine proteinase activity. Therefore, these data suggest that, in the absence of PMN serine protease activity, hCAP-18 can be processed by less-efficient mechanisms. It is known that hCAP-18 in seminal fluid, which is derived from the epithelium of the epididymis, is processed by the aspartic protease gastricin, resulting in ALL-38 (66). Although the LL-37 release by stimulated PMNs from PLS patients migrated at the predicted size on SDS-PAGE gels, we cannot formally exclude the possibility that PLS PMNs release an alternatively processed form of hCAP-18. Alternative processing of hCAP-18 is further suggested by differences that were noted in the intensity of the intermediate-size fragments. Furthermore, we have indirect evidence to suggest that the processing of hCAP-18 in PMNs of PLS patients is associated with enzymes located in the azurophilic granules, since in PMNs from patients the rate of processing was found to diminish with less-potent azurophilic granule secretagogues (11, 65). The question of which enzymes process hCAP-18 in PMNs of PLS patients remains to be elucidated.
Finally, serine proteinases possess antimicrobial activity through direct enzymatic action and/or through peptides derived from the enzymes (3). In addition, research with mouse models has indicated that serine proteinases are required for adequate killing of pathogens such as S. aureus and K. pneumoniae (4, 48, 57, 57). The relatively high prevalence of A. actinomycetemcomitans illustrated the notion that PLS patients may have serious trouble in coping with A. actinomycetemcomitans infections. This microorganism is mainly killed by PMNs via the oxygen independent pathway, e.g., antimicrobial peptides and serine proteinases (51), components that seem to fail in PLS patients. Indeed, our experiments show a reduced killing of A. actinomycetemcomitans for PMNs of PLS patients in an anaerobic environment.
Our knowledge of the key players in periodontal disease is still very limited. Over the last few decades it has become apparent that certain pathogens may be important in the development of the disease (38). The role of the host in the defense against these pathogens has only recently become the subject of research. The findings presented here support the notion that severe early-onset periodontitis in PLS is likely to be caused by malfunctioning of the defense against periodontal pathogens, A. actinomycetemcomitans in particular. It demonstrates, similarly to the Kostmann syndrome, a direct link between an inheritable defect in PMN functioning with difficulty in coping with a specific periodontitis-associated pathogen. Furthermore, it may provide clues in general to understand what exactly causes individuals to be more susceptible to certain pathogens.
Acknowledgments
W. Tigchelaar-Gutter, T. Schoenmaker, D. C. Jansen, J. J. de Soet, and M. Nguyen (Academic Centre for Dentistry Amsterdam, Amsterdam, The Netherlands) and R. Anderson (Pulmonology, Leiden University Medical Centre, The Netherlands) are acknowledged for their assistance. We thank H. De Vree (Periodontology, University of Ghent, Belgium) and J. Kamphoven and A. Wagner (Clinical Genetics, Erasmus University Rotterdam, Rotterdam, The Netherlands) for providing patient material.
Editor: J. B. Bliska
REFERENCES
- 1.Al Khenaizan, S. 2002. Papillon-Lefevre syndrome: the response to acitretin. Int. J. Dermatol. 41:938-941. [DOI] [PubMed] [Google Scholar]
- 2.Almuneef, M., S. Al Khenaizan, S. Al Ajaji, and A. Al Anazi. 2003. Pyogenic liver abscess and Papillon-Lefevre syndrome: not a rare association. Pediatrics 111:e85-e88. [DOI] [PubMed] [Google Scholar]
- 3.Bangalore, N., J. Travis, V. C. Onunka, J. Pohl, and W. M. Shafer. 1990. Identification of the primary antimicrobial domains in human neutrophil cathepsin G. J. Biol. Chem. 265:13584-13588. [PubMed] [Google Scholar]
