

Abstract
SLC25A19 mutations cause Amish lethal microcephaly (MCPHA), which markedly retards brain development and leads to α-ketoglutaric aciduria. Previous data suggested that SLC25A19, also called DNC, is a mitochondrial deoxyribonucleotide transporter. We generated a knockout mouse model of Slc25a19. These animals had 100% prenatal lethality by embryonic day 12. Affected embryos at embryonic day 10.5 have a neural-tube closure defect with ruffling of the neural fold ridges, a yolk sac erythropoietic failure, and elevated α-ketoglutarate in the amniotic fluid. We found that these animals have normal mitochondrial ribo- and deoxyribonucleoside triphosphate levels, suggesting that transport of these molecules is not the primary role of SLC25A19. We identified thiamine pyrophosphate (ThPP) transport as a candidate function of SLC25A19 through homology searching and confirmed it by using transport assays of the recombinant reconstituted protein. The mitochondria of Slc25a19−/− and MCPHA cells have undetectable and markedly reduced ThPP content, respectively. The reduction of ThPP levels causes dysfunction of the α-ketoglutarate dehydrogenase complex, which explains the high levels of this organic acid in MCPHA and suggests that mitochondrial ThPP transport is important for CNS development.
Keywords: development, mitochondrial transporter, mouse model, thiamine pyrophosphate deficiency, neural tube defect
Microcephaly can be caused by many genetic and environmental factors. Amish lethal microcephaly (MCPHA) [Online Mendelian Inheritance in Man (OMIM) 607196; www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM] is inherited in an autosomal recessive pattern and includes severe congenital microcephaly (head circumference >6 SD below the mean), elevated levels (10–100 times) of α-ketoglutaric acid (AKGuria), and death, usually by 6 months of age (1).
Previously, we found a causative mutation in patients with MCPHA (c.530G>C which predicts p.G177A) in SLC25A19, which encodes a mitochondrial membrane carrier superfamily member (OMIM 606521) (2). The SLC25A19 gene also is called DNC, which stands for deoxynucleotide carrier (GenBank accession no. NM_021734) (3). SLC25A19 contains three typical mitochondrial inner mitochondrial membrane carrier motifs (4). The p.G177A substitution was absent in 252 control chromosomes. In addition, this glycine was conserved in 272 of 275 sequences in the Conserved Domain Database (www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml). Functional data showed that SLC25A19 transported deoxynucleotides across membranes in an in vitro assay system (3). Finally, we showed that G177A SLC25A19 had no activity in this assay (2), indicating that MCPHA is associated with a SLC25A19 loss-of-function mutation. These data did not prove that SLC25A19 mutations caused MCPHA, because it was possible that there was a mutation in another gene on the mutant Amish MCPHA haplotype. Although it has been demonstrated that SLC25A19 transported ribo- and deoxyribonucleotides in vitro, it was not known whether that was its primary function. Therefore, we designed experiments to address the issue of causation and the function of SLC25A19. The results show that knockout of Slc25a19 causes CNS defects and AKGuria and that the pathogenic mechanism of SLC25A19 mutations in MCPHA may be a decrease in mitochondrial thiamine pyrophosphate (ThPP) levels.
Results
The strategy used to generate Slc25a19−/− mice is shown in Fig. 1. Homologous recombination of the targeting vector replaced exons 4–6 of Slc25a19 with a NeoR gene. If this transcript was stable, the SLC25A19 protein would be missing amino acids 97–258 and should be nonfunctional. Southern blot analysis showed that 6 of 121 ES cell lines underwent proper recombination (data not shown), and 1 was chosen for blastocyst injection. Three chimeric mice showed germ-line transmission of the mutant allele. The chimeras were mated to black Swiss and 129SvEv female mice. No genetic background differences were noted.
Fig. 1.

Diagram of the Slc25a19 knockout strategy. A 2.5-kb EcoRI (E)/BamHI (B) fragment (5′) and a 3.9-kb SpeI (S)/EcoRI (3′) fragment were cloned into pPNT-far. The construct is missing exons 4–6. Transfected ES cell clones were digested with either BamHI (B) (3′ and neo probes) or NheI (N) (5′ probe) to test for homologous recombination. Thick lines represent mouse sequence, thin lines represent vector sequence, and the arrow represents the NeoR cassette. The stippled boxes represent the Southern blot analysis probes. Black numbered boxes are exons (the translation start site is in exon 2).
Slc25a19+/− animals appeared normal. No homozygous mutant pups were found among 211 pups from Slc25a19+/− intercrosses, suggesting embryonic lethality. Embryos were collected from timed matings, examined, and genotyped. The optimal timing for embryo evaluations was embryonic day (E)10.5; however, mutants were recognizable at E8.5. No Slc25a19−/− embryos were found after E12.5. Most E11.5 mutants were necrotic, indicating death by E11. Fewer than one-quarter of the E10.5 embryos were −/−, suggesting earlier mortality.
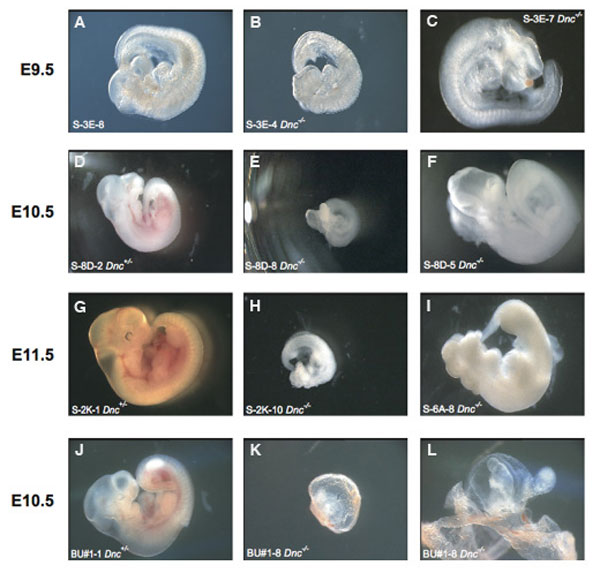
The open neural tube was the most striking feature of the Slc25a19−/− embryos (Fig. 2 and Fig. 8, which is published as supporting information on the PNAS web site). The exencephaly varied but commonly involved the entire brain. The edges of the open neural tube were ruffled and fused at the hindbrain (Figs. 2 and 8). In the trunk, the fused neural tube was convoluted, suggesting irregular relative growth rates of neural to nonneural tissue. Mutant embryos were smaller than unaffected littermates (Fig. 2 A and B). No malformations were noted in the trunk or tail region. Some Slc25a19−/− embryos apparently arrested earlier in development, resulting in amorphic embryos, but this was not further evaluated (Fig. 2D).
Fig. 2.

Examples of unaffected and mutant embryos at E10.5. (A) Unaffected Slc25a19+/− embryo. (B) Slc25a19−/− embryo taken at the same magnification as in A. (C) Higher magnification of embryo in B. Note the ruffling along the open neural tube (yellow arrowheads), point of fusion of neural tube (green arrowhead), and lack of color in heart and blood vessels. (D) Example of an amorphous embryo arrested early in development.
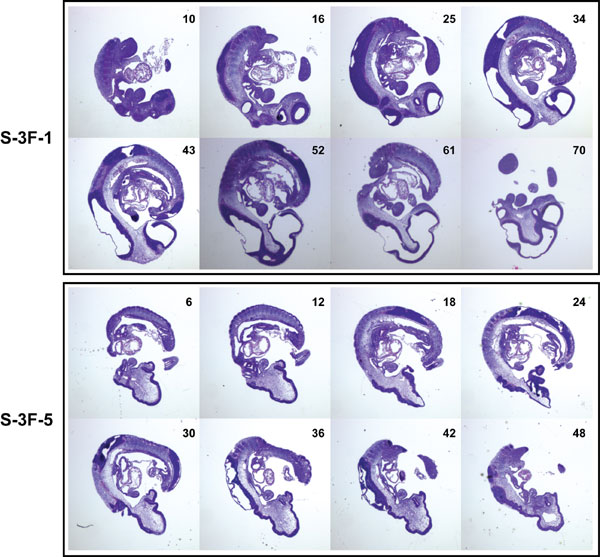
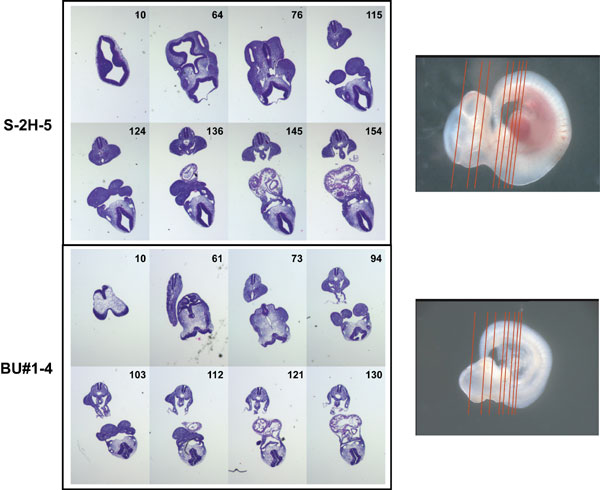
Serial sections showed that loss of Slc25a19 primarily affects anterior structures. Neural ectoderm was present on the outer edges of the forebrain and midbrain regions with no evidence of ventricle formation (Fig. 9, which is published as supporting information on the PNAS web site). Coronal sections show that the neural tube was not fully fused at the level of the heart (Fig. 3 and Fig. 10, which is published as supporting information on the PNAS web site). We conclude that a Slc25a19 loss-of-function mutation causes severe midgestation CNS defects and, occasionally, earlier developmental arrest.
Fig. 3.

Coronal sections of unaffected (Left) and mutant (Right) E10.5 embryos. These images are higher magnifications of the last of the serial sections shown in Fig. 10 (154 and 130, respectively). Yellow arrowheads mark the erythrocytes in the heart and vessels in the section from the unaffected embryo. The rostral neural tube is not fused (green arrows) but is fused caudally (red arrow) in the mutant embryo.