- 4.Belaaouaj, A., R. McCarthy, M. Baumann, Z. Gao, T. J. Ley, S. N. Abraham, and S. D. Shapiro. 1998. Mice lacking neutrophil elastase reveal impaired host defense against gram-negative bacterial sepsis. Nat. Med. 4:615-618. [DOI] [PubMed] [Google Scholar]
- 5.Bimstein, E., J. Lustmann, M. N. Sela, Z. B. Neriah, and W. A. Soskolne. 1990. Periodontitis associated with Papillon-Lefevre syndrome. J. Periodontol. 61:373-377. [DOI] [PubMed] [Google Scholar]
- 6.Borroni, G., A. Pagani, A. Carcaterra, R. Pericoli, P. Gabba, and M. Marconi. 1985. Immunological alterations in a case of Papillon-Lefevre syndrome with recurrent cutaneous infections. Dermatologica 170:27-30. [DOI] [PubMed] [Google Scholar]
- 7.Boutsi, E. A., M. Umeda, T. Nagasawa, N. Laosrisin, and I. Ishikawa. 1997. Follow-up of two cases of Papillon-Lefevre syndrome and presentation of two new cases. Int. J. Periodontics Restorative Dent. 17:334-347. [PubMed] [Google Scholar]
- 8.Brogan, J. M., E. T. Lally, K. Poulsen, M. Kilian, and D. R. Demuth. 1994. Regulation of Actinobacillus actinomycetemcomitans leukotoxin expression: analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infect. Immun. 62:501-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown, R. S., G. L. Hays, C. M. Flaitz, P. A. O'Neill, K. Abramovitch, and R. R. White. 1993. A possible late onset variation of Papillon-Lefevre syndrome: report of 3 cases. J. Periodontol. 64:379-386. [DOI] [PubMed] [Google Scholar]
- 10.Bullon, P., A. Pascual, M. C. Fernandez-Novoa, M. V. Borobio, M. A. Muniain, and F. Camacho. 1993. Late onset Papillon-Lefevre syndrome? A chromosomic, neutrophil function and microbiological study. J. Clin. Periodontol. 20:662-667. [DOI] [PubMed] [Google Scholar]
- 11.Campbell, E. J., E. K. Silverman, and M. A. Campbell. 1989. Elastase and cathepsin-G of human monocytes: quantification of cellular content, release in response to stimuli, and heterogeneity in elastase-mediated proteolytic activity. J. Immunol. 143:2961-2968. [PubMed] [Google Scholar]
- 12.Claesson, R., E. Johansson, and J. Carlsson. 1994. Oxygen-dependent modulation of release and activity of polymorphonuclear leukocyte granule products. Oral Microbiol. Immunol. 9:81-87. [DOI] [PubMed] [Google Scholar]
- 13.Clerehugh, V., D. B. Drucker, G. J. Seymour, and P. S. Bird. 1996. Microbiological and serological investigations of oral lesions in Papillon-Lefevre syndrome. J. Clin. Pathol. 49:255-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole, A. M., J. Shi, A. Ceccarelli, Y. H. Kim, A. Park, and T. Ganz. 2001. Inhibition of neutrophil elastase prevents cathelicidin activation and impairs clearance of bacteria from wounds. Blood 97:297-304. [DOI] [PubMed] [Google Scholar]
- 15.Daniel, M. A., and T. E. Van Dyke. 1996. Alterations in phagocyte function and periodontal infection. J. Periodontol. 67:1070-1075. [DOI] [PubMed] [Google Scholar]
- 16.de Haar, S. F., D. C. Jansen, T. Schoenmaker, H. De Vree, V. Everts, and W. Beertsen. 2004. Loss-of-function mutations in cathepsin C in two families with Papillon-Lefevre syndrome are associated with deficiency of serine proteinases in PMNs. Mutat. Brief 714:1-7. [Online.] http://www3.interscience.wiley.com/homepages/38515/pdf/mutation/714.pdf. [DOI] [PubMed] [Google Scholar]
- 17.De Vree, H., K. Steenackers, and J. A. De Boever. 2000. Periodontal treatment of rapid progressive periodontitis in 2 siblings with Papillon-Lefevre syndrome: 15-year follow-up. J. Clin. Periodontol. 27:354-360. [DOI] [PubMed] [Google Scholar]
- 18.Ebersole, J. L., D. Cappelli, and S. C. Holt. 2001. Periodontal diseases: to protect or not to protect is the question? Acta Odontol. Scand. 59:161-166. [DOI] [PubMed] [Google Scholar]