Sections through the heart (Fig. 3) showed very few erythrocytes in the Slc25a19−/− embryos. Blood smears from the yolk sacs of E10.5 embryos showed that unaffected embryos had many normal erythrocytes, whereas mutants had few erythrocytes and few of those were normal (Fig. 11, which is published as supporting information on the PNAS web site).
To determine whether the mutants had elevated α-ketoglutarate (AKG) as found in humans with MCPHA, we devised techniques to sample amniotic fluid from midgestation mouse embryos. Amniotic fluid AKG was elevated 5× in mutants, compared with controls (Fig. 4). This finding shows that the two major findings in human MCPHA, CNS defects and AKGuria, are caused by a knockout mutation of Slc25a19.
Fig. 4.

AKG levels in amniotic fluid from 25 embryos at E10.5. AKG was measured by using tandem MS and an internal standard. Five embryos were Slc25a19+/+ (triangles), 13 embryos were Slc25a19+/− (squares), and 7 embryos were Slc25a19−/− (circles). The samples represented by each color were measured during the same experiment. The mean values were 6.17 ± 0.72 μmol/liter for the unaffected and 30.8 ± 9.91 μmol/liter for the mutants (P = 0.0155 Mann–Whitney). The data also were dichotomized by using the mean of the entire data set (13.06) and tested with Fisher's exact test (P = 0.0004). The t test could not be used because the data were not normally distributed.
To study the cellular phenotype, murine embryonic fibroblasts (MEFs) were derived from E10.5 embryos and stained with antibodies to mitochondrial HSP70 and subunit I of the cytochrome c oxidase complex to determine whether there were any gross changes in mitochondrial numbers. No differences were observed (data not shown).
As SLC25A19 was known to transport deoxynucleotides (3), we hypothesized that the knockout would disrupt the mitochondrial nucleotide pool and cause depletion or deletion of mitochondrial DNA. We used quantitative PCR to detect mitochondrial DNA depletion in whole embryos, yolk sacs, and MEFs. No consistent differences in the ratio of mitochondrial to nuclear DNA were found (data not shown). We also used Southern blot analysis to screen for mitochondrial deletions, but the results were normal (data not shown). We conclude that there was no evidence for mitochondrial genomic abnormalities in Slc25a19−/− animals.
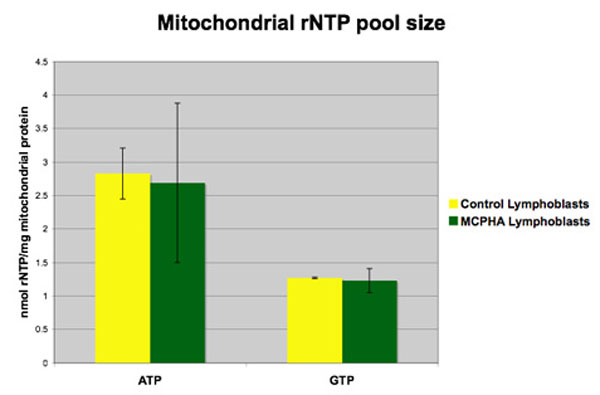
We then speculated that disruption of nucleotide transport might alter mitochondrial function without affecting DNA synthesis or integrity. To test this idea, mitochondria from wild-type and mutant MEFs were assayed directly for dNTP levels (Fig. 5). No significant differences were found in the dATP, dCTP, and dGTP levels. Surprisingly, the dTTP level was slightly higher in the mutant mitochondria. Similar results were obtained with mitochondria isolated from human lymphoblast cell cultures derived from two children with MCPHA and two controls. SLC25A19 previously has been shown to transport ribonucleotides (3), so measurements of mitochondrial rNTP pools from these lymphoblasts also were performed (Fig. 12, which is published as supporting information on the PNAS web site). Because of the limited sensitivity of this assay, only levels of ATP and GTP were obtained. Again, no differences in the level of these nucleotides in patient samples compared with control samples were seen. We conclude that loss-of-function mutations in SLC25A19 do not disrupt mitochondrial deoxynucleotide pools in mice or in humans.
Fig. 5.

Mitochondrial dNTP levels in wild-type and mutant MEFs and human lymphoblasts. For the MEFs, mitochondria were isolated from a wild-type (blue bars) or mutant (red bars) culture on two occasions. dNTP levels were measured in duplicate and averaged (±SD). Lymphoblast nucleotide levels were determined from mitochondria isolated from two control (yellow bars) or patient (green bars) cultures and averaged (±SD) according to affection status.
While this study was underway, a novel mitochondrial transporter, Tpc1p, was discovered in Saccharomyces cerevisiae (5). This protein transports ThPP into the mitochondria in exchange for thiamine monophosphate (ThMP) and is the yeast protein most similar to human SLC25A19. Because of the similarity of SLC25A19 to this yeast protein (28% amino acid identity and 50% similarity), the ability of SLC25A19 to transport ThPP and ThMP was tested by using phospholipid vesicles reconstituted with recombinant wild-type and G177A mutant human SLC25A19. Because SLC25A19 catalyzes only exchange and not uniport of substrates (3), and because ThPP and ThMP are not available in radioactive form, their transport was tested by measuring the uptake of α-35S-dATP into liposomes reconstituted with SLC25A19 that had been preloaded with ThPP or ThMP (Fig. 6). With the wild-type protein, similar α-35S-dATP uptake was found with internal ThPP as with internal dATP. α-35S-dATP also exchanged with internal ThMP, although to a lower extent. In contrast, thiamine was not exchanged. The G177A mutant diminished transport by ≈70% compared with wild type. Partial reduction of the G177A mutant activity, as shown in Fig. 6, was observed when using proteins overexpressed in Escherichia coli at 25°C (see Materials and Methods), and a complete inhibition was observed when expression was done at 37°C (data not shown and ref. 2).
Fig. 6.

Transport assays of wild-type and mutant SLC25A19. Proteoliposomes reconstituted with recombinant wild-type (gray bars) and G177A mutant (white bars) SLC25A19 were preloaded internally with 200 μM each dATP, ThPP, ThMP, or thiamine. Transport was started with 20 μM α-35S-dATP and terminated at 2 min. The values are means ±SD of at least three experiments.
To test whether the loss of SLC25A19 caused changes in levels of ThPP and ThMP, mitochondrial and cytosolic fractions from wild-type and mutant human and mouse cells were isolated. ThPP and ThMP levels were assayed by MS and compared with total cellular protein (Table 1). No ThPP or ThMP was detected in the mitochondrial fraction of Slc25a19−/− MEFs. In the cytosolic fraction, a 2.5× and 3.7× increase in ThPP and ThMP compared with wild-type MEFs was seen, respectively. In the mitochondrial fraction of human lymphoblasts, ThPP and ThMP levels were decreased 10× and 17×, respectively, compared with controls. The ThPP and ThMP were increased 1.4× and 1.8× in the cytosol compared with controls.
Table 1.
Levels of ThPP and ThMP
| Cell type | Mitochondria |
Postmitochondrial supernatant |
||
|---|---|---|---|---|
| ThPP* | ThMP* | ThPP* | ThMP* | |
| Wild-type MEFs | 2.02 ± 0.25 | 0.19 ± 0.02 | 2.75 ± 0.91 | 0.28 ± 0.04 |
| Mutant MEFs | Not detectable | Not detectable | 6.88 ± 0.42† | 1.05 ± 0.05† |
| Control lymphoblasts | 1.03 ± 0.25 | 0.37 ± 0.01 | 3.23 ± 0.26 | 0.22 ± 0.01 |
| Patient lymphoblasts | 0.10 ± 0.01† | 0.02 ± 0.01† | 4.63 ± 0.21† | 0.39 ± 0.12† |
Data represent the mean ± SEM of three independent experiments.
*Data are presented as ng/mg of total cellular protein.
†Values are significantly different from control (P < 0.05).
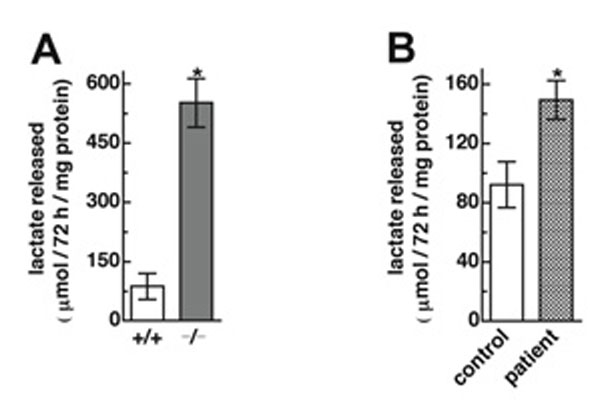
To assess the consequences of lack of ThPP in the mitochondria, the activities of the ThPP-requiring enzyme complexes AKG dehydrogenase (KGDH) and pyruvate dehydrogenase (PDH) from mutant and wild-type mitochondria were assayed directly in an in vitro system. KDGH and PDH complex activities were severely reduced in mutant fibroblasts (Fig. 7 A and C). Addition of ThPP to the assay resulted in full recovery of activity. PDH complex activity also was measured indirectly by acetylcarnitine formation, which confirmed the results of the direct measurement (data not shown). KGDH and PDH complex activities in patient mitochondria also were decreased but less severely (Fig. 7 B and D). The lactate level in the medium was measured in both MEFs and lymphoblasts. In mutant MEFs, a 6.3× increase and in patient lymphoblasts, a 1.6× increase in lactate was seen (Fig. 13, which is published as supporting information on the PNAS web site). We conclude that SLC25A19 transports ThPP. Mitochondrial ThPP levels were undetectable in mutant cells from Slc25a19−/− mice and markedly reduced in human cells with a point mutation. The data suggest that reduced mitochondrial levels of ThPP contribute to the metabolic abnormalities in human MCPHA.