- 19.Ebersole, J. L., L. Kesavalu, S. L. Schneider, R. L. Machen, and S. C. Holt. 1995. Comparative virulence of periodontopathogens in a mouse abscess model. Oral Dis. 1:115-128. [DOI] [PubMed] [Google Scholar]
- 20.Eickholz, P., B. Kugel, S. Pohl, H. Naher, and H. J. Staehle. 2001. Combined mechanical and antibiotic periodontal therapy in a case of Papillon-Lefevre syndrome. J. Periodontol. 72:542-549. [DOI] [PubMed] [Google Scholar]
- 21.Eley, B. M., and S. W. Cox. 2003. Proteolytic and hydrolytic enzymes from putative periodontal pathogens: characterization, molecular genetics, effects on host defenses and tissues and detection in gingival crevice fluid. Periodontology 2000. 31:105-124. [DOI] [PubMed] [Google Scholar]
- 22.Eronat, N., F. Ucar, and G. Kilinc. 1993. Papillon Lefevre syndrome: treatment of two cases with a clinical microbiological and histopathological investigation. J. Clin. Pediatr. Dent. 17:99-104. [PubMed] [Google Scholar]
- 23.Firatli, E., N. Gurel, A. Efeoglu, and S. Badur. 1996. Clinical and immunological findings in 2 siblings with Papillon-Lefevre syndrome. J. Periodontol. 67:1210-1215. [DOI] [PubMed] [Google Scholar]
- 24.Firatli, E., B. Tuzun, and A. Efeoglu. 1996. Papillon-Lefevre syndrome. Analysis of neutrophil chemotaxis. J. Periodontol. 67:617-620. [DOI] [PubMed] [Google Scholar]
- 25.Frohm, M., B. Agerberth, G. Ahangari, M. Stahle-Backdahl, S. Liden, H. Wigzell, and G. H. Gudmundsson. 1997. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J. Biol. Chem. 272:15258-15263. [DOI] [PubMed] [Google Scholar]
- 26.Ghaffer, K. A., F. M. Zahran, H. M. Fahmy, and R. S. Brown. 1999. Papillon-Lefevre syndrome: neutrophil function in 15 cases from 4 families in Egypt. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodont. 88:320-325. [DOI] [PubMed] [Google Scholar]
- 27.Gongora, R., A. Corell, J. R. Regueiro, M. Carasol, C. Rodriguez-Gallego, E. Paz-Artal, M. Timon, L. Allende, and A. Arnaiz-Villena. 1994. Peripheral blood reduction of memory (CD29+, CD45RO+, and “bright” CD2+ and LFA-1+) T lymphocytes in Papillon-Lefevre syndrome. Hum. Immunol. 41:185-192. [DOI] [PubMed] [Google Scholar]
- 28.Hamers, M. N., A. A. Bot, R. S. Weening, H. J. Sips, and D. Roos. 1984. Kinetics and mechanism of the bactericidal action of human neutrophils against Escherichia coli. Blood 64:635-641. [PubMed] [Google Scholar]
- 29.Hart, T. C., P. S. Hart, D. W. Bowden, M. D. Michalec, S. A. Callison, S. J. Walker, Y. Zhang, and E. Firatli. 1999. Mutations of the cathepsin C gene are responsible for Papillon-Lefevre syndrome. J. Med. Genet. 36:881-887. [PMC free article] [PubMed] [Google Scholar]
- 30.Haubek, D., J. M. Dirienzo, E. M. Tinoco, J. Westergaard, N. J. Lopez, C. P. Chung, K. Poulsen, and M. Kilian. 1997. Racial tropism of a highly toxic clone of Actinobacillus actinomycetemcomitans associated with juvenile periodontitis. J. Clin. Microbiol. 35:3037-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haubek, D., K. Poulsen, J. Westergaard, G. Dahlen, and M. Kilian. 1996. Highly toxic clone of Actinobacillus actinomycetemcomitans in geographically widespread cases of juvenile periodontitis in adolescents of African origin. J. Clin. Microbiol. 34:1576-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodge, P., and B. Michalowicz. 2001. Genetic predisposition to periodontitis in children and young adults. Periodontol. 2000. 26:113-134. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa, I., M. Umeda, and N. Laosrisin. 1994. Clinical, bacteriological, and immunological examinations and the treatment process of two Papillon-Lefevre syndrome patients. J. Periodontol. 65:364-371. [DOI] [PubMed] [Google Scholar]
- 34.Johansson, A., R. Claesson, G. Belibasakis, E. Makoveichuk, L. Hanstrom, G. Olivecrona, G. Sandstrom, and S. Kalfas. 2001. Protease inhibitors, the responsible components for the serum-dependent enhancement of Actinobacillus actinomycetemcomitans leukotoxicity. Eur. J. Oral Sci. 109:335-341. [DOI] [PubMed] [Google Scholar]
- 35.Johansson, A., R. Claesson, L. Hanstrom, G. Sandstrom, and S. Kalfas. 2000. Polymorphonuclear leukocyte degranulation induced by leukotoxin from Actinobacillus actinomycetemcomitans. J. Periodontal Res. 35:85-92. [DOI] [PubMed] [Google Scholar]
- 36.Johansson, A., L. Hanstrom, and S. Kalfas. 2000. Inhibition of Actinobacillus actinomycetemcomitans leukotoxicity by bacteria from the subgingival flora. Oral Microbiol. Immunol. 15:218-225. [DOI] [PubMed] [Google Scholar]
- 37.Johansson, A., G. Sandstrom, R. Claesson, L. Hanstrom, and S. Kalfas. 2000. Anaerobic neutrophil-dependent killing of Actinobacillus actinomycetemcomitans in relation to the bacterial leukotoxicity. Eur. J. Oral Sci. 108:136-146. [DOI] [PubMed] [Google Scholar]
- 38.Kadowaki, T., A. Baba, N. Abe, R. Takii, M. Hashimoto, T. Tsukuba, S. Okazaki, Y. Suda, T. Asao, and K. Yamamoto. 2004. Suppression of pathogenicity of Porphyromonas gingivalis by newly developed gingipain inhibitors. Mol. Pharmacol. [DOI] [PubMed]
- 39.Kam, C. M., J. E. Kerrigan, K. M. Dolman, R. Goldschmeding, A. E. dem Borne, and J. C. Powers. 1992. Substrate and inhibitor studies on proteinase 3. FEBS Lett. 297:119-123. [DOI] [PubMed] [Google Scholar]
- 40.Kelk, P., R. Claesson, L. Hanstrom, U. H. Lerner, S. Kalfas, and A. Johansson. 2005. Abundant secretion of bioactive interleukin-1beta by human macrophages induced by Actinobacillus actinomycetemcomitans leukotoxin. Infect. Immun. 73:453-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kleinfelder, J. W., H. H. Topoll, H. R. Preus, R. F. Muller, D. E. Lange, and W. Bocker. 1996. Microbiological and immunohistological findings in a patient with Papillon-Lefevre syndrome. J. Clin. Periodontol. 23:1032-1038. [DOI] [PubMed] [Google Scholar]
- 42.Kressin, S., A. Herforth, S. Preis, V. Wahn, and H. G. Lenard. 1995. Papillon-Lefevre syndrome—successful treatment with a combination of retinoid and concurrent systematic periodontal therapy: case reports. Quintessence Int. 26:795-803. [PubMed] [Google Scholar]
- 43.Lally, E. T., I. R. Kieba, A. Sato, C. L. Green, J. Rosenbloom, J. Korostoff, J. F. Wang, B. J. Shenker, S. Ortlepp, M. K. Robinson, and P. C. Billings. 1997. RTX toxins recognize a beta2 integrin on the surface of human target cells. J. Biol. Chem. 272:30463-30469. [DOI] [PubMed] [Google Scholar]
- 44.Lehrer, R. I., and T. Ganz. 1999. Antimicrobial peptides in mammalian and insect host defence. Curr. Opin. Immunol. 11:23-27. [DOI] [PubMed] [Google Scholar]
- 45.Liu, R., C. Cao, H. Meng, and Z. Tang. 2000. Leukocyte functions in 2 cases of Papillon-Lefevre syndrome. J. Clin. Periodontol. 27:69-73. [DOI] [PubMed] [Google Scholar]
- 46.Lundgren, T., S. Renvert, P. N. Papapanou, and G. Dahlen. 1998. Subgingival microbial profile of Papillon-Lefevre patients assessed by DNA-probes. J. Clin. Periodontol. 25:624-629. [DOI] [PubMed] [Google Scholar]
- 47.Lyberg, T. 1982. Immunological and metabolical studies in two siblings with Papillon-Lefevre syndrome. J. Periodontal Res. 17:563-568. [DOI] [PubMed] [Google Scholar]
- 48.MacIvor, D. M., S. D. Shapiro, C. T. Pham, A. Belaaouaj, S. N. Abraham, and T. J. Ley. 1999. Normal neutrophil function in cathepsin G-deficient mice. Blood 94:4282-4293. [PubMed] [Google Scholar]
- 49.Mandell, G. L. 1974. Bactericidal activity of aerobic and anaerobic polymorphonuclear neutrophils. Infect. Immun. 9:337-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marsh, P. M. M. 1992. Oral microbiology. Chapman & Hall, New York, N.Y.