Fig. 7.

KGDH and PDH complex assays. The KGDH (A and B) and PDH (C and D) complex activities were assayed by measuring NADH formed in the presence or absence of ThPP. (A and C) Wild-type (white bar) and knockout (gray bar) MEFs. (B and D) Human lymphoblasts from a normal individual (white bar) and a MCPHA patient (cross-hatched bar). The data represent the mean ± SEM of three independent experiments in duplicate. ANOVA analysis (two-way) was applied to compare the two groups, with and without ThPP, and ∗∗ denotes significance compared with control (P < 0.05).
Discussion
These data show that Slc25a19 is essential for murine development. All mice homozygous for this knockout mutation die before E12. The homozygous animals are growth-retarded and have abnormal CNS development with an open and convoluted neural tube. The animals also manifest erythropoietic failure, being nearly devoid of erythrocytes at E9.5. In addition, the AKG levels in the amniotic fluid in 5 of 7 affected embryos were elevated in comparison to 18 heterozygous and homozygous wild-type littermates. That the amniotic fluid AKG elevation is less dramatic than the AKGuria in the MCPHA patients is expected because the mouse embryos die before kidney formation and therefore do not concentrate the metabolites. Finally, a subset of embryos appears to undergo an earlier gestational demise because of an unknown mechanism.
Previous data suggested that the function of SLC25A19 was to transport deoxynucleotides across the inner mitochondrial membrane (3). We hypothesized that a loss of this function would lead to abnormally low levels of mitochondrial matrix deoxyribonucleotides and depletion or deletions in mitochondrial DNA (6), with abnormal mitochondria and reduced respiratory function (7, 8). However, there were no gross changes in the number or morphology of the mitochondria, nor did we detect mitochondrial deletions in the knockout mice, as would be expected from abnormal nucleotide levels. Although SLC25A19 may be able to transport dNTPs in vivo, this function is not critical because loss of this gene does not alter dNTP levels in mitochondria of mutant cells. That the physiological role of SLC25A19 is something other than dNTP transport is further supported by a recent article by Lam et al. (9). They showed that SLC25A19 is not associated with mitochondrial DNA depletion caused by dideoxynucleoside analogs used in antiretroviral therapy or with mitochondrial dNTP uptake.
The recent discovery of the Tpc1p gene (5) in yeast (Swiss-Prot entry P53257, YGR096W) suggested another function for SLC25A19. Tpc1p transports ThPP produced in the cytosol into the mitochondria in exchange for mitochondrially generated ThMP, which then is metabolized and converted back to ThPP (5). In an in vitro exchange assay, we found that a protein containing the G177A mutation reduced ThPP and ThMP transport by ≈70% compared with wild-type protein. Mitochondrial ThPP was not detectable in MEFs from knockout mice and markedly reduced in lymphoblasts from patients with MCPHA. We also showed that the mitochondrial ThPP-dependent enzyme complex PDH and KGDH activities were lower in SLC25A19 mutant cells than in controls. The addition of ThPP to the PDH and KGDH complex assay mixtures restored full activity in both the mouse and human cell lines.
We recognized differences comparing the murine Slc25a19 knockout to the human MCPHA phenotype. The embryonic lethality of the Slc25a19−/− animals illustrates the increased severity of the mouse model. Humans with MCPHA are viable and appear to be born at ratios (to unaffected children) that show that the Amish SLC25A19 mutation that causes MCPHA does not cause pregnancy loss at an appreciable rate. The neural tube defect and arrest of brain formation in the mouse as compared with the microcephaly in MCPHA patients also illustrates the increased severity in the mouse model. The murine erythropoietic defect does not mirror a recognized aspect of human MCPHA [patients with MCPHA have normal blood counts (R. I. Kelley, personal communication)].
A likely explanation for the differences in the mouse and human phenotypes is that the human point mutation retains some ThPP transport activity, whereas the murine null mutation does not. That the mitochondrial ThPP levels and activities of mitochondrial ThPP-requiring enzymes were less severely reduced in cells from MCPHA patients than in knockout MEFs, and that the G177A mutant protein retained some activity when expressed at 25°C and not at 37°C, supports this hypothesis. However, alternative explanations must be considered. First, another transporter may have overlapping functions with SLC25A19, and there may be differences in the functional overlap in the two species. It also is possible that SLC25A19 has unrecognized functions in addition to nucleotide and ThPP transport, and that these functions are lost in the knockout mouse but retained in the human MCPHA mutation. Finally, it is possible that the mouse model is not ideal for these experiments. To address these issues, future studies will need to involve knockin constructs that model the human mutation.