- 51.Miyasaki, K. T., M. E. Wilson, A. J. Brunetti, and R. J. Genco. 1986. Oxidative and nonoxidative killing of Actinobacillus actinomycetemcomitans by human neutrophils. Infect. Immun. 53:154-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ong, P. Y., T. Ohtake, C. Brandt, I. Strickland, M. Boguniewicz, T. Ganz, R. L. Gallo, and D. Y. Leung. 2002. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 347:1151-1160. [DOI] [PubMed] [Google Scholar]
- 53.Petit, M. D., T. J. M. van Steenbergen, L. M. Scholte, U. van der Velden, and J. de Graaff. 1993. Epidemiology and transmission of Porphyromonas gingivalis and Actinobacillus actinomycetemcomitans among children and their family members: a report of four surveys. J. Clin. Periodontol. 20:641-650. [DOI] [PubMed] [Google Scholar]
- 54.Pham, C. T. N., J. L. Ivanovich, S. Z. Raptis, B. Zehnbauer, and T. J. Ley. 2004. Papillon-Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J. Immunol. 173:7277-7281. [DOI] [PubMed] [Google Scholar]
- 55.Pratchyapruit, W. O., and P. Kullavanijaya. 2002. Papillon-Lefevre syndrome: a case report. J. Dermatol. 29:329-335. [DOI] [PubMed] [Google Scholar]
- 56.Putsep, K., G. Carlsson, H. G. Boman, and M. Andersson. 2002. Deficiency of antibacterial peptides in patients with Morbus Kostmann: an observation study. Lancet 360:1144-1149. [DOI] [PubMed] [Google Scholar]
- 57.Reeves, E. P., H. Lu, H. L. Jacobs, C. G. Messina, S. Bolsover, G. Gabella, E. O. Potma, A. Warley, J. Roes, and A. W. Segal. 2002. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416:291-297. [DOI] [PubMed] [Google Scholar]
- 58.Richardson, K. C., L. Jarett, and E. H. Finke. 1960. Embedding in epoxy resins for ultrathin sectioning in electron microscopy. Stain Technol. 35:313-325. [DOI] [PubMed] [Google Scholar]
- 59.Robertson, K. L., D. B. Drucker, J. James, A. S. Blinkhorn, S. Hamlet, and P. S. Bird. 2001. A microbiological study of Papillon-Lefevre syndrome in two patients. J. Clin. Pathol. 54:371-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rudiger, S., G. Petersilka, and T. F. Flemmig. 1999. Combined systemic and local antimicrobial therapy of periodontal disease in Papillon-Lefevre syndrome: a report of 4 cases. J. Clin. Periodontol. 26:847-854. [PubMed] [Google Scholar]
- 61.Salyers, A. and White, A. 1994. Bacterial Pathogenesis: a molecular approach. ASM Press, Washington, D.C.
- 62.Schroeder, H. E., R. A. Seger, H. U. Keller, and E. M. Rateitschak-Pluss. 1983. Behavior of neutrophilic granulocytes in a case of Papillon-Lefevre syndrome. J. Clin. Periodontol. 10:618-635. [DOI] [PubMed] [Google Scholar]
- 63.Slots, J., and M. Ting. 1999. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in human periodontal disease: occurrence and treatment. Periodontol. 2000 20:82-121. [DOI] [PubMed] [Google Scholar]
- 64.Smeianov, V., K. Scott, and G. Reid. 2000. Activity of cecropin P1 and FA-LL-37 against urogenital microflora. Microbes Infect. 2:773-777. [DOI] [PubMed] [Google Scholar]
- 65.Sorensen, O. E., P. Follin, A. H. Johnsen, J. Calafat, G. S. Tjabringa, P. S. Hiemstra, and N. Borregaard. 2001. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 97:3951-3959. [DOI] [PubMed] [Google Scholar]
- 66.Sorensen, O. E., L. Gram, A. H. Johnsen, E. Andersson, S. Bangsboll, G. S. Tjabringa, P. S. Hiemstra, J. Malm, A. Egesten, and N. Borregaard. 2003. Processing of seminal plasma hCAP-18 to ALL-38 by gastricsin: a novel mechanism of generating antimicrobial peptides in vagina. J. Biol. Chem. 278:28540-28546. [DOI] [PubMed] [Google Scholar]
- 67.Stabholz, A., N. S. Taichman, and W. A. Soskolne. 1995. Occurrence of Actinobacillus actinomycetemcomitans and anti-leukotoxin antibodies in some members of an extended family affected by Papillon-Lefevre syndrome. J. Periodontol. 66:653-657. [DOI] [PubMed] [Google Scholar]
- 68.Stalder, J. F., T. P. Huu, B. Dreno, A. Vergracht, J. D. Bignon, and P. Litoux. 1989. Defective leucocyte adhesion in Papillon-Lefevre syndrome. Br. J. Dermatol. 121:668-669. [DOI] [PubMed] [Google Scholar]
- 69.Tanaka, D., K. T. Miyasaki, and R. I. Lehrer. 2000. Sensitivity of Actinobacillus actinomycetemcomitans and Capnocytophaga spp. to the bactericidal action of LL-37: a cathelicidin found in human leukocytes and epithelium. Oral Microbiol. Immunol. 15:226-231. [DOI] [PubMed] [Google Scholar]
- 70.Tinanoff, N., J. M. Tanzer, K. S. Kornman, and E. G. Maderazo. 1986. Treatment of the periodontal component of Papillon-Lefevre syndrome. J. Clin. Periodontol. 13:6-10. [DOI] [PubMed] [Google Scholar]
- 71.Tinanoff, N., P. Tempro, and E. G. Maderazo. 1995. Dental treatment of Papillon-Lefevre syndrome: 15-year follow-up. J. Clin. Periodontol. 22:609-612. [DOI] [PubMed] [Google Scholar]
- 72.Toomes, C., J. James, A. J. Wood, C. L. Wu, D. McCormick, N. Lench, C. Hewitt, L. Moynihan, E. Roberts, C. G. Woods, A. Markham, M. Wong, R. Widmer, K. A. Ghaffar, M. Pemberton, I. R. Hussein, S. A. Temtamy, R. Davies, A. P. Read, P. Sloan, M. J. Dixon, and N. S. Thakker. 1999. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat. Genet. 23:421-424. [DOI] [PubMed] [Google Scholar]
- 73.Ullbro, C., C. G. Crossner, T. Lundgren, P. A. Stalblad, and S. Renvert. 2000. Osseointegrated implants in a patient with Papillon-Lefevre syndrome: a 4 1/2-year follow up. J. Clin. Periodontol. 27:951-954. [DOI] [PubMed] [Google Scholar]
- 74.Van Dyke, T. E., M. A. Taubman, J. L. Ebersole, A. D. Haffajee, S. S. Socransky, D. J. Smith, and R. J. Genco. 1984. The Papillon-Lefevre syndrome: neutrophil dysfunction with severe periodontal disease. Clin. Immunol. Immunopathol. 31:419-429. [DOI] [PubMed] [Google Scholar]
- 75.Velazco, C. H., C. Coelho, F. Salazar, A. Contreras, J. Slots, and J. J. Pacheco. 1999. Microbiological features of Papillon-Lefevre syndrome periodontitis. J. Clin. Periodontol. 26:622-627. [DOI] [PubMed] [Google Scholar]
- 76.Wara-aswapati, N., J. Lertsirivorakul, T. Nagasawa, Y. Kawashima, and I. Ishikawa. 2001. Papillon-Lefevre syndrome: serum immunoglobulin G (IgG) subclass antibody response to periodontopathic bacteria. A case report. J. Periodontol. 72:1747-1754. [DOI] [PubMed] [Google Scholar]
- 77.Welch, R. A. 1991. Pore-forming cytolysins of gram-negative bacteria. Mol. Microbiol. 5:521-528. [DOI] [PubMed] [Google Scholar]
- 78.Zanetti, M., R. Gennaro, and D. Romeo. 1995. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 374:1-5. [DOI] [PubMed] [Google Scholar]