Oxidative phosphorylation is important in early embryogenesis (10–13). Blocking the tricarboxylic acid cycle by targeted disruption of the murine dihydrolipoamide dehydrogenase (Dld) gene [encoding the E3 component of PDH, KGDH, and the branched-chain α-ketoacid dehydrogenase (BCKDH) complexes], or the gene for PDH E1α subunit (Pdha1), causes developmental arrest and lethality by E8.5 or E12.5, respectively (14, 15). We speculate that the growth retardation of the Slc25a19−/− embryos is caused by decreased flux through the PDH and KGDH complex reactions. The structural brain malformations of Slc25a19−/− embryos support the hypothesis that the developing brain has a higher need for oxidative metabolism than other organs do (16). In addition to the reduction of energy produced by oxidative metabolism in the Slc25a19−/− embryos and MCPHA patients caused by reduced PDH and KGDH complex activities, reduced Krebs cycle intermediates also could play a role in the phenotype of the mutants. In fact, the lower level of succinyl-CoA in the mitochondria, a product of the KGDH complex, may be the cause of erythropoietic failure because heme biosynthesis requires condensation of succinyl-CoA and glycine. Heme deficiency caused by disruption of the murine gene for this enzyme in erythroid cells (ALAS-E) caused anemia and death by E11.5 (17). In contrast to the Slc25a19−/− mutant, the ALAS-E-null embryos had some immature primitive nonhemoglobinized erythrocytes, suggesting that reduction of heme synthesis cannot solely be responsible for the lack of red cell development in the Slc25a19−/− embryos. This issue will need to be addressed by future studies.
In summary, we have made a mouse model for human Amish microcephaly by knocking out the murine Slc25a19 gene, an ortholog of human SLC25A19 or DNC. The mutant has a CNS and metabolic phenotype that is similar to the human disease. Loss of murine SLC25A19 function does not cause mitochondrial dNTP pool reduction, as previously predicted, but instead reduces mitochondrial ThPP. The reduction in ThPP inhibits the activity of key metabolic enzymes, including the KGDH complex, which explains the AKGuria in MCPHA and emphasizes the role of oxidative metabolism in early embryogenesis.
Materials and Methods
Generation of Slc25a19−/− Animals.
The pPNTDNC targeting construct (Fig. 1) was linearized with NotI. TC1 cells (2 × 107) were transfected with 25 μg of linear DNA in 1 ml of PBS with a Gene Pulser (Bio-Rad, Hercules, CA). Cells were changed at 24 h to medium with G418 and 5-iodo-2′-fluoro-2′deoxy-1-β-d-arabino-furanosyl-uracil (FIAU). Seven days later, colonies were selected and expanded for freezing and DNA analysis. Approximately 15 targeted ES cells were injected into each C57B6/J blastocyst. The blastocysts were implanted into 2.5-days postcoitum pseudopregnant NIH Swiss–Webster mice. Chimeras were mated to Black Swiss mice at 6 weeks of age to assess germ-line transmission.
Embryo Procedures.
Embryos were harvested at E8.5–E13.5, and yolk sacs were collected for genotyping. Most embryos were fixed in 4% paraformaldehyde overnight at 4°C, transferred to 100% methanol, and stored at −20°C. Paraffin embedding, sectioning, and hematoxylin and eosin staining of embryos were performed by Histoserve (Germantown, MD).
To collect fetal blood, an embryo in its yolk sac was put in a 24-well culture dish with 1 ml of PBS. The yolk sac and amnion were removed, allowing the yolk sac vessel blood to spill into the PBS. Cells were centrifuged, spread on slides, stained with Wright–Giemsa, and examined by microscopy.
To collect amniotic fluid, E10.5 embryos were dissected with the yolk sac intact. Embryos were transferred to a dry dish and rolled to remove PBS to avoid diluting the amniotic fluid. Amniotic fluid was collected with a 1-ml syringe and a 32-gauge needle. To minimize clogging, the needle was inserted into the amniotic cavity with the opening away from the embryo, and fluid was slowly drawn into the syringe.
Cell Culture.
MEFs were collected from E10.5 embryos, and yolk sacs were used for genotyping. Embryos were minced with forceps and put in a 12-well cell dish with DMEM with 10% FBS, penicillin/streptomycin, and glutamine. Adherent cells were expanded by using standard techniques. DNA was isolated periodically and genotyped. Lymphoblasts from two MCPHA patients (provided by R. I. Kelley, Kennedy Krieger Institute, Baltimore, MD) were grown in RPMI medium 1640 with 10% FBS, penicillin/streptomycin, and glutamine.
AKG Assay.
Aliquots (2–5 μl) of mouse amniotic fluid were pipetted in a glass tube, followed by 0.5 ml of water and 0.05 nmol of 2H4-AKG (Eurisotop, Saint-Aubin Cedex, France), for an internal standard. AKG was derivatized by adding 10 μl of 6 M HCl and 60 μl of 10 mg/ml pentafluorobenzylhydroxylamine for 30 min at 60°C. The derivatives were extracted by using a solid-phase extraction cartridge and eluted with 0.6 ml of methanol in an HPLC vial. Ten microliters of eluate was injected on a liquid chromatography-tandem MS system (API 3000; Applied Biosystems, Foster City, CA) with a C18 analytical column, and elution was done with a binary linear gradient. The tandem MS was used in the negative mode, and AKG and the internal standards were measured in the multiple-reaction mode.
Nucleotide Levels.
Mitochondrial nucleotide pools were prepared as per Song et al. (6). Briefly, MEFs were harvested in 20 150-mm dishes by trypsinization. Human lymphoblasts were collected from suspension culture in five T100 flasks. Mitochondrial isolation and preparation of extracts were as described in ref. 6. rNTP pools were determined by HPLC using a Hewlett–Packard 1050 system with 260-nm detection. Extracts were injected into an Altex Partisil 10 SAX ion-exchange column equilibrated in 75 mM NH4PO4 (pH 3.6). rNTPs were eluted with a NH4PO4 gradient buffer ranging from 0.075 M to 1.0 M (pH 3.6) at 1.2 ml/min. The ribonucleotides were identified by retention times and quantitated by peak area in comparison to authentic standards.
Overexpression and Purification of SLC25A19 and SLC25A19 Mutant.
Wild-type and G177A mutant SLC25A19 were produced as inclusion bodies in E. coli BL21 (DE3) as described in ref. 18, except that induction was carried out for 20 h at 25°C. Inclusion bodies were purified as described in ref. 5, except that the Triton buffer contained Hepes, pH 7.2 (instead of Pipes).
Reconstitution of SLC25A19 Proteins into Liposomes and Transport Measurements.
The recombinant proteins were reconstituted into liposomes in the presence of substrates, as described in ref. 19. External substrate was removed from proteoliposomes on Sephadex G-75 columns and preequilibrated with 50 mM NaCl and 10 mM Pipes at pH 6.8. Transport was started by adding 35S-dATP (PerkinElmer, Wellesley, MA) to proteoliposomes and terminated by addition of 0.1 mM p-chloromercuribenzene sulfonate (the “inhibitor-stop” method; see ref. 18). The amount of wild-type and SLC25A19 mutant proteins incorporated into liposomes varied from 16% to 27% of the protein added to the reconstitution mixture.
ThPP and ThMP Determination by MS.
ThPP and ThMP extraction from mitochondria and postmitochondrial supernatant was performed essentially as described by Pontarin et al. (20). Cells were suspended in 0.2 M mannitol/0.07 M sucrose/0.2 mM EGTA/0.5% BSA/10 mM Tris·HCl (pH 7.4) and homogenized and centrifuged as described in ref. 19. The mitochondrial extract was lyophilized overnight and frozen. The postmitochondrial supernatant was treated with 100% methanol, centrifuged, lyophilized overnight, and frozen. The samples were dissolved in water, filtered, and subjected to liquid chromatography-MS analysis.
A Quattro Premier mass spectrometer with an Alliance 2695 HPLC system (Waters, Milford, MA) was used for electrospray-ionization liquid chromatography-tandem MS analysis of ThPP and ThMP in the positive-ion mode. The multiple reaction monitoring transition monitored for ThPP was m/z 425.2 > 122.1 and for ThMP was m/z 345.2 > 122.1. Chromatographic resolution of ThPP and ThMP was achieved by using a Hypercarb porous graphitic carbon column (4.6 × 100 mm, 5-μm particle size; Thermo Electron, Waltham, MA) eluted with a linear gradient from 100% water with 0.1% TFA (initial phase) to 10% water containing 0.1% TFA/90% acetonitrile. HPLC flow was 1 ml/min and split to 200 μl/min before entering the MS source. Calibration curves were set at four concentrations by using standards processed under the same conditions as the samples. The best fit was determined by using regression analysis of the peak analyte area.
PDH and KGDH Complex Assays.
Mitochondria were isolated from human lymphoblasts and murine fibroblasts by using a kit (Pierce, Rockford, IL) with the Halt Protease Inhibitor Mixture (Pierce, product no. 78415) according to the manufacturer's instructions. PDH and KGDH complex activities were assayed essentially as by Munujos et al. (21). Lymphoblast and fibroblast mitochondria were preincubated in 10 mM Tris·HCl at pH 7.2 for 30 min at 30°C, solubilized with 0.1% (wt/vol) Triton X-100, and centrifuged at 10,000 × g for 10 min. Seventy microliters of supernatant (180–200 μg of protein) was added to 430 μl of a mixture containing 100 mM Tris·HCl at pH 7.6, 4 mM CoA, 4 mM NAD+, and 40 μM rotenone with or without 4 mM ThPP. The assay was started with 8 mM AKG or 8 mM pyruvate, and the formation of NADH followed at 340 nm.
Lactate Release in Extracellular Medium.
A total of 2 × 106 human lymphoblasts were grown in 10 ml of medium for 72 h, then spun down at 1,500 × g for 10 min. Then, 1 × 106 mouse embryo fibroblasts were grown in 75-cm2 flasks for 72 h (close to confluence); the medium was collected and centrifuged to remove cell debris. The lactate in the supernatants from both cell types was assayed as per Gutmann and Wahlefeld (22).
Supplementary Material
Acknowledgments
We thank G. Elliot, C. Rivas, R. Nussbaum, D. Bodine, L. Wheeler, W. Pavan, C. Yang, S. Loftus, D. Larson, J. Fekecs, and R. I. Kelley for their technical expertise, advice, and encouragement. This work was supported by funds from Intramural Research Program of the National Human Genome Research Institute Grant LS-45039 from the Army Research Office (to C.K.M.), the Ministero dell'Università e della Ricerca, Ministero della Salute, Ministero dell'Università e della Ricerca Grant L.488/92 (cluster 03), and European Commission Contract LSHM-CT-2004-503116.
Glossary
Abbreviations
- MCPHA
Amish lethal microcephaly
- En
embryonic day n
- AKG
α-ketoglutarate
- ThPP
thiamine pyrophosphate
- AKGuria
α-ketoglutaric acid
- MEF
murine embryonic fibroblast
- ThMP
thiamine monophosphate
- KGDH
AKG dehydrogenase
- PDH
pyruvate dehydrogenase.
Footnotes
The authors declare no conflict of interest.
References
- 1.Kelley RI, Robinson D, Puffenberger EG, Strauss KA, Morton DH. Am J Med Genet. 2002;112:318–326. doi: 10.1002/ajmg.10529. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg MJ, Agarwala R, Bouffard G, Davis J, Fiermonte G, Hilliard MS, Koch T, Kalikin LM, Makalowska I, Morton DH, et al. Nat Genet. 2002;32:175–179. doi: 10.1038/ng948. [DOI] [PubMed] [Google Scholar]
- 3.Dolce V, Fiermonte G, Runswick MJ, Palmieri F, Walker JE. Proc Natl Acad Sci USA. 2001;98:2284–2288. doi: 10.1073/pnas.031430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmieri F. Pflügers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 5.Marobbio CM, Vozza A, Harding M, Bisaccia F, Palmieri F, Walker JE. EMBO J. 2002;21:5653–5661. doi: 10.1093/emboj/cdf583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song S, Wheeler LJ, Mathews CK. J Biol Chem. 2003;278:43893–43896. doi: 10.1074/jbc.C300401200. [DOI] [PubMed] [Google Scholar]
- 7.Holmuhamedov E, Jahangir A, Bienengraeber M, Lewis LD, Terzic A. Mitochondrion. 2003;3:13–19. doi: 10.1016/S1567-7249(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 8.Miranda S, Foncea R, Guerrero J, Leighton F. Biochem Biophys Res Commun. 1999;258:44–49. doi: 10.1006/bbrc.1999.0580. [DOI] [PubMed] [Google Scholar]
- 9.Lam W, Chen C, Ruan S, Leung CH, Cheng YC. Mol Pharmacol. 2005;67:408–416. doi: 10.1124/mol.104.007120. [DOI] [PubMed] [Google Scholar]
- 10.Johnson MT, Mahmood S, Patel MS. J Biol Chem. 2003;278:31457–31460. doi: 10.1074/jbc.R300002200. [DOI] [PubMed] [Google Scholar]
- 11.Hance N, Ekstrand MI, Trifunovic A. Hum Mol Genet. 2005;14:1775–1783. doi: 10.1093/hmg/ddi184. [DOI] [PubMed] [Google Scholar]
- 12.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 13.Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, Williams RS. Cell. 2000;101:389–399. doi: 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- 14.Johnson MT, Yang HS, Magnuson T, Patel MS. Proc Natl Acad Sci USA. 1997;94:14512–14517. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson MT, Mahmood S, Hyatt SL, Yang HS, Soloway PD, Hanson RW, Patel MS. Mol Genet Metab. 2001;74:293–302. doi: 10.1006/mgme.2001.3249. [DOI] [PubMed] [Google Scholar]
- 16.Brown GK, Otero LJ, LeGris M, Brown RM. J Med Genet. 1994;31:875–879. doi: 10.1136/jmg.31.11.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakajima O, Takahashi S, Harigae H, Furuyama K, Hayashi N, Sassa S, Yamamoto M. EMBO J. 1999;18:6282–6289. doi: 10.1093/emboj/18.22.6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fiermonte G, Walker JE, Palmieri F. Biochem J. 1993;294:293–299. doi: 10.1042/bj2940293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmieri F, Indiveri C, Bisaccia F, Iacobazzi V. Methods Enzymol. 1995;260:349–369. doi: 10.1016/0076-6879(95)60150-3. [DOI] [PubMed] [Google Scholar]
- 20.Pontarin G, Gallinaro L, Ferraro P, Reichard P, Bianchi V. Proc Natl Acad Sci USA. 2003;100:12159–12164. doi: 10.1073/pnas.1635259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munujos P, Coll-Canti J, Beleta J, Gonzalez-Sastre F, Gella FJ. Clin Chim Acta. 1996;255:13–25. doi: 10.1016/0009-8981(96)06385-1. [DOI] [PubMed] [Google Scholar]
- 22.Gutmann I, Wahlefeld A. In: Methods in Enzymatic Analysis. Bergmeyer HU, editor. New York: Academic; 1974. pp. 1464–1468. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}